
Pathogenic Insights into DNA Mismatch Repair (MMR) Genes–Proteins and Microsatellite Instability: Focus on Adrenocortical Carcinoma and Beyond
 , , and
, , and
Abstract
:1. Introduction
Aim
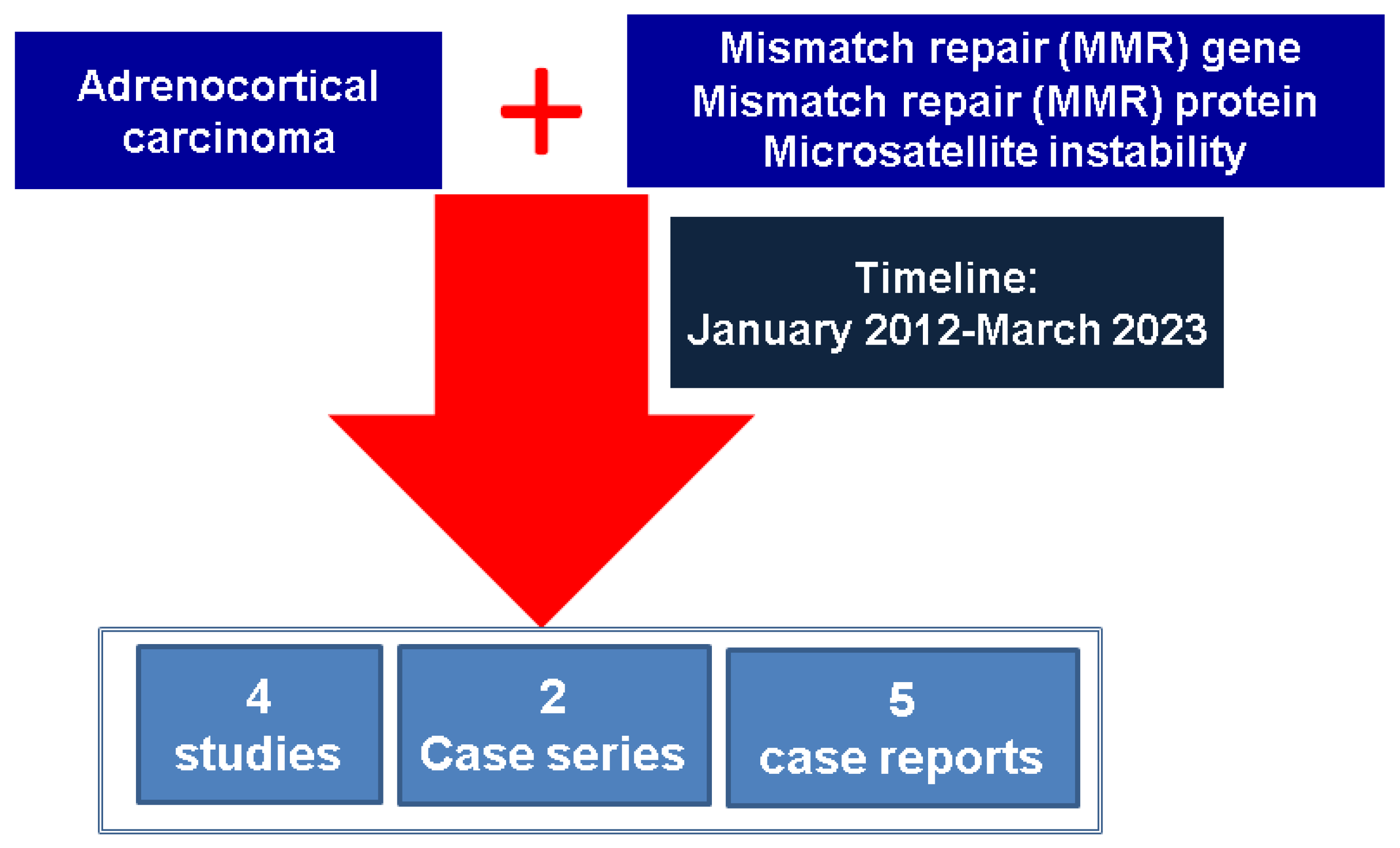
2. Materials and Methods
3. Results: MMR System and Endocrine Approach
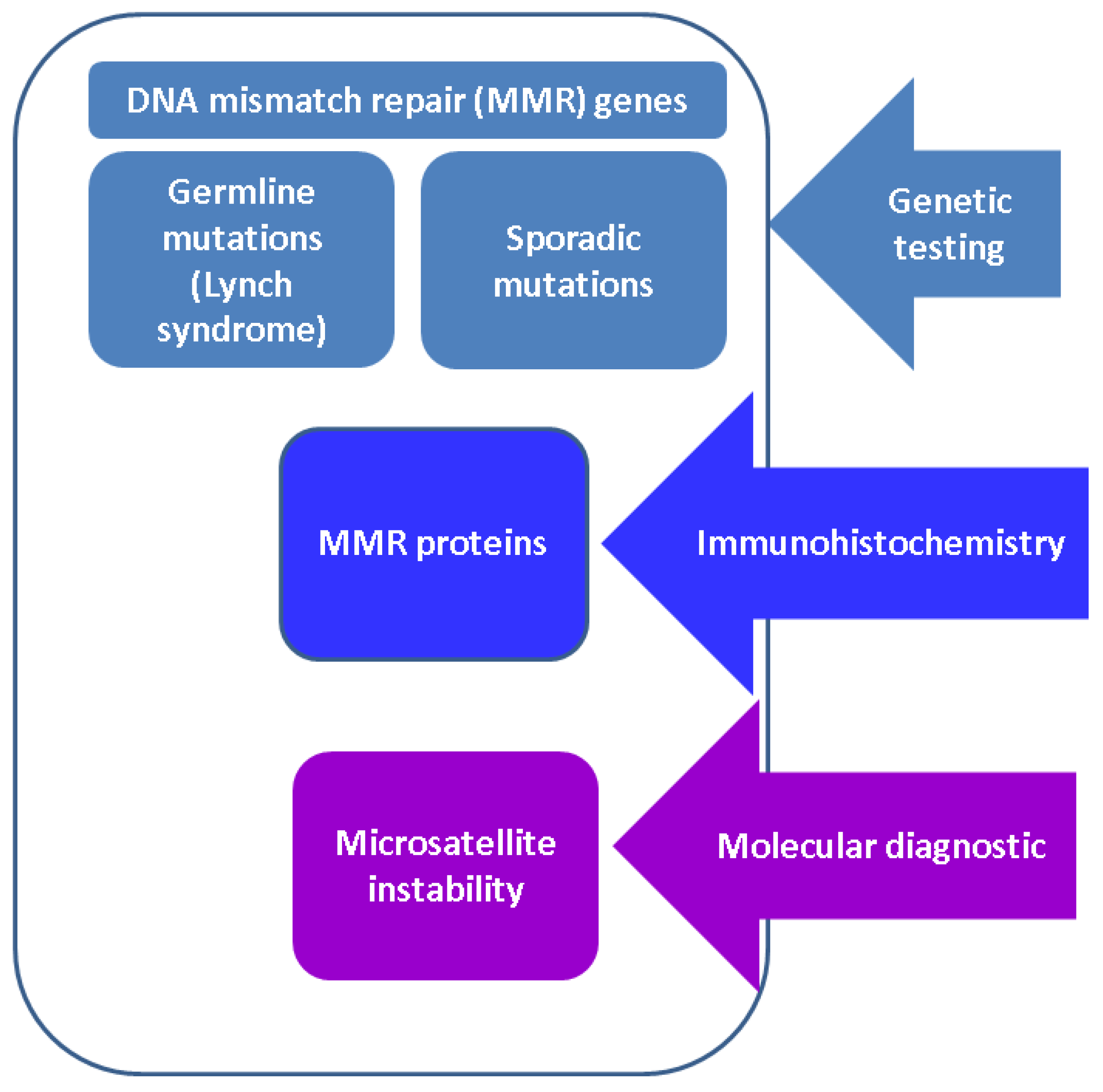
3.1. DNA MMR Genes: Focus on LS
3.2. ACC and MMR Status
4. Discussion
4.1. TC and MMR/MSI Involvement
4.2. NEN and MMR/MSI Assessment
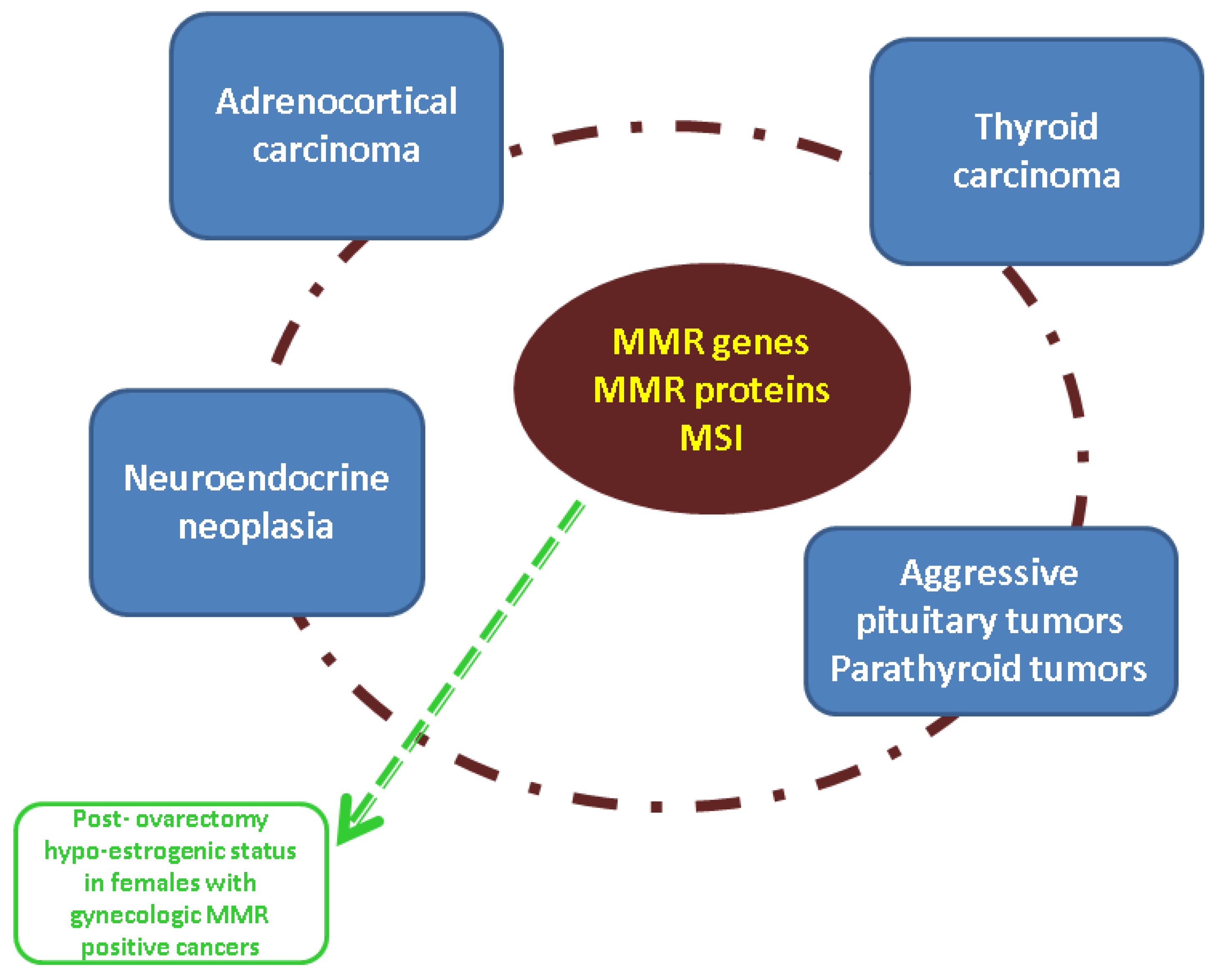
4.3. Integrating MMR Assessment into the Larger Panel of Endocrine Tumors
4.4. Endocrine Considerations in Patients with Germline MMR Mutations
4.5. Current Data on the MMR-MSI System in ACCs: Is There a Next Step?
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACC | adrenocortical carcinoma |
| APC | adenomatous polyposis coli |
| BC | breast cancer |
| CRC | colorectal cancer |
| EC | endometrial cancer |
| FNMTC | familial non-medullary thyroid cancer |
| HNPCC | hereditary non-polyposis colorectal cancer syndrome |
| IHC | immunohistochemistry |
| ICPI | immune checkpoint inhibitors |
| LS | Lynch syndrome |
| LOH | loss of heterozygosity |
| MMR | mismatch repair |
| MSI | microsatellite instability |
| MEN | multiple endocrine neoplasia |
| MiNEN | mixed neuroendocrine non-neuroendocrine neoplasm |
| NEN | neuroendocrine neoplasia |
| NGS | next-generation sequencing |
| OC | ovarian cancer |
| PCR | polymerase chain reaction |
| SNPs | single nucleotide polymorphisms |
| TC | thyroid cancer |
| WES | whole-exome sequencing |
References
- Wang, Q. Cancer predisposition genes: Molecular mechanisms and clinical impact on personalized cancer care: Examples of Lynch and HBOC syndromes. Acta Pharmacol. Sin. 2016, 37, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Latham, A.; Srinivasan, P.; Kemel, Y.; Shia, J.; Bandlamudi, C.; Mandelker, D.; Middha, S.; Hechtman, J.; Zehir, A.; Dubard-Gault, M.; et al. Microsatellite Instability Is Associated with the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol. 2019, 37, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Lynch, P.M.; Lanspa, S.J.; Snyder, C.L.; Lynch, J.F.; Boland, C.R. Review of the Lynch syndrome: History, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 2009, 76, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World J. Gastroenterol. 2015, 21, 9253–9261. [Google Scholar] [CrossRef]
- Li, X.; Liu, G.; Wu, W. Recent advances in Lynch syndrome. Exp. Hematol. Oncol. 2021, 10, 37. [Google Scholar] [CrossRef]
- Olave, M.C.; Graham, R.P. Mismatch repair deficiency: The what, how and why it is important. Genes Chromosom. Cancer 2021, 61, 314–321. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, Y.; Peng, G. Mismatch repair-based stratification for immune checkpoint blockade therapy. Am. J. Cancer Res. 2018, 8, 1977–1988. [Google Scholar]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; ten Broeke, S.W.; Plazzer, J.-P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef]
- Lynch, H.T.; Shaw, T.G. Practical genetics of colorectal cancer. Chin. Clin. Oncol. 2013, 2, 12–26. [Google Scholar] [CrossRef]
- Tomita, N.; Ishida, H.; Tanakaya, K.; Yamaguchi, T.; Kumamoto, K.; Tanaka, T.; Hinoi, T.; Miyakura, Y.; Hasegawa, H.; Takayama, T.; et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2020 for the Clinical Practice of Hereditary Colorectal Cancer. Int. J. Clin. Oncol. 2021, 26, 1353–1419. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, L.; Vakiani, E.; Shia, J. Detecting mismatch repair deficiency in solid neoplasms: Immunohistochemistry, microsatellite instability, or both? Mod. Pathol. 2022, 35, 1515–1528. [Google Scholar] [CrossRef] [PubMed]
- Lalli, E. ‘You cannot expect miracles to happen overnight’: Patience pays off when you wish to establish a new adrenocortical carcinoma cell line. Eur. J. Endocrinol. 2021, 185, C9–C11. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann. Transl. Med. 2020, 8, 1685. [Google Scholar] [CrossRef] [PubMed]
- Biller, L.H.; Creedon, S.A.; Klehm, M.; Yurgelun, M.B. Lynch Syndrome-Associated Cancers beyond Colorectal Cancer. Gastrointest. Endosc. Clin. N. Am. 2021, 32, 75–93. [Google Scholar] [CrossRef]
- Da Costa, W.H.; Jabboure, G.; Da Cunha, I.W. Urological cancer related to familial syndromes. Int. Braz. J. Urol. 2017, 43, 192–201. [Google Scholar] [CrossRef]
- Vanoli, A.; Grillo, F.; Furlan, D.; Arpa, G.; Grami, O.; Guerini, C.; Riboni, R.; Mastracci, L.; Di Sabatino, A. Small Bowel Epithelial Precursor Lesions: A Focus on Molecular Alterations. Int. J. Mol. Sci. 2021, 22, 4388. [Google Scholar] [CrossRef]
