
Molecular Genetics of Thrombotic Myeloproliferative Neoplasms: Implications in Precision Oncology
 , , and
, , and
Abstract
:1. Introduction
2. Gene Mutations in Thrombotic MPN
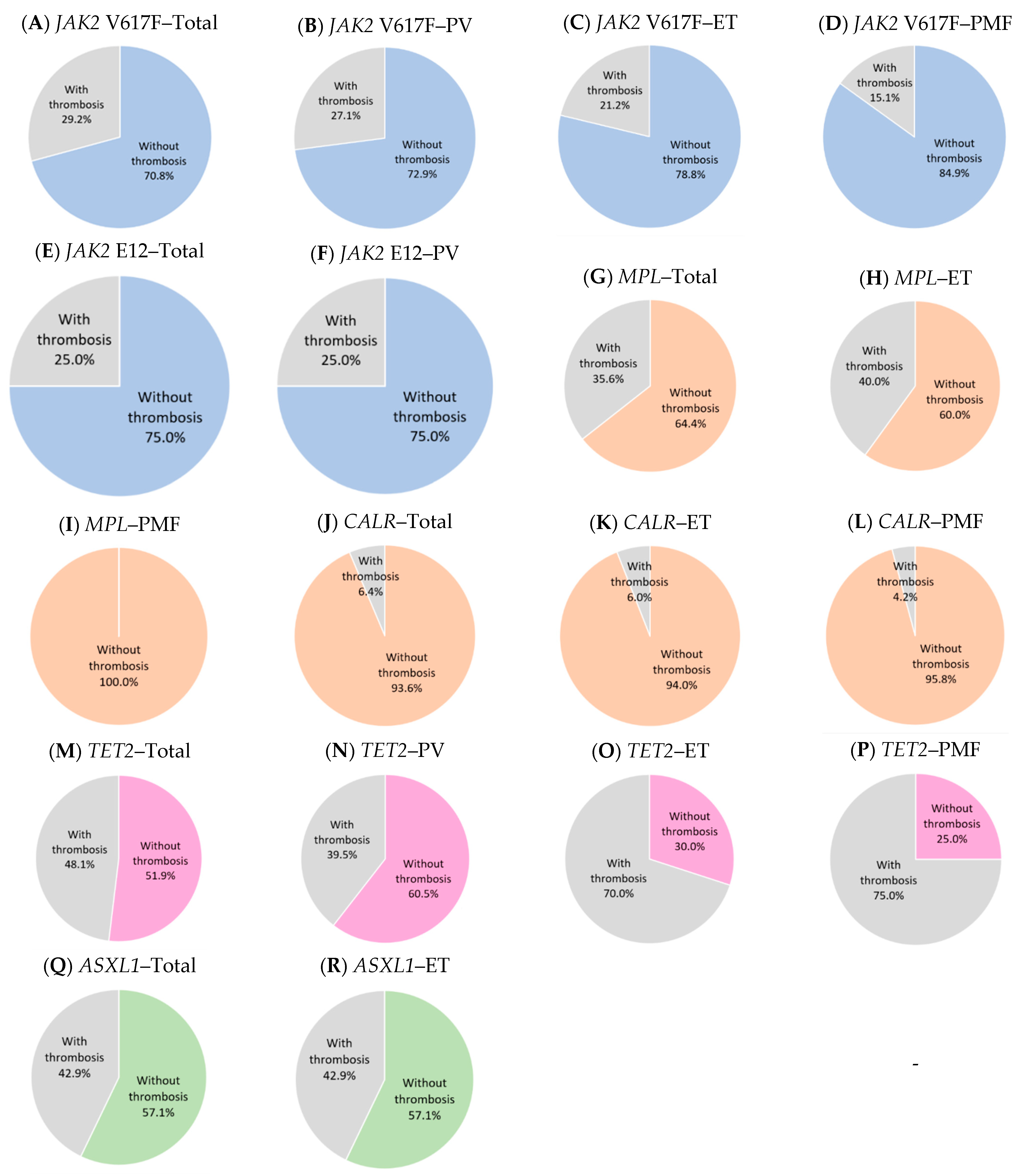
2.1. JAK2 V617F
2.2. JAK2 Exon 12
2.3. CALR
2.4. MPL
2.5. TET2
2.6. ASXL1

{kind=link}
{kind=link}
{kind=link}
| PV [138] | ET [138] | PMF [139] | |
|---|---|---|---|
| Prevalence of MPN | 49.2% | 34.7% | 14.4% |
| Driver mutations | |||
| JAK2 | 98% | 52% | 62% |
| CALR | 0% | 26% | 22% |
| MPL | 0% | 4% | 5% |
| Co-occurring mutations | |||
| ‡TET2 | 22% | 16% | 15% |
| ‡ASXL1 | 12% | 11% | 48% |
| * Prevalence of thrombotic at diagnosis | 28.6% | 20.7% | 9.5% |
| * Prevalence of bleeding at diagnosis | 6.9% | 7.3% | 8.9% |
3. Triple-Negative MPN
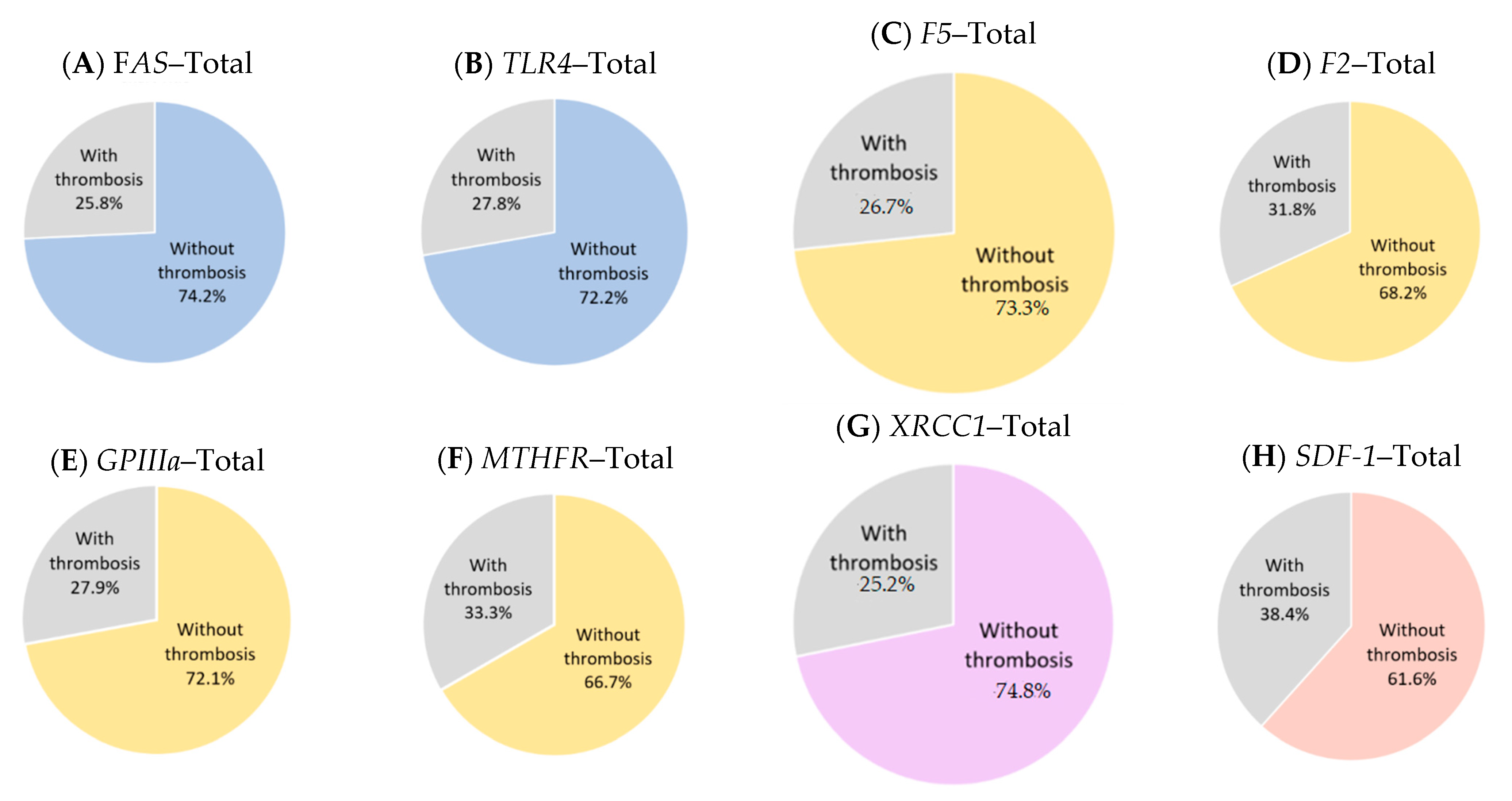
4. Gene Polymorphisms in Thrombotic MPN
5. Epigenetic Changes in Thrombotic MPN
6. Risk Assessment of Thrombosis in MPN According to Different Gene Mutations
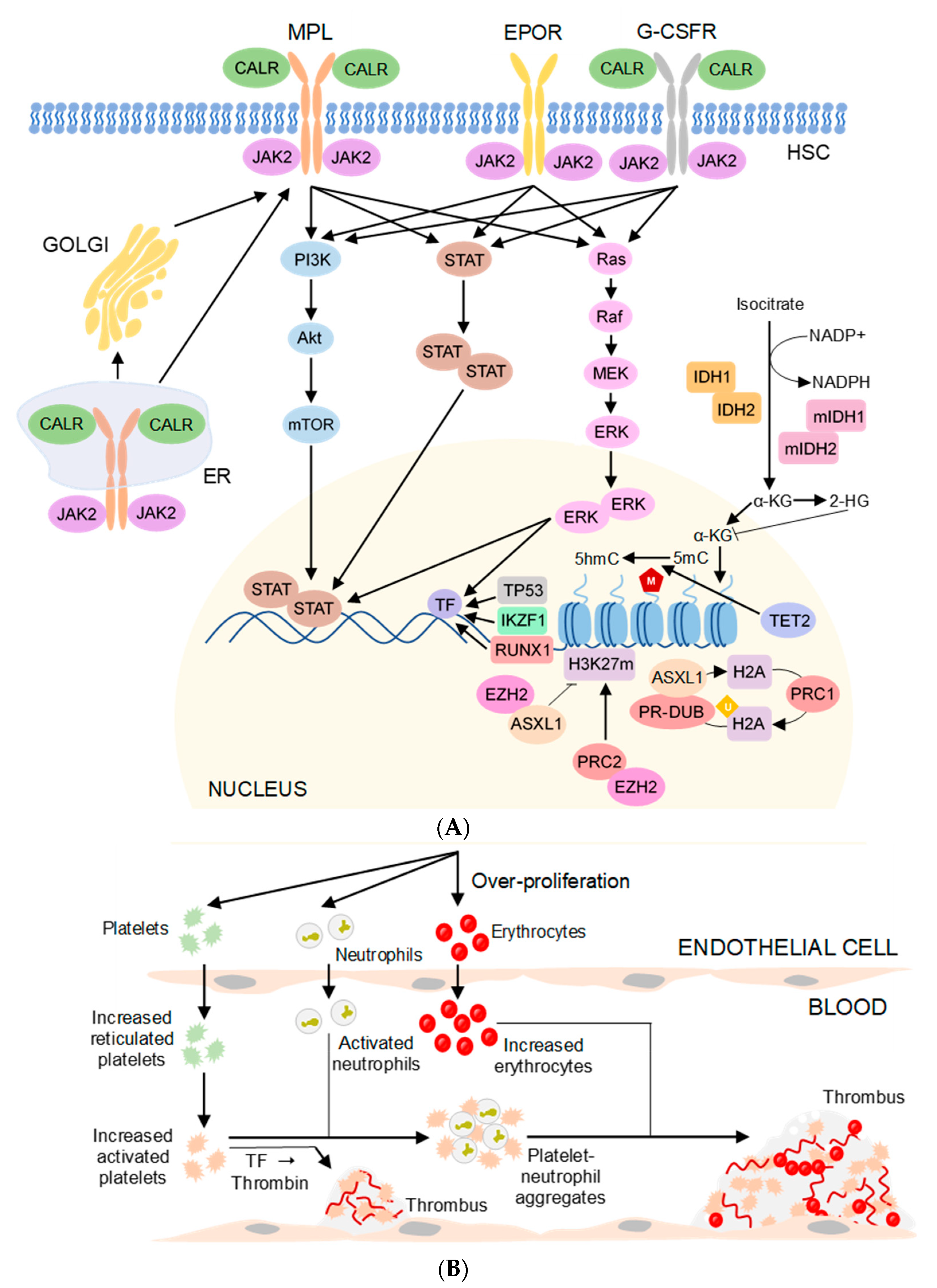
7. Pathogenesis in Thrombotic MPN
8. Currently Available Drugs and Therapies for Thrombotic MPN
8.1. Hydroxyurea
8.2. Interferon-Alpha
8.3. Anagrelide
8.4. Ruxolitinib
8.5. Momelotinib
8.6. Fedratinib
8.7. Pacritinib
8.8. Busulfan
8.9. Aspirin
8.10. Anticoagulants
8.11. Phlebotomy
8.12. Radiophosphorus and Chlorambucil
8.13. Allogenic Stem-Cell Transplantation
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Liisborg, C.; Hasselbalch, H.C.; Sorensen, T.L. Ocular manifestations in patients with Philadelphia-negative myeloproliferative neoplasms. Cancers 2020, 12, 573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 94, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Pardanani, A. Essential thrombocythemia. N. Engl. J. Med. 2019, 381, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 1551–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumi, E.; Sant’Antonio, E.; Boveri, E.; Pietra, D.; Cavalloni, C.; Roncoroni, E.; Astori, C.; Arcaini, L. Diagnosis and management of prefibrotic myelofibrosis. Expert Rev. Hematol. 2018, 11, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Kim, J.; Byun, J.M.; Hong, J.; Koh, Y.; Shin, D.-Y.; Kim, I.; Yoon, S.-S.; Park, H.; Bang, S.-M. Incidence, characteristics and risk factors of thromboembolic events in East Asian patients with BCR-ABL1 negative myeloproliferative neoplasms. Sci. Rep. 2021, 11, 17819. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Elvers, M.; Schafer, A.I. The pathobiology of thrombosis, microvascular disease, and hemorrhage in the myeloproliferative neoplasms. Blood 2021, 137, 2152–2160. [Google Scholar] [CrossRef]
- Hultcrantz, M.; Bjorkholm, M.; Dickman, P.W.; Landgren, O.; Derolf, A.R.; Kristinsson, S.Y.; Andersson, T.M.L. Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: A population-based cohort study. Ann. Intern. Med. 2018, 168, 317–325. [Google Scholar] [CrossRef]
- Song, I.-C.; Yeon, S.H.; Lee, M.-W.; Ryu, H.; Lee, H.-J.; Yun, H.-J.; Kim, S.Y.; Jo, D.-Y. Thrombotic and hemorrhagic events in 2016 World Health Organization-defined Philadelphia-negative myeloproliferative neoplasm. Korean J. Intern. Med. 2021, 36, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Rungjirajittranon, T.; Owattanapanich, W.; Ungprasert, P.; Siritanaratkul, N.; Ruchutrakool, T. A systematic review and meta-analysis of the prevalence of thrombosis and bleeding at diagnosis of Philadelphia-negative myeloproliferative neoplasms. BMC Cancer 2019, 19, 184. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.A.; Bjerrum, O.W.; Ranjan, A.; Skov, V.; Kruse, T.A.; Thomassen, M.; Skytthe, A.; Hasselbalch, H.C.; Christensen, K. Myeloproliferative neoplasms in Danish twins. Acta Haematol. 2018, 139, 195–198. [Google Scholar] [CrossRef]
- Huang, X.; Wu, J.; Deng, X.; Xu, X.; Zhang, X.; Ma, W.; Hu, T.; Yang, J.; Guan, M.; Tang, G. Mutation profiles of classic myeloproliferative neoplasms detected by a customized next-generation sequencing-based 50-gene panel. J. Bio-X Res. 2020, 3, 13–20. [Google Scholar] [CrossRef]
- Loscocco, G.G.; Guglielmelli, P.; Vannucchi, A.M. Impact of Mutational Profile on the Management of Myeloproliferative Neoplasms: A Short Review of the Emerging Data. Onco Targets 2020, 13, 12367–12382. [Google Scholar] [CrossRef] [PubMed]
- Kubesova, B.; Pavlova, S.; Malcikova, J.; Kabathova, J.; Radova, L.; Tom, N.; Tichy, B.; Plevova, K.; Kantorova, B.; Fiedorova, K.; et al. Low-burden TP53 mutations in chronic phase of myeloproliferative neoplasms: Association with age, hydroxyurea administration, disease type and JAK2 mutational status. Leukemia 2018, 32, 450–461. [Google Scholar] [CrossRef]
- Makarik, T.; Sabirov, K.; Kitsenko, E.; Treglazova, S.; Stepanova, E.; Fevraleva, I.; Kulikov, S.; Sudarikov, A. PB2041: Evaluation of the prognostic value of thrombophilia markers in patients with myeloproliferative neoplasms. HemaSphere 2022, 6, 1912–1913. [Google Scholar] [CrossRef]
- King, S.A. MPN-500 The Characteristics of Patients With Unprovoked Venous Thrombotic Events With JAK2 GGCC (46/1) Haplotype Treated in Tertiary Care Center in Saudi Arabia. Clin. Lymphoma Myeloma Leuk. 2022, 22, S340–S341. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [Green Version]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Mejía-Ochoa, M.; Acevedo Toro, P.A.; Cardona-Arias, J.A. Systematization of analytical studies of polycythemia vera, essential thrombocythemia and primary myelofibrosis, and a meta-analysis of the frequency of JAK2, CALR and MPL mutations: 2000–2018. BMC Cancer 2019, 19, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plo, I.; Vainchenker, W. Molecular and genetic bases of myeloproliferative disorders: Questions and perspectives. Clin. Lymphoma Myeloma 2009, 9 (Suppl. S3), S329–S339. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Huang, L.J.; Lodish, H.F. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J. Biol. Chem. 2008, 283, 5258–5266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Sazawal, S.; Chhikara, S.; Mahapatra, M.; Saxena, R. Association of JAK2V617F mutation with thrombosis in Indian patients with Philadelphia negative chronic myeloproliferative neoplasms. Indian J. Pathol. Microbiol. 2018, 61, 371–374. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Wan, S.; Zhao, H.; Sun, W.; Tian, D. Relationship between JAK2 V617F gene mutation and vascular embolism diseases. J. Leuk Lymphoma 2015, 24, 431–432. [Google Scholar]
- Heller, P.G.; Lev, P.R.; Salim, J.P.; Kornblihtt, L.I.; Goette, N.P.; Chazarreta, C.D.; Glembotsky, A.C.; Vassallu, P.S.; Marta, R.F.; Molinas, F.C. JAK2V617F mutation in platelets from essential thrombocythemia patients: Correlation with clinical features and analysis of STAT5 phosphorylation status. Eur. J. Haematol. 2006, 77, 210–216. [Google Scholar] [CrossRef]
- Randi, M.L.; Ruzzon, E.; Tezza, F.; Scapin, M.; Duner, E.; Scandellari, R.; Fabris, F. JAK2V617F mutation is common in old patients with polycythemia vera and essential thrombocythemia. Aging Clin. Exp. Res. 2011, 23, 17–21. [Google Scholar] [CrossRef]
- Basquiera, A.L.; Soria, N.W.; Ryser, R.; Salguero, M.; Moiraghi, B.; Sackmann, F.; Sturich, A.G.; Borello, A.; Berretta, A.; Bonafe, M.; et al. Clinical significance of V617F mutation of the JAK2 gene in patients with chronic myeloproliferative disorders. Hematology 2009, 14, 323–330. [Google Scholar] [CrossRef]