- Mete, O.; Erickson, L.A.; Juhlin, C.C.; de Krijger, R.R.; Sasano, H.; Volante, M.; Papotti, M.G. Overview of the 2022 WHO Classification of Adrenal Cortical Tumors. Endocr. Pathol. 2022, 33, 155–196. [Google Scholar] [CrossRef]
- Petr, E.J.; Else, T. Adrenocortical carcinoma (ACC): When and why should we consider germline testing? La Presse Médicale 2018, 47, e119–e125. [Google Scholar] [CrossRef]
- Else, T.; Rodriguez-Galindo, C. 5th International ACC Symposium: Hereditary Predisposition to Childhood ACC and the Associated Molecular Phenotype. Horm. Cancer 2016, 7, 36–39. [Google Scholar] [CrossRef]
- Kiseljak-Vassiliades, K.; Zhang, Y.; Bagby, S.M.; Kar, A.; Pozdeyev, N.; Xu, M.; Gowan, K.; Sharma, V.; Raeburn, C.D.; Albuja-Cruz, M.; et al. Development of new preclinical models to advance adrenocortical carcinoma research. Endocr.-Relat. Cancer 2018, 25, 437–451. [Google Scholar] [CrossRef]
- Raygada, M.; Raffeld, M.; Bernstein, A.; Miettinen, M.; Glod, J.; Hughes, M.S.; Reilly, K.; Widemann, B.; Del Rivero, J. Case report of adrenocortical carcinoma associated with double germline mutations in MSH2 and RET. Am. J. Med. Genet. Part A 2021, 185, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Domènech, M.; Grau, E.; Solanes, A.; Izquierdo, A.; Del Valle, J.; Carrato, C.; Pineda, M.; Dueñas, N.; Pujol, M.; Lázaro, C.; et al. Characteristics of Adrenocortical Carcinoma Associated with Lynch Syndrome. J. Clin. Endocrinol. Metab. 2021, 106, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Cheng, H.; Zhang, Y.; Zhou, Y. Identification of NDRG Family Member 4 (NDRG4) and CDC28 Protein Kinase Regulatory Subunit 2 (CKS2) as Key Prognostic Genes in Adrenocortical Carcinoma by Transcriptomic Analysis. Experiment 2021, 27, e928523. [Google Scholar] [CrossRef]
- Luo, G.; Chen, G.; Chen, P.; Zhou, J. Pan-cancer analysis of histone methyltransferase KMT2D with potential implications for prognosis and immunotherapy in human cancer. Comb. Chem. High Throughput Screen. 2022, 26, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Raymond, V.M.; Everett, J.N.; Furtado, L.V.; Gustafson, S.L.; Jungbluth, C.R.; Gruber, S.B.; Hammer, G.D.; Stoffel, E.M.; Greenson, J.K.; Giordano, T.J.; et al. Adrenocortical Carcinoma Is a Lynch Syndrome–Associated Cancer. J. Clin. Oncol. 2013, 31, 3012–3018. [Google Scholar] [CrossRef] [PubMed]
- Brondani, V.B.; Montenegro, L.; Lacombe, A.M.F.; Magalhães, B.M.; Nishi, M.Y.; Funari, M.F.d.A.; Narcizo, A.d.M.; Cardoso, L.C.; Siqueira, S.A.C.; Zerbini, M.C.N.; et al. High Prevalence of Alterations in DNA Mismatch Repair Genes of Lynch Syndrome in Pediatric Patients with Adrenocortical Tumors Carrying a Germline Mutation on TP53. Cancers 2020, 12, 621. [Google Scholar] [CrossRef]
- Jouinot, A.; Bertherat, J. Diseases Predisposing to Adrenocortical Malignancy (Li–Fraumeni Syndrome, Beckwith–Wiedemann Syndrome, and Carney Complex). Genet. Endocr. Dis. Syndr. 2019, 111, 149–169. [Google Scholar] [CrossRef]
- Challis, B.G.; Kandasamy, N.; Powlson, A.S.; Koulouri, O.; Annamalai, A.K.; Happerfield, L.; Marker, A.J.; Arends, M.J.; Nik-Zainal, S.; Gurnell, M. Familial Adrenocortical Carcinoma in Association with Lynch Syndrome. J. Clin. Endocrinol. Metab. 2016, 101, 2269–2272. [Google Scholar] [CrossRef]
- Kaur, R.J.; Pichurin, P.N.; Hines, J.M.; Singh, R.J.; Grebe, S.K.; Bancos, I. Adrenal Cortical Carcinoma Associated with Lynch Syndrome: A Case Report and Review of Literature. J. Endocr. Soc. 2019, 3, 784–790. [Google Scholar] [CrossRef]
- Wright, J.P.; Montgomery, K.W.; Tierney, J.; Gilbert, J.; Solórzano, C.C.; Idrees, K. Ectopic, retroperitoneal adrenocortical carcinoma in the setting of Lynch syndrome. Fam. Cancer 2018, 17, 381–385. [Google Scholar] [CrossRef]
- Araújo, A.N.; Bugalho, M.J. Advanced Adrenocortical Carcinoma: Current Perspectives on Medical Treatment. Horm. Metab. Res. 2021, 53, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Nevgi, A.; Klein, O.; Cheung, A.S. Sustained remission of Lynch syndrome-associated metastatic adrenocortical carcinoma following checkpoint inhibitor therapy-associated multiorgan autoimmunity. Clin. Endocrinol. 2020, 93, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Ardolino, L.; Hansen, A.; Ackland, S.; Joshua, A. Advanced Adrenocortical Carcinoma (ACC): A Review with Focus on Second-Line Therapies. Horm. Cancer 2020, 11, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Casey, R.; Giger, O.; Seetho, I.; Marker, A.; Pitfield, D.; Boyle, L.; Gurnell, M.; Shaw, A.; Tischkowitz, M.; Maher, E.; et al. Rapid disease progression in a patient with mismatch repair-deficient and cortisol secreting adrenocortical carcinoma treated with pembrolizumab. Semin. Oncol. 2018, 45, 151–155. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Fishbein, L.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Ross, J.S.; Darabi, S.; Demeure, M.J.; Kar, A.; Foust, L.J.; et al. Targeted genomic analysis of 364 adrenocortical carcinomas. Endocr.-Relat. Cancer 2021, 28, 671–681. [Google Scholar] [CrossRef]