- Ohyashiki, K.; Kiguchi, T.; Ito, Y.; Fujimoto, H.; Gotoh, A.; Tauchi, T.; Miyazawa, K.; Kimura, Y.; Ohyashiki, J.H. Leukocytosis is linked to thrombosis at diagnosis, while JAK2 V617F mutation is associated with thrombosis during the course of essential thrombocythemia. Int. J. Hematol. 2008, 87, 446–448. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Tanaka, H.; Luis, E.J.; Sakai, K.; Kumode, T.; Sano, K.; Serizawa, K.; Rai, S.; Morita, Y.; Hanamoto, H.; et al. Elevated plasma levels of procoagulant microparticles are a novel risk factor for thrombosis in patients with myeloproliferative neoplasms. Int. J. Hematol. 2017, 106, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 2012, 120, 5128–5133; quiz 5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, Y.; Seki, R.; Kanajii, T.; Nohara, M.; Koteda, S.; Kawaguchi, K.; Nomura, K.; Nakamura, T.; Morishige, S.; Oku, E. Association between thromboembolic events and the JAK2 V617F mutation in myeloproliferative neoplasms cancer. Kurume Med. J. 2014, 60, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speletas, M.; Katodritou, E.; Daiou, C.; Mandala, E.; Papadakis, E.; Kioumi, A.; Ritis, K.; Korantzis, I. Correlations of JAK2-V617F mutation with clinical and laboratory findings in patients with myeloproliferative disorders. Leuk Res. 2007, 31, 1053–1062. [Google Scholar] [CrossRef]
- Andrikovics, H.; Krahling, T.; Balassa, K.; Halm, G.; Bors, A.; Koszarska, M.; Batai, A.; Dolgos, J.; Csomor, J.; Egyed, M.; et al. Distinct clinical characteristics of myeloproliferative neoplasms with calreticulin mutations. Haematologica 2014, 99, 1184–1190. [Google Scholar] [CrossRef] [Green Version]
- Tafesh, L.; Musgrave, K.; Roberts, W.; Plews, D.; Carey, P.; Biss, T. Myeloproliferative neoplasms in children and adolescents and thrombosis at unusual sites: The role of driver mutations. J. Pediatr Hematol. 2019, 41, 490–493. [Google Scholar] [CrossRef]
- Penka, M.; Schwarz, J.; Doubek, M.; Dulicek, P.; Indrak, K.; Brychtova, Y.; Hlusí, A.; Kissova, J.; Mayer, J.; Pavlik, T. JAK2 mutation and additional thrombophilic markers predispose to thrombosis in myeloproliferative diseases with thrombocythemia. Blood 2008, 112, 5257. [Google Scholar] [CrossRef]
- Ivanyi, J.L.; Marton, E.; Plander, M. Significance of the JAK2V617F mutation in patients with chronic myeloproliferative neoplasia. Orv. Hetil. 2011, 152, 1795–1803. [Google Scholar] [CrossRef]
- Garces-Eisele, J.; Gonzalez-Carrillo, M.L.; Reyes-Nunez, V.; Ruiz-Arguelles, G.J. Primary thrombophilia in Mexico VII: The V617F mutation of JAK2 is not a frequent cause of thrombosis. Hematology 2008, 13, 244–246. [Google Scholar] [CrossRef]
- Mahjoub, S.; Baccouche, H.; Sahnoun, M.; Kaabi, H.; Manai, Z.; Slama, H.; Ben Romdhane, N. The JAK2 mutation in myeloproliferative disorders: A predictive factor of thrombosis. Tunis. Med. 2015, 93, 474–477. [Google Scholar]
- Guo, X.; Yang, L.; Yan, K.; Yang, R.; Lin, J. Relationship between JAK2-V617F gene mutation in peripheral blood mononuclear cells and thrombotic events in patients with myeloproliferative neoplasms. J. Mod. Lab. Med. 2017, 32, 143–145. [Google Scholar]
- De Stefano, V.; Za, T.; Rossi, E.; Vannucchi, A.M.; Ruggeri, M.; Elli, E.; Mico, C.; Tieghi, A.; Cacciola, R.R.; Santoro, C.; et al. Increased risk of recurrent thrombosis in patients with essential thrombocythemia carrying the homozygous JAK2 V617F mutation. Ann. Hematol. 2010, 89, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gango, A.; Mozes, R.; Boha, Z.; Kajtar, B.; Timar, B.; Kiraly, P.A.; Kiss, R.; Fesus, V.; Nagy, N.; Demeter, J.; et al. Quantitative assessment of JAK2 V617F and CALR mutations in Philadelphia negative myeloproliferative neoplasms. Leuk Res. 2018, 65, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.G.; Choi, H.W.; Lee, J.H.; Choi, Y.J.; Choi, H.J.; Shin, J.H.; Suh, S.P.; Szardenings, M.; Kim, H.R.; Shin, M.G. Coexistence of JAK2 and CALR mutations and their clinical implications in patients with essential thrombocythemia. Oncotarget 2016, 7, 57036–57049. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.S.; Kim, Y.K.; Jung, S.I.; Jung, H.R.; Chung, I.S. Correlations between Janus kinase 2 V617F allele burdens and clinicohematologic parameters in myeloproliferative neoplasms. Ann. Lab. Med. 2012, 32, 385–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogan, I.; Chap, D.; Hoffman, R.; Axelman, E.; Brenner, B.; Nadir, Y. JAK-2 V617F mutation increases heparanase procoagulant activity. Thromb. Haemost. 2016, 115, 73–80. [Google Scholar] [CrossRef]
- Patriarca, A.; Pompetti, F.; Malizia, R.; Iuliani, O.; Di Marzio, I.; Spadano, A.; Dragani, A. Is the absence of JAK2V617F mutation a risk factor for bleeding in essential thrombocythemia? An analysis of 106 patients. Blood Transfus. 2010, 8, 21. [Google Scholar]
- Ayer, M.; Menken, I.; Yamak, M.; Ayer, F.A.; Kirkizlar, O.; Burak Aktuglu, M. The impact of mean platelet volume (MPV) and JAK-2 mutation on thrombosis in chronic myeloproliferative diseases. Indian J. Hematol. Blood Transfus. 2017, 33, 181–187. [Google Scholar] [CrossRef]
- Ilhan, G.; Karakus, S.; Sahin, F.I. JAK 2V617F mutation: Frequency and relation to clinical and laboratory features of BCR-ABL negative myeloproliferative diseases. Uhod-Uluslar Hematol. 2012, 22, 77–84. [Google Scholar] [CrossRef]
- Hattori, N.; Fukuchi, K.; Nakashima, H.; Maeda, T.; Adachi, D.; Saito, B.; Yanagisawa, K.; Matsuda, I.; Nakamaki, T.; Gomi, K.; et al. Megakaryopoiesis and platelet function in polycythemia vera and essential thrombocythemia patients with JAK2 V617F mutation. Int. J. Hematol. 2008, 88, 181–188. [Google Scholar] [CrossRef]
- Carobbio, A.; Finazzi, G.; Guerini, V.; Spinelli, O.; Delaini, F.; Marchioli, R.; Borrelli, G.; Rambaldi, A.; Barbui, T. Leukocytosis is a risk factor for thrombosis in essential thrombocythemia: Interaction with treatment, standard risk factors, and JAK2 mutation status. Blood 2007, 109, 2310–2313. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Longo, G.; Pancrazzi, A.; Ponziani, V.; Bogani, C.; Ferrini, P.R.; Rambaldi, A.; Guerini, V.; et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007, 21, 1952–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowczyk, M.; Wojtaszewska, M.; Lewandowski, K.; Gil, L.; Lewandowska, M.; Lehmann-Kopydlowska, A.; Kroll-Balcerzak, R.; Balcerzak, A.; Iwola, M.; Michalak, M.; et al. The JAK2 V617F mutational status and allele burden may be related with the risk of venous thromboembolic events in patients with Philadelphia-negative myeloproliferative neoplasms. Thromb. Res. 2015, 135, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Horvat, I.; Boban, A.; Zadro, R.; Antolic, M.R.; Serventi-Seiwerth, R.; Roncevic, P.; Radman, I.; Sertic, D.; Vodanovic, M.; Pulanic, D.; et al. Influence of blood count, cardiovascular risks, inherited thrombophilia, and JAK2 V617F burden allele on type of thrombosis in patients with Philadelphia chromosome negative myeloproliferative neoplasms. Clin. Lymphoma Myeloma Leuk. 2019, 19, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.S.; Pallisgaard, N.; Moller, M.B.; Hasselbalch, H.C. High prevalence of arterial thrombosis in JAK2 mutated essential thrombocythaemia: Independence of the V617F allele burden. Hematology 2008, 13, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.M.; Lee, J.S.; Ahn, J.Y.; Lee, J.; Hyun, M.; Kim, B.; Park, M.; Chi, H.S.; Kim, H.; Kim, H.; et al. Vascular events in Korean patients with myeloproliferative neoplasms and their relationship to JAK2 mutation. Thromb. Haemost. 2009, 101, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Uyanik, M.S.; Baysal, M.; Pamuk, G.E.; Maden, M.; Akker, M.; Umit, E.G.; Demir, M.; Aydogdu, E. Is JAK2V617F mutation the only factor for thrombosis in Philadelphia-negative chronic myeloproliferative neoplasms? Indian J. Hematol. Blood Transfus. 2016, 32, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Yonal, I.; Pinarbasi, B.; Hindilerden, F.; Hancer, V.S.; Nalcaci, M.; Kaymakoglu, S.; Diz-Kucukkaya, R. The clinical significance of JAK2V617F mutation for Philadelphia-negative chronic myeloproliferative neoplasms in patients with splanchnic vein thrombosis. J. Thromb. Thrombolysis 2012, 34, 388–396. [Google Scholar] [CrossRef]
- Karakose, S.; Oruc, N.; Zengin, M.; Akarca, U.S.; Ersoz, G. Diagnostic value of the JAK2 V617F mutation for latent chronic myeloproliferative disorders in patients with Budd-Chiari syndrome and/or portal vein thrombosis. Turk. J. Gastroenterol. 2015, 26, 42–48. [Google Scholar] [CrossRef]
- Colaizzo, D.; Amitrano, L.; Guardascione, M.A.; Tiscia, G.L.; D’Andrea, G.; Longo, V.A.; Grandone, E.; Margaglione, M. Outcome of patients with splanchnic venous thrombosis presenting without overt MPN: A role for the JAK2 V617F mutation re-evaluation. Thromb. Res. 2013, 132, e99–e104. [Google Scholar] [CrossRef]
- Xavier, S.G.; Gadelha, T.; Pimenta, G.; Eugenio, A.M.; Ribeiro, D.D.; Gomes, F.M.; Bonamino, M.; Zalcberg, I.R.; Spector, N. JAK2V617F mutation in patients with splanchnic vein thrombosis. Dig. Dis. Sci. 2010, 55, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Lippert, E.; Girodon, F.; Dobo, I.; Fouassier, M.; Masliah, C.; Praloran, V.; Hermouet, S. Latent myeloproliferative disorder revealed by the JAK2-V617F mutation and endogenous megakaryocytic colonies in patients with splanchnic vein thrombosis. Blood 2006, 108, 3223–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dentali, F.; Squizzato, A.; Brivio, L.; Appio, L.; Campiotti, L.; Crowther, M.; Grandi, A.M.; Ageno, W. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: A meta-analysis. Blood 2009, 113, 5617–5623. [Google Scholar] [CrossRef] [PubMed]
- Zerjavic, K.; Zagradisnik, B.; Herodez, S.S.; Lokar, L.; Krasevac, M.G.; Vokac, N.K. Is the JAK2 V617F mutation a hallmark for different forms of thrombosis? Acta Haematol. 2010, 124, 49–56. [Google Scholar] [CrossRef]
- Mattar, M.M.; Nassef, S.; El Husseiny, N.M.; El Masry, M.R.; Salah, M.; Morad, M.A.; Gawad, A.A. Incidence of silent thrombosis in patients younger than 60 years with myeloproliferative neoplasms: Single-center egyptian study. Clin. Lymphoma Myeloma Leuk 2019, 19, E425–E429. [Google Scholar] [CrossRef]
- Gruppo Italiano Studio Policitemia. Polycythemia vera: The natural history of 1213 patients followed for 20 years. Ann. Intern. Med. 1995, 123, 656–664. [Google Scholar] [CrossRef]
- De Stefano, V.; Teofili, L.; Leone, G.; Michiels, J.J. Spontaneous erythroid colony formation as the clue to an underlying myeloproliferative disorder in patients with Budd-Chiari syndrome or portal vein thrombosis. Semin. Thromb. Hemost. 1997, 23, 411–418. [Google Scholar] [CrossRef]
- Janssen, H.L.A.; Garcia-Pagan, J.C.; Elias, E.; Mentha, G.; Hadengue, A.; Valla, D.C.; Disorders, E.G.S.V. Budd-Chiari syndrome: A review by an expert panel. J. Hepatol. 2003, 38, 364–371. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Cervantes, F.; Leebeek, F.W.; Marzac, C.; Cassinat, B.; Chevret, S.; Cazals-Hatem, D.; Plessier, A.; Garcia-Pagan, J.C.; Darwish Murad, S.; et al. The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: A report on 241 cases. Blood 2008, 111, 4922–4929. [Google Scholar] [CrossRef] [Green Version]
- P’ng, S.; Carnley, B.; Baker, R.; Kontorinis, N.; Cheng, W. Undiagnosed myeloproliferative disease in cases of intra-abdominal thrombosis: The utility of the JAK2 617F mutation. Clin. Gastroenterol. Hepatol. 2008, 6, 472–475. [Google Scholar] [CrossRef]
- Primignani, M.; Barosi, G.; Bergamaschi, G.; Gianelli, U.; Fabris, F.; Reati, R.; Dell’Era, A.; Bucciarelli, P.; Mannucci, P.M. Role of the JAK2 mutation in the diagnosis of chronic myeloproliferative disorders in splanchnic vein thrombosis. Hepatology 2006, 44, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhang, C.; Han, G.; Zhang, W.; He, C.; Yin, Z.; Liu, Z.; Bai, W.; Li, R.; Bai, M.; et al. Prevalence of the JAK2V617F mutation in Chinese patients with Budd-Chiari syndrome and portal vein thrombosis: A prospective study. J. Gastroenterol. Hepatol. 2012, 27, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Sharma, A.; Sazawal, S.; Ahuja, A.; Upadhyay, A.; Mahapatra, M.; Saxena, R. Prevalence of JAK2V617F mutation in deep venous thrombosis patients and its clinical significance as a thrombophilic risk factor: Indian perspective. Clin. Appl. Thromb. Hem. 2015, 21, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Yoo, E.H.; Jang, J.H.; Park, K.J.; Gwak, G.Y.; Kim, H.J.; Kim, S.H.; Kim, D.K. Prevalence of overt myeloproliferative neoplasms and JAK2 V617F mutation in Korean patients with splanchnic vein thrombosis. Int. J. Lab. Hematol. 2011, 33, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yang, Z.; Bai, M.; Shi, X.; Han, G.; Fan, D. Meta-analysis: The significance of screening for JAK2V617F mutation in Budd–Chiari syndrome and portal venous system thrombosis. Aliment. Pharm. 2011, 33, 1087–1103. [Google Scholar] [CrossRef] [PubMed]
- Sarid, N.; Eshel, R.; Rahamim, E.; Carmiel, M.; Kirgner, I.; Shpringer, M.; Trestman, S.; Marilus, R.; Perry, C.; Polliack, A.; et al. JAK2 mutation: An aid in the diagnosis of occult myeloproliferative neoplasms in patients with major intraabdominal vein thrombosis and normal blood counts. Isr. Med. Assoc. J. 2013, 15, 698–700. [Google Scholar] [PubMed]
- Xavier, S.; Gadelha, T.; Rezende, S.; Zalcberg, I.; Spector, N. JAK2V617F mutation in patients with thrombosis: To screen or not to screen? Int. J. Lab. Hematol. 2011, 33, 117–124. [Google Scholar] [CrossRef]
- Smalberg, J.H.; De Maat, M.P.M.; Leebeek, F.W.G. Absence of the JAK2 V617F mutation in patients with arterial thrombosis without overt myeloproliferative disease. J. Thromb. Haemost. 2008, 6, 1606–1607. [Google Scholar] [CrossRef]
- Passamonti, S.M.; Biguzzi, E.; Cazzola, M.; Franchi, F.; Gianniello, F.; Bucciarelli, P.; Pietra, D.; Mannucci, P.M.; Martinelli, I. The JAK2 V617F mutation in patients with cerebral venous thrombosis. J. Thromb. Haemost. 2012, 10, 998–1003. [Google Scholar] [CrossRef]
- Ball, S.; Thein, K.Z.; Maiti, A.; Nugent, K. Thrombosis in Philadelphia negative classical myeloproliferative neoplasms: A narrative review on epidemiology, risk assessment, and pathophysiologic mechanisms. J. Thromb. Thrombolysis 2018, 45, 516–528. [Google Scholar] [CrossRef]
- Linnemann, B.; Kraft, C.; Roskos, M.; Zgouras, D.; Lindhoff-Last, E. Inferior vena cava thrombosis and its relationship with the JAK2V617F mutation and chronic myeloproliferative disease. Thromb. Res. 2012, 129, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Amarapurkar, D.; Punamiya, S.; Patel, N.; Parekh, S.; Mehta, S.; Shah, N. Prevalence of JAK2V617F mutation in intra-abdominal venous thrombosis. Trop. Gastroenterol. 2012, 32, 279–284. [Google Scholar]