- Cerquetti, L.; Bucci, B.; Carpinelli, G.; Lardo, P.; Proietti, A.; Saporito, R.; Rindi, G.; Petrangeli, E.; Toscano, V.; Stigliano, A. Antineoplastic Effect of a Combined Mitotane Treatment/Ionizing Radiation in Adrenocortical Carcinoma: A Preclinical Study. Cancers 2019, 11, 1768. [Google Scholar] [CrossRef]
- Aswath, K.; Welch, J.; Gubbi, S.; Veeraraghavan, P.; Avadhanula, S.; Gara, S.K.; Dikoglu, E.; Merino, M.; Raffeld, M.; Xi, L.; et al. Co-Occurrence of Familial Non-Medullary Thyroid Cancer (FNMTC) and Hereditary Non-Polyposis Colorectal Cancer (HNPCC) Associated Tumors—A Cohort Study. Front. Endocrinol. 2021, 12, 653401. [Google Scholar] [CrossRef]
- Santos, L.S.; Silva, S.N.; Gil, O.M.; Ferreira, T.C.; Limbert, E.; Rueff, J. Mismatch repair single nucleotide polymorphisms and thyroid cancer susceptibility. Oncol. Lett. 2018, 15, 6715–6726. [Google Scholar] [CrossRef]
- Kim, C.S.; Mandel, S.J. Contemporary Management of Thyroid Nodules. Annu. Rev. Med. 2022, 73, 517–528. [Google Scholar] [CrossRef]
- Luo, M.; Huang, Y.; Li, Y.; Zhang, Y. Metastatic rectal cancer to papillary thyroid carcinoma: A case report and review of literature. BMC Gastroenterol. 2020, 20, 136–138. [Google Scholar] [CrossRef]
- Fazekas-Lavu, M.; Parker, A.; Spigelman, A.D.; Scott, R.J.; Epstein, R.J.; Jensen, M.; Samaras, K. Thyroid cancer in a patient with Lynch syndrome—Case report and literature review. Ther. Clin. Risk Manag. 2017, 13, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Pozdeyev, N.; Rose, M.M.; Bowles, D.W.; Schweppe, R. Molecular therapeutics for anaplastic thyroid cancer. Semin. Cancer Biol. 2020, 61, 23–29. [Google Scholar] [CrossRef]
- Javid, M.; Sasanakietkul, T.; Nicolson, N.G.; Gibson, C.E.; Callender, G.G.; Korah, R.; Carling, T. DNA Mismatch Repair Deficiency Promotes Genomic Instability in a Subset of Papillary Thyroid Cancers. World J. Surg. 2018, 42, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Masago, K. Alteration of DNA mismatch repair capacity underlying the co-occurrence of non-small-cell lung cancer and nonmedullary thyroid cancer. Sci. Rep. 2021, 11, 3597. [Google Scholar] [CrossRef]
- Johnson, J.M.; Chen, J.; Ali, S.M.; Dardi, I.K.; Tuluc, M.; Cognetti, D.; Campling, B.; Sama, A.R. Molecular Profiling of Synchronous Colon Cancers and Anaplastic Thyroid Cancer in a Patient with Lynch Syndrome. J. Gastrointest. Cancer 2018, 49, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Genutis, L.K.; Tomsic, J.; Bundschuh, R.A.; Brock, P.L.; Williams, M.D.; Roychowdhury, S.; Reeser, J.W.; Frankel, W.L.; Alsomali, M.; Routbort, M.J.; et al. Microsatellite Instability Occurs in a Subset of Follicular Thyroid Cancers. Thyroid 2019, 29, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.L.; Schmid, K.W.; Czapiewski, P. The prevalence of DNA microsatellite instability in anaplastic thyroid carcinoma—Systematic review and discussion of current therapeutic options. Contemp. Oncol./Współczesna Onkol. 2021, 25, 213–223. [Google Scholar] [CrossRef]
- Wong, K.S.; Lorch, J.H.; Alexander, E.K.; Nehs, M.A.; Nowak, J.A.; Hornick, J.L.; Barletta, J.A. Clinicopathologic Features of Mismatch Repair-Deficient Anaplastic Thyroid Carcinomas. Thyroid 2019, 29, 666–673. [Google Scholar] [CrossRef]
- Pelizzo, M.; Pennelli, G.; Zane, M.; Galuppini, F.; Colletti, P.; Boschin, I.M.; Rubello, D. Papillary thyroid carcinoma (PTC) in Lynch syndrome: Report of two cases and discussion on Lynch syndrome behaviour and genetics. Biomed. Pharmacother. 2015, 74, 9–16. [Google Scholar] [CrossRef]
- Romaniuk, A.; Lyndin, M.; Smiyanov, V.; Sikora, V.; Rieznik, A.; Kuzenko, Y.; Budko, H.; Moskalenko, Y.; Karpenko, L.; Gladchenko, O. Primary multiple tumor with affection of the thyroid gland, uterus, urinary bladder, mammary gland and other organs. Pathol.-Res. Pract. 2017, 213, 574–579. [Google Scholar] [CrossRef]