- Owens, C.D. JAK2 V617F mutation, mesenteric vein thrombosis, and myeloproliferative disorders. J. Vasc. Surg. 2010, 52, 205–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, D.W.; Patel, R.K.; Lea, N.C.; Westbrook, R.H.; O’Grady, J.G.; Heaton, N.D.; Pagliuca, A.; Mufti, G.J.; Heneghan, M.A. The prevalence of the activating JAK2 tyrosine kinase mutation in chronic porto-splenomesenteric venous thrombosis. Aliment. Pharm. 2010, 31, 1330–1336. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, E.; Sun, Q.; Ma, J.; Li, Q.; Cao, Z.; Wang, J.; Jia, Y.; Zhang, H.; Song, Z.; et al. The prevalence of JAK2, MPL, and CALR mutations in chinese patients with BCR-ABL1-negative myeloproliferative neoplasms. Am. J. Clin. Pathol. 2015, 144, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Im, K.; Park, S.N.; Kwon, J.; Kim, J.A.; Lee, D.S. CALR, JAK2, and MPL mutation profiles in patients with four different subtypes of myeloproliferative neoplasms: Primary myelofibrosis, essential thrombocythemia, polycythemia vera, and myeloproliferative neoplasm, unclassifiable. Am. J. Clin. Pathol. 2015, 143, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Qiao, C.; Sun, C.; Ouyang, Y.; Wang, J.J.; Qian, S.X.; Li, J.Y.; Zhang, S.J. Clinical importance of different calreticulin gene mutation types in wild-type JAK2 essential thrombocythemia and myelofibrosis patients. Haematologica 2014, 99, e182–e184. [Google Scholar] [CrossRef] [Green Version]
- Schnittger, S.; Bacher, U.; Eder, C.; Dicker, F.; Alpermann, T.; Grossmann, V.; Kohlmann, A.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular analyses of 15,542 patients with suspected BCR-ABL1-negative myeloproliferative disorders allow to develop a stepwise diagnostic workflow. Haematologica 2012, 97, 1582–1585. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Thiele, J.; Vardiman, J.W. The 2008 World Health Organization classification system for myeloproliferative neoplasms: Order out of chaos. Cancer 2009, 115, 3842–3847. [Google Scholar] [CrossRef]
- Kouroupi, E.; Zoi, K.; Parquet, N.; Zoi, C.; Kiladjian, J.J.; Grigoraki, V.; Vainchenker, W.; Lellouche, F.; Marzac, C.; Schlageter, M.H.; et al. Mutations in exon 12 of JAK2 are mainly found in JAK2 V617F-negative polycythaemia vera patients. Br. J. Haematol. 2008, 142, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Pietra, D.; Li, S.; Brisci, A.; Passamonti, F.; Rumi, E.; Theocharides, A.; Ferrari, M.; Gisslinger, H.; Kralovics, R.; Cremonesi, L.; et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 2008, 111, 1686–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Kralovics, R.; De Libero, G.; Theocharides, A.; Gisslinger, H.; Skoda, R.C. Clonal heterogeneity in polycythemia vera patients with JAK2 exon12 and JAK2-V617F mutations. Blood 2008, 111, 3863–3866. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef]
- Fiorini, A.; Chiusolo, P.; Rossi, E.; Za, T.; De Ritis, D.G.; Ciminello, A.; Leone, G.; De Stefano, V. Absence of the JAK2 exon 12 mutations in patients with splanchnic venous thrombosis and without overt myeloproliferative neoplasms. Am. J. Hematol. 2009, 84, 126–127. [Google Scholar] [CrossRef]
- Raghavan, M.; Wijeyesakere, S.J.; Peters, L.R.; Del Cid, N. Calreticulin in the immune system: Ins and outs. Trends Immunol. 2013, 34, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.; Duggan, B.; Atkinson, E.A.; Famulski, K.S.; Nemer, M.; Bleackley, R.C.; Michalak, M. Modulation of gene expression by calreticulin binding to the glucocorticoid receptor. Nature 1994, 367, 476–480. [Google Scholar] [CrossRef]
- De Stefano, V.; Qi, X.; Betti, S.; Rossi, E. Splanchnic vein thrombosis and myeloproliferative neoplasms: Molecular-driven diagnosis and long-term treatment. Thromb. Haemost. 2016, 115, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Mansier, O.; Prouzet-Mauleon, V.; Jegou, G.; Barroso, K.; Raymundo, D.P.; Chauveau, A.; Dumas, P.Y.; Lagarde, V.; Turcq, B.; Pasquet, J.M.; et al. The expression of myeloproliferative neoplasm-associated calreticulin variants depends on the functionality of ER-associated degradation. Cancers 2019, 11, 1921. [Google Scholar] [CrossRef] [Green Version]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Rotunno, G.; Mannarelli, C.; Guglielmelli, P.; Pacilli, A.; Pancrazzi, A.; Pieri, L.; Fanelli, T.; Bosi, A.; Vannucchi, A.M. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014, 123, 1552–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poisson, J.; Plessier, A.; Kiladjian, J.J.; Turon, F.; Cassinat, B.; Andreoli, A.; De Raucourt, E.; Goria, O.; Zekrini, K.; Bureau, C.; et al. Selective testing for calreticulin gene mutations in patients with splanchnic vein thrombosis: A prospective cohort study. J. Hepatol. 2017, 67, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Turon, F.; Cervantes, F.; Colomer, D.; Baiges, A.; Hernandez-Gea, V.; Garcia-Pagan, J.C. Role of calreticulin mutations in the aetiological diagnosis of splanchnic vein thrombosis. J. Hepatol. 2015, 62, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef]
- Martino, B.; Mammi, C.; Labate, C.; Antonia, M.E.; Ronco, F.; Recchia, A.G.; Ielo, D.; Alati, C.; Oliva, E.; Lagana, C.; et al. On the absence of calreticulin (CALR) mutations in chronic myeloprolierative neoplasms (MPNs) with splanchnic venous thrombosis (SVT): Experience from a single institution. Blood 2014, 124, 3198. [Google Scholar] [CrossRef]
- Le Coniat, M.; Souyri, M.; Vigon, I.; Wendling, F.; Tambourin, P.; Berger, R. The human homolog of the myeloproliferative virus maps to chromosome band 1p34. Hum. Genet. 1989, 83, 194–196. [Google Scholar] [CrossRef]
- Vigon, I.; Mornon, J.P.; Cocault, L.; Mitjavila, M.T.; Tambourin, P.; Gisselbrecht, S.; Souyri, M. Molecular cloning and characterization of MPL, the human homolog of the v-mpl oncogene: Identification of a member of the hematopoietic growth factor receptor superfamily. Proc. Natl. Acad. Sci. USA 1992, 89, 5640–5644. [Google Scholar] [CrossRef] [Green Version]
- Plo, I.; Bellanne-Chantelot, C.; Mosca, M.; Mazzi, S.; Marty, C.; Vainchenker, W. Genetic alterations of the thrombopoietin/MPL/JAK2 axis impacting megakaryopoiesis. Front. Endocrinol. 2017, 8, 234. [Google Scholar] [CrossRef]
- Teofili, L.; Giona, F.; Martini, M.; Cenci, T.; Guidi, F.; Torti, L.; Palumbo, G.; Amendola, A.; Foa, R.; Larocca, L.M. Markers of myeloproliferative diseases in childhood polycythemia vera and essential thrombocythemia. J. Clin. Oncol. 2007, 25, 1048–1053. [Google Scholar] [CrossRef]
- Buxhofer-Ausch, V.; Olcaydu, D.; Gisslinger, B.; Schalling, M.; Frantal, S.; Thiele, J.; Mullauer, L.; Kvasnicka, H.M.; Watzke, H.; Kralovics, R.; et al. Decanucleotide insertion polymorphism of F7 significantly influences the risk of thrombosis in patients with essential thrombocythemia. Eur. J. Haematol. 2014, 93, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Green, A.R. The myeloproliferative disorders. N. Engl. J. Med. 2006, 355, 2452–2466. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.A.; Campbell, P.J.; Scott, L.M.; Bench, A.J.; Erber, W.N.; Bareford, D.; Wilkins, B.S.; Reilly, J.T.; Hasselbalch, H.C.; Bowman, R.; et al. MPL mutations in myeloproliferative disorders: Analysis of the PT-1 cohort. Blood 2008, 112, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Akpinar, T.S.; Hancer, V.S.; Nalcaci, M.; Diz-Kucukkaya, R. MPL W515L/K mutations in chronic myeloproliferative neoplasms. Turk. J. Haematol. 2013, 30, 8–12. [Google Scholar] [CrossRef]
- Tefferi, A.; Levine, R.L.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Finke, C.M.; Mullally, A.; Li, C.Y.; Pardanani, A.; et al. Frequent TET2 mutations in systemic mastocytosis: Clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia 2009, 23, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Jeong, J.J.; Gu, X.; Nie, J.; Sundaravel, S.; Liu, H.; Kuo, W.L.; Bhagat, T.D.; Pradhan, K.; Cao, J.; Nischal, S.; et al. Cytokine-regulated phosphorylation and activation of TET2 by JAK2 in hematopoiesis. Cancer Discov. 2019, 9, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S. Dysregulation of TET2 in hematologic malignancies. Int. J. Hematol. 2017, 105, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of TET2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.S.; Jeon, D.S.; Kim, J.R.; Ryoo, N.H.; Suh, J.S. Analysis of the Ten-Eleven Translocation 2 (TET2) gene mutation in myeloproliferative neoplasms. Ann. Clin. Lab. Sci. 2014, 44, 173–179. [Google Scholar] [PubMed]
- Tefferi, A.; Pardanani, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Gangat, N.; Finke, C.M.; Schwager, S.; Mullally, A.; et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009, 23, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Colaizzo, D.; Tiscia, G.L.; Pisanelli, D.; Bafunno, V.; Amitrano, L.; Grandone, E.; Guardascione, M.A.; Margaglione, M. New TET2 gene mutations in patients with myeloproliferative neoplasms and splanchnic vein thrombosis. J. Thromb. Haemost. 2010, 8, 1142–1144. [Google Scholar] [CrossRef]
- Segura-Díaz, A.; Stuckey, R.; Florido, Y.; González-Martín, J.M.; López-Rodríguez, J.F.; Sánchez-Sosa, S.; González-Pérez, E.; Sáez Perdomo, M.N.; Perera, M.d.M.; de la Iglesia, S. Thrombotic risk detection in patients with polycythemia vera: The predictive role of DNMT3A/TET2/ASXL1 mutations. Cancers 2020, 12, 934. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.L.; Berger, J.; Randazzo, F.; Brock, H.W. A human homolog of Additional sex combs, ADDITIONAL SEX COMBS-LIKE 1, maps to chromosome 20q11. Gene 2003, 306, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Functional and cancer genomics of ASXL family members. Br. J. Cancer 2013, 109, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurnari, C.; Falconi, G.; De Bellis, E.; Voso, M.T.; Fabiani, E. The role of forkhead box proteins in acute myeloid leukemia. Cancers 2019, 11, 865. [Google Scholar] [CrossRef] [Green Version]
- Viny, A.D.; Levine, R.L. Genetics of myeloproliferative neoplasms. Cancer J. 2014, 20, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Milosevic, J.D.; Kralovics, R. Genetic and epigenetic alterations of myeloproliferative disorders. Int. J. Hematol. 2013, 97, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbuccia, N.; Murati, A.; Trouplin, V.; Brecqueville, M.; Adelaide, J.; Rey, J.; Vainchenker, W.; Bernard, O.A.; Chaffanet, M.; Vey, N.; et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009, 23, 2183–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, O.; Pardanani, A.; Patel, J.; Wadleigh, M.; Lasho, T.; Heguy, A.; Beran, M.; Gilliland, D.G.; Levine, R.L.; Tefferi, A. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia 2011, 25, 1200–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.B.; Sun, M.; He, C.K.; Ju, M.K.; Zhou, F.L.; Wu, S.Y.; Zhou, Y.; Liu, L.; Shen, H.; Huang, T.T.; et al. ASXL1 mutations in Chinese patients with essential thrombocythemia. Exp. Med. 2018, 15, 4149–4156. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Marin, F.; Bellosillo, B.; Martinez-Aviles, L.; Soler, G.; Carbonell, P.; Luengo-Gil, G.; Caparros, E.; Torregrosa, J.M.; Besses, C.; Vicente, V. Leukemic transformation driven by an ASXL1 mutation after a JAK2V617F-positive primary myelofibrosis: Clonal evolution and hierarchy revealed by next-generation sequencing. J. Hematol. Oncol. 2013, 6, 68. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Courtier, F.; Garnier, S.; Carbuccia, N.; Guille, A.; Adélaide, J.; Chaffanet, M.; Hirsch, P.; Paz, D.L.; Slama, B.; Vey, N.; et al. Targeted molecular characterization shows differences between primary and secondary myelofibrosis. Genes Chromosomes Cancer 2020, 59, 30–39. [Google Scholar] [CrossRef]
- Hobbs, G.S.; Rampal, R.K. Clinical and molecular genetic characterization of myelofibrosis. Curr. Opin. Hematol. 2015, 22, 177–183. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martinez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Krahling, T.; Balassa, K.; Kiss, K.P.; Bors, A.; Batai, A.; Halm, G.; Egyed, M.; Fekete, S.; Remenyi, P.; Masszi, T.; et al. Co-occurrence of myeloproliferative neoplasms and solid tumors is attributed to a synergism between cytoreductive therapy and the common TERT polymorphism rs2736100. Cancer Epidemiol. Biomark. Prev. 2016, 25, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira Cristina, S.; Polo, B.; Lacerda, J.F. Somatic mutations in Philadelphia chromosome-negative myeloproliferative neoplasms. Semin. Hematol. 2018, 55, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finazzi, M.C.; Carobbio, A.; Cervantes, F.; Isola, I.M.; Vannucchi, A.M.; Guglielmelli, P.; Rambaldi, A.; Finazzi, G.; Barosi, G.; Barbui, T. CALR mutation, MPL mutation and triple negativity identify patients with the lowest vascular risk in primary myelofibrosis. Leukemia 2015, 29, 1209–1210. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Gau, J.P.; Chou, H.J.; You, J.Y.; Huang, C.E.; Chen, Y.Y.; Lung, J.; Chou, Y.S.; Leu, Y.W.; Lu, C.H.; et al. Frequencies, clinical characteristics, and outcome of somatic CALR mutations in JAK2-unmutated essential thrombocythemia. Ann. Hematol. 2014, 93, 2029–2036. [Google Scholar] [CrossRef]
- Gangat, N.; Wassie, E.A.; Lasho, T.L.; Finke, C.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.; Wolanskyj, A.P.; Maffioli, M.; Casalone, R.; et al. Mutations and thrombosis in essential thrombocythemia: Prognostic interaction with age and thrombosis history. Eur. J. Haematol. 2015, 94, 31–36. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V.; Lopez, J.A.; Gray, L.A.; Padilla, A.; Borthakur, G.; Roberts, S.C.; Pruthi, R.K.; Tefferi, A. Hemostatic gene polymorphisms and the prevalence of thrombotic complications in polycythemia vera and essential thrombocythemia. Blood Coagul Fibrinolysis 2004, 15, 21–24. [Google Scholar] [CrossRef]
- Randi, M.L.; Lombardi, A.M.; Scapin, M.; Tezza, F.; Scandellari, R.; Ruzzon, E.; Duner, E.; Fabris, F. Haemostatic proteins gene polymorphisms in patients with unusual vein thrombosis and Ph-myeloproliferative disorders. Thromb. Haemost. 2007, 98, 702–704. [Google Scholar]
- Dambrauskiene, R.; Gerbutavicius, R.; Ugenskiene, R.; Jankauskaite, R.; Savukaityte, A.; Simoliuniene, R.; Rudzianskiene, M.; Gerbutaviciene, R.; Juozaityte, E. Genetic polymorphisms of hemostatic factors and thrombotic risk in non BCR-ABL myeloproliferative neoplasms: A pilot study. Balkan J. Med. Genet. 2017, 20, 35–42. [Google Scholar] [CrossRef]
- Azevedo, A.P.; Silva, S.N.; Reichert, A.; Lima, F.; Junior, E.; Rueff, J. Effects of polymorphic DNA genes involved in BER and caspase pathways on the clinical outcome of myeloproliferative neoplasms under treatment with hydroxyurea. Mol. Med. Rep. 2018, 18, 5243–5255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soyer, N.; Küçükarslan, A.S.; Sahin, F.; Çekdemir, D.; Kosova, B.; Eroglu, Z.; Töbü, M.; Tombuloglu, M.; Çagirgan, S.; Dönmez, A. Factor V G1691A (Leiden) and prothrombin G20210A gene mutation status, and thrombosis in patients with chronic myeloproliferative disorders/Kronik myeloproliferatif hastalik tanili hastalarda Factor V 1691A (Leiden) ve protrombin G20210A gen mutasyonu ve tromboz. Turk. J. Hematol. 2011, 28, 306. [Google Scholar]