- Feng, A.L.; Le, A.; Johnson, D.N.; Varvares, M.A. Multiple simultaneous head and neck cancers in Lynch syndrome: Case report and literature review. Laryngoscope 2018, 128, 2759–2761. [Google Scholar] [CrossRef] [PubMed]
- Pande, M.; Wei, C.; Chen, J.; Amos, C.I.; Lynch, P.M.; Lu, K.H.; Lucio, L.A.; Boyd-Rogers, S.G.; Bannon, S.A.; Mork, M.E.; et al. Cancer spectrum in DNA mismatch repair gene mutation carriers: Results from a hospital based Lynch syndrome registry. Fam. Cancer 2012, 11, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Caso, R.; Beamer, M.; Lofthus, A.D.; Sosin, M. Integrating surgery and genetic testing for the modern surgeon. Ann. Transl. Med. 2017, 5, 399. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S.; Gil, O.M.; Silva, S.N.; Gomes, B.C.; Ferreira, T.C.; Limbert, E.; Rueff, J. Micronuclei Formation upon Radioiodine Therapy for Well-Differentiated Thyroid Cancer: The Influence of DNA Repair Genes Variants. Genes 2020, 11, 1083. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, N.; Ghemigian, A.; Carsote, M.; Albu, S.E.; Terzea, D.; Valea, A. Thyroid nodules after initial evaluation by primary health care practitioners: An ultrasound pictorial essay. Arch. Balk. Med. Union. 2016, 51, 434–438. [Google Scholar]
- Verrienti, A.; Carbone, A.; Sponziello, M.; Pecce, V.; Cito, D.S.; Bruno, R. Papillary thyroid carcinoma as first and isolated neoplastic disease in a Lynch syndrome family member with a germline MLH1 mutation. Endocrine 2022, 77, 199–202. [Google Scholar] [CrossRef]
- Qiao, P.-P.; Tian, K.-S.; Han, L.-T.; Ma, B.; Shen, C.-K.; Zhao, R.-Y.; Zhang, Y.; Wei, W.-J.; Chen, X.-P. Correlation of mismatch repair deficiency with clinicopathological features and programmed death-ligand 1 expression in thyroid carcinoma. Endocrine 2022, 76, 660–670. [Google Scholar] [CrossRef]
- Pontoppidan, K.; Mathew, R.P.; Moran, G.W. Acute dysphagia after a normal endoscopy: Think outside the box. Clin. Med. 2013, 13, 315–316. [Google Scholar] [CrossRef]
- Mojtová, E.; Hanajíková, J.; Hamidová, O.; Bognár, G.; Dyttert, D.; Grigerová, M.; Kečkéš, Š.; Podoba, J. An incidental finding of pheochromocytoma in a 33-year-old patient with Lynch syndrome. Vnitr. Lek. 2020, 66, 80–84. [Google Scholar] [CrossRef]
- Riff, B.P.; Katona, B.; Wilkerson, M.; Nathanson, K.L.; Metz, D.C. HNPCC-Associated Pheochromocytoma. Pancreas 2015, 44, 676–678. [Google Scholar] [CrossRef]
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges (Review). Oncol. Lett. 2019, 17, 3048–3054. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Von Salomé, J.; Aravidis, C.; Silander, G.; Askmalm, M.S.; Henriksson, I.; Gebre-Medhin, S.; Frödin, J.-E.; Björck, E.; Lagerstedt-Robinson, K.; et al. A retrospective study of extracolonic, non-endometrial cancer in Swedish Lynch syndrome families. Hered. Cancer Clin. Pract. 2018, 16, 16. [Google Scholar] [CrossRef] [PubMed]
- Cox, V.L.; Bamashmos, A.A.S.; Foo, W.C.; Gupta, S.; Yedururi, S.; Garg, N.; Kang, H.C. Lynch Syndrome: Genomics Update and Imaging Review. Radiographics 2018, 38, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Farha, N.; Hrabe, J.; Sleiman, J.; Beard, J.; Lyu, R.; Bhatt, A.; Church, J.; Heald, B.; Liska, D.; Mankaney, G.; et al. Clinically actionable findings on surveillance EGD in asymptomatic patients with Lynch syndrome. Gastrointest. Endosc. 2022, 95, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Kidambi, T.D.; Pedley, C.; Bergsland, E.K.; Terdiman, J.P.; Blanco, A. Lower gastrointestinal neuroendocrine neoplasms associated with hereditary cancer syndromes: A case series. Fam. Cancer 2017, 110, 223–543. [Google Scholar] [CrossRef]
- Sekine, R.; Shimazu, K.; Nakano, D.; Yamaguchi, T.; Suzuki, Y.; Fukuda, K.; Yoshida, T.; Taguchi, D.; Iijima, K.; Nanjyo, H.; et al. A novel Lynch syndrome pedigree bearing germ-line MSH2 missense mutation c.1808A>T (Asp603Val). Jpn. J. Clin. Oncol. 2022, 52, 81–85. [Google Scholar] [CrossRef]
- Sorscher, S.; Saroya, B. A molecularly confirmed neuroendocrine tumor resulting from Lynch Syndrome. J. Gastrointest. Oncol. 2013, 4, 95–96. [Google Scholar] [CrossRef]
- Lou, L.; Lv, F.; Wu, X.; Li, Y.; Zhang, X. Clinical implications of mismatch repair deficiency screening in patients with mixed neuroendocrine non-neuroendocrine neoplasms (MiNEN). Eur. J. Surg. Oncol. (EJSO) 2020, 47, 323–330. [Google Scholar] [CrossRef]
- Morani, A.C.; Hanafy, A.K.; Ramani, N.S.; Katabathina, V.S.; Yedururi, S.; Dasyam, A.K.; Prasad, S.R. Hereditary and Sporadic Pancreatic Ductal Adenocarcinoma: Current Update on Genetics and Imaging. Radiol. Imaging Cancer 2020, 2, e190020. [Google Scholar] [CrossRef]
- Grant, R.C.; Denroche, R.; Jang, G.H.; Nowak, K.M.; Zhang, A.; Borgida, A.; Holter, S.; Topham, J.T.; Wilson, J.; Dodd, A.; et al. Clinical and genomic characterisation of mismatch repair deficient pancreatic adenocarcinoma. Gut 2020, 70, 1894–1903. [Google Scholar] [CrossRef]
- Aslanian, H.R.; Lee, J.H.; Canto, M.I. AGA Clinical Practice Update on Pancreas Cancer Screening in High-Risk Individuals: Expert Review. Gastroenterology 2020, 159, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Katabathina, V.S.; Buddha, S.; Rajebi, H.; Shah, J.N.; Morani, A.C.; Lubner, M.G.; Dasyam, A.; Nazarullah, A.; Menias, C.O.; Prasad, S.R. Pancreas in Hereditary Syndromes: Cross-sectional Imaging Spectrum. RadioGraphics 2021, 41, 1082–1102. [Google Scholar] [CrossRef] [PubMed]
- Pittman, M.E.; Brosens, L.A.; Wood, L.D. Genetic Syndromes with Pancreatic Manifestations. Surg. Pathol. Clin. 2016, 9, 705–715. [Google Scholar] [CrossRef]
- Karamurzin, Y.; Zeng, Z.; Stadler, Z.K.; Zhang, L.; Ouansafi, I.; Al-Ahmadie, H.A.; Sempoux, C.; Saltz, L.B.; Soslow, R.A.; O’Reilly, E.M.; et al. Unusual DNA mismatch repair–deficient tumors in Lynch syndrome: A report of new cases and review of the literature. Hum. Pathol. 2012, 43, 1677–1687. [Google Scholar] [CrossRef]
- Barrera, A.S.; Pla, S.S.; Maña, C.M.B.; Salas, R.C.; Monforte, N.G.; González, N.B.; Monzonis, A.R.; Navarro, F.J.A.; Cueto, M.R.B.; Borobia, F.G. Pancreatic non-functioning neuroendocrine tumor: A new entity genetically related to Lynch syndrome. J. Gastrointest. Oncol. 2017, 8, E73–E79. [Google Scholar] [CrossRef] [PubMed]
- Ban, X.; Mo, S.; Lu, Z.; Jia, C.; Shao, H.; Chang, X.; Mao, X.; Zhang, Y.; Pang, J.; Zhang, Y.; et al. Expression and methylation status of MMR and MGMT in well-differentiated pancreatic neuroendocrine tumors and potential clinical applications. Endocrine 2022, 77, 538–545. [Google Scholar] [CrossRef]
- Koumarianou, A.; Kaltsas, G.A.; Chatzellis, E.; Kyriakopoulos, G.; Kolomodi, D.; Alexandraki, K.I. Immunotherapeutics at the spearhead: Current status in targeting neuroendocrine neoplasms. Endocrine 2021, 73, 232–239. [Google Scholar] [CrossRef]
- Rekhi, B.; Menon, S.; Deodhar, K.K.; Ghosh, J.; Chopra, S.; Maheshwari, A. Clinicopathological features of 50 mismatch repair (MMR)-deficient endometrial carcinomas, tested by immunohistochemistry: A single institutional feasibility study, India. Ann. Diagn. Pathol. 2020, 47, 151558. [Google Scholar] [CrossRef]
- Yousef, I.; Siyam, F.; Layfield, L.; Freter, C.; Sowers, J.R. Cervical neuroendocrine tumor in a young female with Lynch Syndrome. Neuro Endocrinol. Lett. 2014, 35, 89–94. [Google Scholar]
- Shetty, I.; Fuller, S.; Raygada, M.; Merino, M.J.; Thomas, B.J.; Widemann, B.C.; Reilly, K.M.; Pacak, K.; Del Rivero, J. Adrenocortical carcinoma masquerading as pheochromocytoma: A histopathologic dilemma. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020, 19-0147. [Google Scholar] [CrossRef]
- Calsina, B.; Piñeiro-Yáñez, E.; Martínez-Montes, Á.M.; Caleiras, E.; Fernández-Sanromán, Á.; Monteagudo, M.; Torres-Pérez, R.; Fustero-Torre, C.; Pulgarín-Alfaro, M.; Gil, E.; et al. Genomic and immune landscape Of metastatic pheochromocytoma and paraganglioma. Nat. Commun. 2023, 14, 1122. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Kai, K.; Sato, S.; Nishida, H.; Hirakawa, K.; Nasu, K.; Narahara, H. Mixed large and small cell neuroendocrine carcinoma and endometrioid carcinoma of the endometrium with high microsatellite instability: A case report and literature review. SAGE Open Med. Case Rep. 2021, 9, 2050313X21999200. [Google Scholar] [CrossRef] [PubMed]
- Teodosescu, A.; Chan, I.; Elder, J.; Wu, M. A correlation study of mismatch repair immunohistochemical protein expression of pancreatic solid tumors in cytology cell blocks and matching surgical specimens. Diagn. Cytopathol. 2021, 49, 700–705. [Google Scholar] [CrossRef]
- Riccò, B.; Salati, M.; Bonetti, L.R.; Dominici, M.; Luppi, G. PD-1 blockade in deficient mismatch repair mixed adenoneuroendocrine carcinoma of the stomach: New hope for an orphan disease. Tumori J. 2020, 106, NP57–NP62. [Google Scholar] [CrossRef]
- Luong, T.V.; Nisa, Z.; Watkins, J.; Hayes, A.R. Should immunohistochemical expression of mismatch repair (MMR) proteins and microsatellite instability (MSI) analysis be routinely performed for poorly differentiated colorectal neuroendocrine carcinomas? Endocrinol. Diabetes Metab. Case Rep. 2020, 2020, 20-0058. [Google Scholar] [CrossRef]