- Ruggeri, M.; Gisslinger, H.; Tosetto, A.; Rintelen, C.; Mannhalter, C.; Pabinger, I.; Heis, N.; Castaman, G.; Missiaglia, E.; Lechner, K.; et al. Factor V Leiden mutation carriership and venous thromboembolism in polycythemia vera and essential thrombocythemia. Am. J. Hematol. 2002, 71, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; de Nully Brown, P.; Thorsen, S.; Hasselbalch, H.C. Frequent occurrence of anticardiolipin antibodies, Factor V Leiden mutation, and perturbed endothelial function in chronic myeloproliferative disorders. Am. J. Hematol. 2002, 69, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Maral, S.; Acar, M.; Balcik, O.S.; Uctepe, E.; Hatipoglu, O.F.; Akdeniz, D.; Altun, H.U.; Kosar, A.; Gunduz, M.; Gunduz, E. Matrix metalloproteinases 2 and 9 polymorphism in patients with myeloproliferative diseases: A STROBE-compliant observational study. Medicine 2015, 94, e732. [Google Scholar] [CrossRef] [PubMed]
- Horvat, I.; Antolić, M.R.; Rončević, P.; Serventi-Seiwerth, R.; Zadro, R. Thrombosis in MPN patients and gene frequency of human platelet antigens. In Proceedings of the MPN & MPNr-EuroNet Tenth Meeting “Biology of sporadic and Hereditary Myeloproliferative Disease”, Zagreb, Croatia, 9–10 April 2015; p. 50. [Google Scholar]
- Papadakis, E.; Papageorgiou, V.; Tsepanis, K.; Theocharidou, D.; Papadopoulos, V.K.; Georgiou, E.; Efraimidou, S.; Kioumi, A. Impact of inherited thrombophilia factors on thrombotic risk in patients with newly diagnosed BCR-ABL (-) myeloproliferative disorders: Finally a role of MTHFR-C677T polymorphism? Blood 2012, 120, 5065. [Google Scholar] [CrossRef]
- Gerli, G.; Vanelli, C.; Turri, O.; Erario, M.; Gardellini, A.; Pugliano, M.; Biondi, M.L. SDF1-3’A gene polymorphism is associated with chronic myeloproliferative disease and thrombotic events. Clin. Chem. 2005, 51, 2411–2414. [Google Scholar] [CrossRef]
- Tognon, R.; Nunes Nde, S.; Castro, F.A. Apoptosis deregulation in myeloproliferative neoplasms. Einstein (Sao Paulo) 2013, 11, 540–544. [Google Scholar] [CrossRef] [Green Version]
- De Melo Campos, P.; Machado-Neto, J.A.; Eide, C.A.; Savage, S.L.; Scopim-Ribeiro, R.; da Silva Souza Duarte, A.; Favaro, P.; Lorand-Metze, I.; Costa, F.F.; Tognon, C.E.; et al. IRS2 silencing increases apoptosis and potentiates the effects of ruxolitinib in JAK2V617F-positive myeloproliferative neoplasms. Oncotarget 2016, 7, 6948–6959. [Google Scholar] [CrossRef] [Green Version]
- Ozdemirkiran, F.G.; Nalbantoglu, S.; Gokgoz, Z.; Payzin, B.K.; Vural, F.; Cagirgan, S.; Berdeli, A. FAS/FASL gene polymorphisms in Turkish patients with chronic myeloproliferative disorders. Arch. Med. Sci. 2017, 13, 426–432. [Google Scholar] [CrossRef] [Green Version]
- Fagniez, O.; Tertian, G.; Dreyfus, M.; Ducreux, D.; Adams, D.; Denier, C. Hematological disorders related cerebral infarctions are mostly multifocal. J. Neurol. Sci. 2011, 304, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Gorukmez, O.; Sag, S.O.; Gorukmez, O.; Ture, M.; Topak, A.; Sahinturk, S.; Ozkaya, G.; Gulten, T.; Ali, R.; Yakut, T. Association of the ACE I/D gene polymorphisms with JAK2V617F-positive polycythemia vera and essential thrombocythemia. Genet. Test. Mol. Biomark. 2015, 19, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Speletas, M.; Liadaki, K.; Kalala, F.; Daiou, C.; Katodritou, E.; Mandala, E.; Korantzis, I.; Ritis, K.; Zintzaras, E.; Germenis, A.E. TLR4 single nucleotide polymorphisms and thrombosis risk in patients with myeloproliferative disorders. Thromb. Res. 2008, 122, 27–32. [Google Scholar] [CrossRef]
- Métivier, R.; Gallais, R.; Tiffoche, C.; Le Péron, C.; Jurkowska, R.Z.; Carmouche, R.P.; Ibberson, D.; Barath, P.; Demay, F.; Reid, G. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008, 452, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Zee, B.M.; Levin, R.S.; Xu, B.; LeRoy, G.; Wingreen, N.S.; Garcia, B.A. In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 2010, 285, 3341–3350. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Xu, M.; Zhu, B. Epigenetic inheritance mediated by histone lysine methylation: Maintaining transcriptional states without the precise restoration of marks? Philos. Trans. R Soc. Lond B Biol. Sci. 2013, 368, 20110332. [Google Scholar] [CrossRef] [Green Version]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Schaub, F.X.; Looser, R.; Li, S.; Hui, H.S.; Lehmann, T.; Tichelli, A.; Skoda, R.C. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 2010, 115, 2003–2007. [Google Scholar] [CrossRef] [Green Version]
- Hussein, K.; Abdel-Wahab, O.; Lasho, T.L.; Van Dyke, D.L.; Levine, R.L.; Hanson, C.A.; Pardanani, A.; Tefferi, A. Cytogenetic correlates of TET2 mutations in 199 patients with myeloproliferative neoplasms. Am. J. Hematol. 2010, 85, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Green, A.; Beer, P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N. Engl. J. Med. 2010, 362, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Mai, M.; McClure, R.F.; Tefferi, A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia 2010, 24, 1146–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Jager, R.; Gisslinger, H.; Passamonti, F.; Rumi, E.; Berg, T.; Gisslinger, B.; Pietra, D.; Harutyunyan, A.; Klampfl, T.; Olcaydu, D.; et al. Deletions of the transcription factor Ikaros in myeloproliferative neoplasms. Leukemia 2010, 24, 1290–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nischal, S.; Zhou, L.; Yu, Y.T.; Bhagat, T.; Mo, Y.K.; Heuck, C.; Suzuki, M.; Maqbool, S.; Pardanani, A.; Greally, J.; et al. Epigenomic profiling of myeloproliferative diseases reveal idiopathic myelofibrosis as an epigenetically distinct subgroup and highlights the epigenetic effects of JAK2V617F mutation. Blood 2010, 116, 276. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Gottgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Mascarenhas, J.; Roper, N.; Chaurasia, P.; Hoffman, R. Epigenetic abnormalities in myeloproliferative neoplasms: A target for novel therapeutic strategies. Clin. Epigenetics 2011, 2, 197–212. [Google Scholar] [CrossRef] [Green Version]
- Ihalainen, J.; Juvonen, E.; Savolainen, E.R.; Ruutu, T.; Palotie, A. Calcitonin gene methylation in chronic myeloproliferative disorders. Leukemia 1994, 8, 230–235. [Google Scholar]
- Aviram, A.; Witenberg, B.; Shaklai, M.; Blickstein, D. Detection of methylated ABL1 promoter in philadelphia-negative myeloproliferative disorders. Blood Cells Mol. Dis. 2003, 30, 100–106. [Google Scholar] [CrossRef]
- Bennemann, K.; Schubert, C.; Wilop, S.; Brummendorf, T.H.; Galm, O.; Jost, E. Epigenetic downregulation of secreted frizzled-related proteins in Philadelphia positive and Philadelphia negative myeloproliferative neoplasms. Blood 2010, 116, 4647. [Google Scholar] [CrossRef]
- Suzuki, R.; Onizuka, M.; Kojima, M.; Shimada, M.; Tsuboi, K.; Ogawa, Y.; Kawada, H.; Ando, K. Infrequent hypermethylation of WIF-1 promoter in BCR/ABL-negative myeloproliferative disorders. Tokai J. Exp. Clin. Med. 2007, 32, 131–135. [Google Scholar] [PubMed]
- Capello, D.; Deambrogi, C.; Rossi, D.; Lischetti, T.; Piranda, D.; Cerri, M.; Spina, V.; Rasi, S.; Gaidano, G.; Lunghi, M. Epigenetic inactivation of suppressors of cytokine signalling in Philadelphia-negative chronic myeloproliferative disorders. Br. J. Haematol. 2008, 141, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Jost, E.; do O, N.; Dahl, E.; Maintz, C.E.; Jousten, P.; Habets, L.; Wilop, S.; Herman, J.G.; Osieka, R.; Galm, O. Epigenetic alterations complement mutation of JAK2 tyrosine kinase in patients with BCR/ABL-negative myeloproliferative disorders. Leukemia 2007, 21, 505–510. [Google Scholar] [CrossRef] [Green Version]