- Sherman, S.K.; Schuitevoerder, D.; Chan, C.H.F.; Turaga, K.K. Metastatic Colorectal Cancers with Mismatch Repair Deficiency Result in Worse Survival Regardless of Peritoneal Metastases. Ann. Surg. Oncol. 2020, 27, 5074–5083. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Slodkowska, E.; Parra-Herran, C.; Mirkovic, J. PD-L1,RB1 and mismatch repair protein immunohistochemical expression in neuroendocrine carcinoma, small cell type, of the uterine cervix. Histopathology 2019, 74, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Liu, I.H.; Ford, J.M.; Kunz, P.L. DNA-repair defects in pancreatic neuroendocrine tumors and potential clinical applications. Cancer Treat. Rev. 2015, 44, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, D.; Joost, P.; Aravidis, C.; Stenmark, M.A.; Backman, A.-S.; Melin, B.; Von Salomé, J.; Zagoras, T.; Gebre-Medhin, S.; Burman, P. Corticotroph Pituitary Carcinoma in a Patient With Lynch Syndrome (LS) and Pituitary Tumors in a Nationwide LS Cohort. J. Clin. Endocrinol. Metab. 2017, 102, 3928–3932. [Google Scholar] [CrossRef]
- Teuber, J.; Reinhardt, A.; Reuss, D.; Hähnel, S.; Unterberg, A.; Beynon, C. Aggressive pituitary adenoma in the context of Lynch syndrome: A case report and literature review on this rare coincidence. Br. J. Neurosurg. 2021, 1–6. [Google Scholar] [CrossRef]
- Voisin, M.R.; Almeida, J.P.; Perez-Ordonez, B.; Zadeh, G. Recurrent Undifferentiated Carcinoma of the Sella in a Patient with Lynch Syndrome. World Neurosurg. 2019, 132, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Uraki, S.; Ariyasu, H.; Doi, A.; Furuta, H.; Nishi, M.; Sugano, K.; Inoshita, N.; Nakao, N.; Yamada, S.; Akamizu, T. Atypical pituitary adenoma with MEN1 somatic mutation associated with abnormalities of DNA mismatch repair genes; MLH1 germline mutation and MSH6 somatic mutation. Endocr. J. 2017, 64, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Loughrey, P.; Baker, G.; Herron, B.; Cooke, S.; Iacovazzo, D.; Lindsay, J.; Korbonits, M. Invasive ACTH-producing pituitary gland neoplasm secondary to MSH2 mutation. Cancer Genet. 2021, 256–257, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Uraki, S.; Ariyasu, H.; Doi, A.; Kawai, S.; Takeshima, K.; Morita, S.; Fukai, J.; Fujita, K.; Furuta, H.; Nishi, M.; et al. Reduced Expression of Mismatch Repair Genes MSH6/MSH2 Directly Promotes Pituitary Tumor Growth via the ATR–Chk1 Pathway. J. Clin. Endocrinol. Metab. 2018, 103, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Airi, R.; Sherman, M. Microsatellite instability driven metastatic parathyroid carcinoma managed with the anti-PD1 immunotherapy, pembrolizumab. BMJ Case Rep. 2020, 13, e235293. [Google Scholar] [CrossRef]
- Andreasson, A.; Sulaiman, L.; Vale, S.D.; Martins, J.M.; Ferreira, F.; Miltenberger-Miltenyi, G.; Batista, L.; Haglund, F.; Björck, E.; Nilsson, I.-L.; et al. Molecular characterization of parathyroid tumors from two patients with hereditary colorectal cancer syndromes. Fam. Cancer 2012, 11, 355–362. [Google Scholar] [CrossRef]
- Kostov, S.; Watrowski, R.; Kornovski, Y.; Dzhenkov, D.; Slavchev, S.; Ivanova, Y.; Yordanov, A. Hereditary Gynecologic Cancer Syndromes—A Narrative Review. OncoTargets Ther. 2022, 15, 381–405. [Google Scholar] [CrossRef]
- Aguirre, E.; Grana, B.; Boudet, M.; Balmaña, J. Screening for Lynch Syndrome among Patients with Newly Diagnosed Endometrial Cancer: A Comprehensive Review. Tumori J. 2016, 102, 548–554. [Google Scholar] [CrossRef]
- Zhao, S.; Chen, L.; Zang, Y.; Liu, W.; Liu, S.; Teng, F.; Xue, F.; Wang, Y. Endometrial cancer in Lynch syndrome. Int. J. Cancer 2022, 150, 7–17. [Google Scholar] [CrossRef]
- Gallon, R.; Gawthorpe, P.; Phelps, R.L.; Hayes, C.; Borthwick, G.M.; Santibanez-Koref, M.; Jackson, M.S.; Burn, J. How Should We Test for Lynch Syndrome? A Review of Current Guidelines and Future Strategies. Cancers 2021, 13, 406. [Google Scholar] [CrossRef]
- Corrado, G.; Marchetti, C.; Trozzi, R.; Scambia, G.; Fagotti, A. Fertility preservation in patients with BRCA mutations or Lynch syndrome. Int. J. Gynecol. Cancer 2021, 31, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, M.; Heald, B.; Yanda, C.; Kelly, E.D.; Grobmyer, S.; Eng, C.; Kalady, M.; Pederson, H. Investigating the Link between Lynch Syndrome and Breast Cancer. Eur. J. Breast Health 2020, 16, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Sajjadi, E.; Venetis, K.; Piciotti, R.; Invernizzi, M.; Guerini-Rocco, E.; Haricharan, S.; Fusco, N. Mismatch repair-deficient hormone receptor-positive breast cancers: Biology and pathological characterization. Cancer Cell Int. 2021, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.; Brennan, D.; Rees, M.; Hickey, M. Management of menopausal symptoms and ovarian function preservation in women with gynecological cancer. Int. J. Gynecol. Cancer 2021, 31, 352–359. [Google Scholar] [CrossRef]
- Fedda, F.A.; Euscher, E.D.; Ramalingam, P.; Malpica, A. Prophylactic Risk-reducing Hysterectomies and Bilateral Salpingo-oophorectomies in Patients With Lynch Syndrome: A Clinicopathologic Study of 29 Cases and Review of the Literature. Int. J. Gynecol. Pathol. 2020, 39, 313–320. [Google Scholar] [CrossRef]
- Etchegary, H.; Dicks, E.; Tamutis, L.; Dawson, L. Quality of life following prophylactic gynecological surgery: Experiences of female Lynch mutation carriers. Fam. Cancer 2018, 17, 53–61. [Google Scholar] [CrossRef]
- Holter, S.; Hall, M.J.; Hampel, H.; Jasperson, K.; Kupfer, S.S.; Haidle, J.L.; Mork, M.E.; Palaniapppan, S.; Senter, L.; Stoffel, E.M.; et al. Risk assessment and genetic counseling for Lynch syndrome—Practice resource of the National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Gastrointestinal Cancer. J. Genet. Couns. 2022, 31, 568–583. [Google Scholar] [CrossRef]
- Stupart, D.; Win, A.K.; Winship, I.M.; Jenkins, M. Fertility after young-onset colorectal cancer: A study of subjects with Lynch syndrome. Color. Dis. 2015, 17, 787–793. [Google Scholar] [CrossRef]
- Terribas, E.; Bonache, S.; García-Arévalo, M.; Sánchez, J.; Franco, E.; Bassas, L.; Larriba, S. Changes in the Expression Profile of the Meiosis-Involved Mismatch Repair Genes in Impaired Human Spermatogenesis. J. Androl. 2010, 31, 346–357. [Google Scholar] [CrossRef]
- Al-Obaidy, K.I.; Trevino, K.E.; Idrees, M.T. Clinicopathologic Characterization of Bilateral Testicular Germ Cell Tumors with Immunohistochemical Evaluation of Mismatch Repair and BRAF (V600E) Genes Mutations. Int. J. Surg. Pathol. 2019, 27, 619–623. [Google Scholar] [CrossRef]
- Rudolph, C.; Melau, C.; Nielsen, J.E.; Jensen, K.V.; Liu, D.; Pena-Diaz, J.; Meyts, E.R.-D.; Rasmussen, L.J.; Jørgensen, A. Involvement of the DNA mismatch repair system in cisplatin sensitivity of testicular germ cell tumours. Cell. Oncol. 2017, 40, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Armaiz-Pena, G.; Dahia, P.L.M.; Lu, Y.; Toledo, R.A.; Varghese, J.; Habra, M.A. Endocrine and Neuroendocrine Tumors Special Issue-Checkpoint Inhibitors for Adrenocortical Carcinoma and Metastatic Pheochromocytoma and Paraganglioma: Do They Work? Cancers 2022, 14, 467. [Google Scholar] [CrossRef] [PubMed]
- Petris, R.; Valea, A.; Rentea, D.E.; Gheorghe, A.M.; Ghemigian, A.; Carsote, M.; Petrova, E.; Sandru, F.; Haldan, A.; Nistor, C.E. Pitfalls of adrenal tumors’ management in real-life medicine: A cases series. Manag. Health 2022, 2, 19–26. [Google Scholar]
- Habra, M.A.; Stephen, B.; Campbell, M.; Hess, K.; Tapia, C.; Xu, M.; Rodon Ahnert, J.; Jimenez, C.; Lee, J.E.; Perrier, N.D.; et al. Phase II clinical trial of pembrolizumab efficacy and safety in advanced adrenocortical carcinoma. J. Immunother. Cancer 2019, 7, 253. [Google Scholar] [CrossRef]
- Shen, C.; Wang, Y. Ferroptosis Biomarkers for Predicting Prognosis and Immunotherapy Efficacy in Adrenocortical Carcinoma. Arch. Med. Res. 2023, 54, 45–55. [Google Scholar] [CrossRef]
- Guan, Y.; Yue, S.; Chen, Y.; Pan, Y.; An, L.; Du, H.; Liang, C. Molecular Cluster Mining of Adrenocortical Carcinoma via Multi-Omics Data Analysis Aids Precise Clinical Therapy. Cells 2022, 11, 3784. [Google Scholar] [CrossRef]
- Ławnicka, H. Current Prospects for Adrenocortical Carcinoma Pharmacotherapy. Recent Patents Anti-Cancer Drug Discov. 2023, 18, 29–37. [Google Scholar] [CrossRef]
- Bates, M.F.; Sorensen, M.J. Genetic Testing for Adrenal Tumors—What the Contemporary Surgeon Should Know. Surg. Oncol. Clin. N. Am. 2023, 32, 303–313. [Google Scholar] [CrossRef]
- Riedmeier, M.; Thompson, L.D.R.; Molina, C.A.F.; Decarolis, B.; Härtel, C.; Schlegel, P.-G.; Fassnacht, M.; Wiegering, V. Prognostic value of the Weiss and Wieneke (AFIP) scoring systems in pediatric ACC—A mini review. Endocrine-Related Cancer 2023, 30, e220259. [Google Scholar] [CrossRef]
- Sneha, L.; Saminathan, T.; Dhivyalakshmi, J.; Joseph, L. Precocious puberty in a child: A rare cause and review of literature. J. Fam. Med. Prim. Care 2022, 11, 6523. [Google Scholar] [CrossRef]