- Fourouclas, N.; Li, J.; Gilby, D.C.; Campbell, P.J.; Beer, P.A.; Boyd, E.M.; Goodeve, A.C.; Bareford, D.; Harrison, C.N.; Reilly, J.T.; et al. Methylation of the suppressor of cytokine signaling 3 gene (SOCS3) in myeloproliferative disorders. Haematologica 2008, 93, 1635–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Mercado, M.; Cebrian, V.; Euba, B.; Garcia-Granero, M.; Calasanz, M.J.; Novo, F.J.; Vizmanos, J.L.; Garcia-Delgado, M. Methylation status of SOCS1 and SOCS3 in BCR-ABL negative and JAK2V617F negative chronic myeloproliferative neoplasms. Leuk Res. 2008, 32, 1638–1640. [Google Scholar] [CrossRef]
- Jelinek, J.; Li, J.; Mnjoyan, Z.; Issa, J.P.; Prchal, J.T.; Afshar-Kharghan, V. Epigenetic control of PRV-1 expression on neutrophils. Exp. Hematol. 2007, 35, 1677–1683. [Google Scholar] [CrossRef]
- Bogani, C.; Ponziani, V.; Guglielmelli, P.; Desterke, C.; Rosti, V.; Bosi, A.; Le Bousse-Kerdiles, M.C.; Barosi, G.; Vannucchi, A.M.; Myeloproliferative Disorders Research C. Hypermethylation of CXCR4 promoter in CD34+ cells from patients with primary myelofibrosis. Stem Cells 2008, 26, 1920–1930. [Google Scholar] [CrossRef]
- Jones, L.C.; Tefferi, A.; Idos, G.E.; Kumagai, T.; Hofmann, W.K.; Koeffler, H.P. RARbeta2 is a candidate tumor suppressor gene in myelofibrosis with myeloid metaplasia. Oncogene 2004, 23, 7846–7853. [Google Scholar] [CrossRef] [Green Version]
- Teofili, L.; Martini, M.; Iachininoto, M.G.; Capodimonti, S.; Nuzzolo, E.R.; Torti, L.; Cenci, T.; Larocca, L.M.; Leone, G. Endothelial progenitor cells are clonal and exhibit the JAK2(V617F) mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms. Blood 2011, 117, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Stein, B.L.; Williams, D.M.; Wang, N.-Y.; Rogers, O.; Isaacs, M.A.; Pemmaraju, N.; Spivak, J.L.; Moliterno, A.R. Sex differences in the JAK2V617F allele burden in chronic myeloproliferative disorders. Haematologica 2010, 95, 1090–1097. [Google Scholar] [CrossRef] [Green Version]
- De Stefano, V.; Za, T.; Rossi, E.; Vannucchi, A.M.; Ruggeri, M.; Elli, E.; Mico, C.; Tieghi, A.; Cacciola, R.R.; Santoro, C.; et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: Incidence, risk factors, and effect of treatments. Haematologica 2008, 93, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Gugliotta, L.; Iurlo, A.; Gugliotta, G.; Tieghi, A.; Specchia, G.; Gaidano, G.; Scalzulli, P.R.; Rumi, E.; Dragani, A.; Martinelli, V.; et al. Unbiased pro-thrombotic features at diagnosis in 977 thrombocythemic patients with Philadelphia-negative chronic myeloproliferative neoplasms. Leuk Res. 2016, 46, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2015, 90, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Vannucchi, A.M.; Buxhofer-Ausch, V.; De Stefano, V.; Betti, S.; Rambaldi, A.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015, 5, e369. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, E.; Guglielmelli, P.; Poli, G.; Bogani, C.; Pancrazzi, A.; Longo, G.; Ponziani, V.; Tozzi, L.; Pieri, L.; Santini, V.; et al. Influence of JAK2V617F allele burden on phenotype in essential thrombocythemia. Haematologica 2008, 93, 41–48. [Google Scholar] [CrossRef]
- Falchi, L.; Kantarjian, H.M.; Verstovsek, S. Assessing the thrombotic risk of patients with essential thrombocythemia in the genomic era. Leukemia 2017, 31, 1845–1854. [Google Scholar] [CrossRef]
- Popov, V.M.; Onisai, M.; Găman, M.; Vladareanu, A.M. The role of JAK2 mutation in thrombotic complications of chronic myeloproliferative neoplasms. Hematology 2014, 1, 106–113. [Google Scholar]
- Kelliher, S.; Falanga, A. Thrombosis in myeloproliferative neoplasms: A clinical and pathophysiological perspective. Thromb. Update 2021, 5, 100081. [Google Scholar] [CrossRef]
- Marin Oyarzún, C.P.; Heller, P.G. Platelets as Mediators of Thromboinflammation in Chronic Myeloproliferative Neoplasms. Front. Immunol. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangaraju, R.; Song, J.; Kim, S.J.; Tashi, T.; Reeves, B.N.; Sundar, K.M.; Thiagarajan, P.; Prchal, J.T. Thrombotic, inflammatory, and HIF-regulated genes and thrombosis risk in polycythemia vera and essential thrombocythemia. Blood Adv. 2020, 4, 1115–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treliński, J.; Chojnowski, K.; Cebula-Obrzut, B.; Smolewski, P. Impaired apoptosis of megakaryocytes and bone marrow mononuclear cells in essential thrombocythemia: Correlation with JAK2V617F mutational status and cytoreductive therapy. Med. Oncol. 2012, 29, 2388–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landolfi, R.; Marchioli, R.; Kutti, J.; Gisslinger, H.; Tognoni, G.; Patrono, C.; Barbui, T. Efficacy and safety of low-dose aspirin in polycythemia vera. N. Engl. J. Med. 2004, 350, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Fruchtman, S.M.; Mack, K.; Kaplan, M.E.; Peterson, P.; Berk, P.D. From efficacy to safety: A Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin. Hematol. 1997, 34, 17–23. [Google Scholar]
- Bjorkholm, M.; Derolf, A.R.; Hultcrantz, M.; Kristinsson, S.Y.; Ekstrand, C.; Goldin, L.R.; Andreasson, B.; Birgegard, G.; Linder, O.; Malm, C.; et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J. Clin. Oncol. 2011, 29, 2410–2415. [Google Scholar] [CrossRef] [Green Version]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Masciulli, A.; Mennitto, M.R.; Barbui, T. The CYTO-PV: A large-scale trial testing the intensity of CYTOreductive therapy to prevent cardiovascular events in patients with polycythemia vera. Thrombosis 2011, 2011, 794240. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Vannucchi, A.M.; Finazzi, G.; Finazzi, M.C.; Masciulli, A.; Carobbio, A.; Ghirardi, A.; Tognoni, G. A reappraisal of the benefit-risk profile of hydroxyurea in polycythemia vera: A propensity-matched study. Am. J. Hematol. 2017, 92, 1131–1136. [Google Scholar] [CrossRef] [Green Version]
- Greenfield, G.; McMullin, M.F. Splanchnic venous thrombosis in JAK2 V617F mutation positive myeloproliferative neoplasms—Long term follow-up of a regional case series. Thromb. J. 2018, 16, 33. [Google Scholar] [CrossRef]
- Harrison, C.N.; Campbell, P.J.; Buck, G.; Wheatley, K.; East, C.L.; Bareford, D.; Wilkins, B.S.; van der Walt, J.D.; Reilly, J.T.; Grigg, A.P.; et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N. Engl. J. Med. 2005, 353, 33–45. [Google Scholar] [CrossRef]
- Campbell, P.J.; Bareford, D.; Erber, W.N.; Wilkins, B.S.; Wright, P.; Buck, G.; Wheatley, K.; Harrison, C.N.; Green, A.R. Reticulin accumulation in essential thrombocythemia: Prognostic significance and relationship to therapy. J. Clin. Oncol. 2009, 27, 2991–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbui, T.; Finazzi, M.C.; Finazzi, G. Front-line therapy in polycythemia vera and essential thrombocythemia. Blood Rev. 2012, 26, 205–211. [Google Scholar] [CrossRef]
- Alvarez-Larrán, A.; Sant’Antonio, E.; Harrison, C.; Kiladjian, J.-J.; Griesshammer, M.; Mesa, R.; Ianotto, J.C.; Palandri, F.; Hernández-Boluda, J.C.; Birgegård, G.; et al. Unmet clinical needs in the management of CALR-mutated essential thrombocythaemia: A consensus-based proposal from the European LeukemiaNet. Lancet Haematol. 2021, 8, e658–e665. [Google Scholar] [CrossRef]