- Brönimann, S.; Garstka, N.; Remzi, M. Treatment of adrenocortical carcinoma: Oncological and endocrine outcomes. Curr. Opin. Urol. 2023, 33, 50–58. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| First Author Reference | Year of Publication | Type of Study | Participants | MMR Gene, MMR Proteins, and Microsatellite Instability Status |
|---|---|---|---|---|
| Studies | ||||
| Domènech. M. [22] | 2021 | Cohort study | 634 individuals with LS coming from 220 families | 0.47% prevalence of ACC (N = 3) All individuals carried a MSH2 germline mutation 3 subjects with ACC: loss of expression of MSH2 and MSH6 proteins |
| Pozdeyev, N. [35] | 2021 | Cohort study | 364 individuals with ACC (median age of 52 y) | 13.7% of them with MMR genes mutations |
| Brondani, V.B. [26] | 2020 | Retrospective study | 36 children with ACC caring TP53 p.Arg337His germline mutation (southern Brazil) | 3 cases (8.57%) with MMR anomalies: 2 patients with MLH1 mutation 1 patient with MSH6 mutation |
| Raymond, V.M. [25] | 2013 | Cohort study Retrospective study | 94 patients with ACC (prospective gene counseling) 135 individuals with LS (University of Michigan Cancer Genetics Registry) (retrospective analysis) | 3.2% prevalence of LS (3 cases) 2 cases with ACC IHC available for 4 ACC: 4/4—MSI 3/4—similar MMR protein-gene configuration The prevalence of LS among patients with ACC is similar with LS prevalence among patients with CRC and EC |
| Case series | ||||
| Challis, B.G. [28] | 2016 | Case series | 1 family with LS | One family with adult ACC Proband: 1 female (54 y) with ACC (prior diagnostic of colorectal cancer and ovarian cancer) Her mother: ACC with fatal outcome Her sister: colorectal cancer and EC |
| Karamurzin, Y. [74] | 2012 | Case series | 4 cases with LS | 1 case with ACC (1 case with pancreatic well-differentiated NET) |
| Case reports | ||||
| Raygada, M. [21] | 2021 | Case report | 1 female with LS (44 y) | ACC Loss of heterozygosity of MSH2 gene (c.211+1G>T) This is the first case with a second germline mutation: RET gene mutation (c.2410G>A; p.Val804Met), without clinical manifestation of MEN2A syndrome |
| Shetty, I. [80] | 2020 | Case report | 1 female case with LS (57 y) | Diagnosis of ACC at 51 y Pathogenic mutation (p.Q46X) in MSH2 gene IHC: loss of expression for MSH2 and MSH6 proteins |
| Kaur, R.J. [29] | 2019 | Case report | 1 female case with LS (65 y) | Diagnosis of ACC at 65 y MSH6 mutation |
| Casey, R.T. [34] | 2018 | Case report | 1 female case with LS (58 y) | Diagnosis of ACC at 58 y Pathogenic mutation in MSH2 (p.Asn263fs) This the first case of LS-ACC treated with PD-1 blockade |
| Wright, J.P. [30] | 2018 | Case report | 1 male case with LS (68 y) | Diagnosis of ACC at 68 y MSH2 mutation Ectopic presentation and hormonally inactive ACC |
| Parameter | Outcome |
|---|---|
| level of evidence ACC-MMR/MSI | low (however, ACC is an orphan disease) |
| 2 types of approaches |
|
| potential applications of MMR/MSI in other endocrine tumors | yes: thyroid cancer and neuroendocrine neoplasia |
| Lynch syndrome (germline MMR mutation) and endocrine issues |
|
| results: 10-year analysis ACC-MMR/MSI | 11 studies |
| patients with Lynch syndrome and ACC | n = 2 N1 = 3 ACC /634 LS N2 = 2 ACC/135 LS |
| patients with ACC and MMR status | n = 3 |
| N1 = 364: 13.7% with non-germline MMR mutations N2 = 36: 8.57% with non-germline MMR mutations N3 = 94: 3.2% with germline MMR mutations | |
| 2 case series with Lynch syndrome | 1 new case of ACC/ per paper |
| 5 case reports | 1 new case of ACC/per paper |
| first report of PD-1 blockade for ACC | in 2018 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carsote, M.; Turturea, I.F.; Turturea, M.R.; Valea, A.; Nistor, C.; Gheorghisan-Galateanu, A.-A. Pathogenic Insights into DNA Mismatch Repair (MMR) Genes–Proteins and Microsatellite Instability: Focus on Adrenocortical Carcinoma and Beyond. Diagnostics 2023, 13, 1867. https://doi.org/10.3390/diagnostics13111867
Carsote M, Turturea IF, Turturea MR, Valea A, Nistor C, Gheorghisan-Galateanu A-A. Pathogenic Insights into DNA Mismatch Repair (MMR) Genes–Proteins and Microsatellite Instability: Focus on Adrenocortical Carcinoma and Beyond. Diagnostics. 2023; 13(11):1867. https://doi.org/10.3390/diagnostics13111867
Chicago/Turabian StyleCarsote, Mara, Ionut Florin Turturea, Maria Roxana Turturea, Ana Valea, Claudiu Nistor, and Ancuta-Augustina Gheorghisan-Galateanu. 2023. "Pathogenic Insights into DNA Mismatch Repair (MMR) Genes–Proteins and Microsatellite Instability: Focus on Adrenocortical Carcinoma and Beyond" Diagnostics 13, no. 11: 1867. https://doi.org/10.3390/diagnostics13111867