- Besses, C.; Álvarez-Larrán, A.; Martínez-Avilés, L.; Mojal, S.; Longarón, R.; Salar, A.; Florensa, L.; Serrano, S.; Bellosillo, B. Modulation of JAK2 V617F allele burden dynamics by hydroxycarbamide in polycythaemia vera and essential thrombocythaemia patients. Br. J. Haematol. 2011, 152, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Senín, A.; Fernández-Rodríguez, C.; Bellosillo, B.; Camacho, L.; Longarón, R.; Angona, A.; Besses, C.; Álvarez-Larrán, A. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann. Hematol. 2018, 97, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Griesshammer, M.; Gisslinger, H.; Mesa, R. Current and future treatment options for polycythemia vera. Ann. Hematol. 2015, 94, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Kiladjian, J.J.; Cassinat, B.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Menot, M.L.; Massonnet, G.; Dutel, J.L.; Ghomari, K.; et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon alpha-2a. Blood 2006, 108, 2037–2040. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhang, L. Thrombosis in myeloproliferative neoplasms with JAK2V617F mutation. Clin. Appl. Thromb. 2013, 19, 374–381. [Google Scholar] [CrossRef]
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.-J.; Hehlmann, R.; et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: Revised management recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, M.; Vannucchi, A.M.; Griesshammer, M.; Harrison, C.; Koschmieder, S.; Gisslinger, H.; Álvarez-Larrán, A.; De Stefano, V.; Guglielmelli, P.; Palandri, F.; et al. Appropriate management of polycythaemia vera with cytoreductive drug therapy: European LeukemiaNet 2021 recommendations. Lancet Haematol. 2022, 9, e301–e311. [Google Scholar] [CrossRef]
- Barbui, T.; Finazzi, G. When and how to treat essential thrombocythemia. N. Engl. J. Med. 2005, 353, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Pieri, L.; Paoli, C.; Arena, U.; Marra, F.; Mori, F.; Zucchini, M.; Colagrande, S.; Castellani, A.; Masciulli, A.; Rosti, V.; et al. Safety and efficacy of ruxolitinib in splanchnic vein thrombosis associated with myeloproliferative neoplasms. Am. J. Hematol. 2017, 92, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintás-Cardama, A.; Vaddi, K.; Liu, P.; Manshouri, T.; Li, J.; Scherle, P.A.; Caulder, E.; Wen, X.; Li, Y.; Waeltz, P. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010, 115, 3109–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, G.; McPherson, S.; Mills, K.; McMullin, M.F. The ruxolitinib effect: Understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J. Transl. Med. 2018, 16, 360. [Google Scholar] [CrossRef]
- Shallis, R.M.; Podoltsev, N.A. Emerging agents and regimens for polycythemia vera and essential thrombocythemia. Biomark. Res. 2021, 9, 40. [Google Scholar] [CrossRef]
- Tefferi, A.; Barraco, D.; Lasho, T.L.; Shah, S.; Begna, K.H.; Al-Kali, A.; Hogan, W.J.; Litzow, M.R.; Hanson, C.A.; Ketterling, R.P.; et al. Momelotinib therapy for myelofibrosis: A 7-year follow-up. Blood Cancer J. 2018, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: A randomized clinical trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Harrison, C.N.; Mesa, R.A.; Jamieson, C.; Hood, J.; Bykowski, J.; Zuccoli, G.; Brewer, J. Case series of potential Wernicke’s encephalopathy in patients treated with fedratinib. Blood 2017, 130, 4197. [Google Scholar] [CrossRef]
- Verstovsek, S.; Odenike, O.; Singer, J.W.; Granston, T.; Al-Fayoumi, S.; Deeg, H.J. Phase 1/2 study of pacritinib, a next generation JAK2/FLT3 inhibitor, in myelofibrosis or other myeloid malignancies. J. Hematol. Oncol. 2016, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascarenhas, J.; Virtgaym, E.; Stal, M.; Blacklock, H.; Gerds, A.T.; Mesa, R.; Ganly, P.; Snyder, D.; Tabbara, I.; Tremblay, D.; et al. Outcomes of patients with myelofibrosis treated with compassionate use pacritinib: A sponsor-independent international study. Ann. Hematol. 2018, 97, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Barosi, G.; Birgegard, G.; Cervantes, F.; Finazzi, G.; Griesshammer, M.; Harrison, C.; Hasselbalch, H.C.; Hehlmann, R.; Hoffman, R.; et al. Philadelphia-negative classical myeloproliferative neoplasms: Critical concepts and management recommendations from European LeukemiaNet. J. Clin. Oncol. 2011, 29, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finazzi, G.; Caruso, V.; Marchioli, R.; Capnist, G.; Chisesi, T.; Finelli, C.; Gugliotta, L.; Landolfi, R.; Kutti, J.; Gisslinger, H.; et al. Acute leukemia in polycythemia vera: An analysis of 1638 patients enrolled in a prospective observational study. Blood 2005, 105, 2664–2670. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Ruggeri, M.; Rodeghiero, F.; Barbui, T. Second malignancies in patients with essential thrombocythaemia treated with busulphan and hydroxyurea: Long-term follow-up of a randomized clinical trial. Br. J. Haematol. 2000, 110, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Larrán, A.; Martínez-Avilés, L.; Hernández-Boluda, J.C.; Ferrer-Marín, F.; Antelo, M.L.; Burgaleta, C.; Mata, M.I.; Xicoy, B.; Martínez-Trillos, A.; Gómez-Casares, M.T.; et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann. Hematol. 2014, 93, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Larran, A.; Pereira, A.; Guglielmelli, P.; Hernandez-Boluda, J.C.; Arellano-Rodrigo, E.; Ferrer-Marin, F.; Samah, A.; Griesshammer, M.; Kerguelen, A.; Andreasson, B.; et al. Antiplatelet therapy versus observation in low-risk essential thrombocythemia with a CALR mutation. Haematologica 2016, 101, 926–931. [Google Scholar] [CrossRef]
- De Stefano, V.; Vannucchi, A.M.; Ruggeri, M.; Cervantes, F.; Alvarez-Larran, A.; Iurlo, A.; Randi, M.L.; Pieri, L.; Rossi, E.; Guglielmelli, P.; et al. Splanchnic vein thrombosis in myeloproliferative neoplasms: Risk factors for recurrences in a cohort of 181 patients. Blood Cancer J. 2016, 6, e493. [Google Scholar] [CrossRef] [Green Version]
- Finazzi, G.; De Stefano, V.; Barbui, T. Splanchnic vein thrombosis in myeloproliferative neoplasms: Treatment algorithm 2018. Blood Cancer J. 2018, 8, 64. [Google Scholar] [CrossRef] [Green Version]
- Schieppati, F.; Falanga, A. Evidence-Based Minireview: Are DOACs an alternative to vitamin K antagonists for treatment of venous thromboembolism in patients with MPN? Hematology 2021, 2021, 448–452. [Google Scholar] [CrossRef]
- Krecak, I.; Lucijanic, M.; Verstovsek, S. Advances in Risk Stratification and Treatment of Polycythemia Vera and Essential Thrombocythemia. Curr. Hematol. Malig. Rep. 2022, 17, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Berk, P.; Goldberg, J.; Donovan, P.; Fruchtman, S.; Berlin, N.; Wasserman, L. Therapeutic recommendations in polycythemia vera based on Polycythemia Vera Study Group protocols. Semin. Hematol. 1986, 23, 132–143. [Google Scholar] [PubMed]
- Pearson, T.; Wetherley-Mein, G. Vascular occlusive episodes and venous hæmatocrit in primary proliferative polycythæmlx. Lancet 1978, 312, 1219–1222. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Barbui, T. How I treat patients with polycythemia vera. Blood 2007, 109, 5104–5111. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Passamonti, F.; Accorsi, P.; Pane, F.; Vannucchi, A.M.; Velati, C.; Gale, R.P.; Tura, S.; Barosi, G. Evidence- and consensus-based recommendations for phlebotomy in polycythemia vera. Leukemia 2018, 32, 2077–2081. [Google Scholar] [CrossRef]



| Type of MPN | Polycythaemia Vera (PV) | Essential Thrombocythaemia (ET) | Primary Myelofibrosis (PMF) |
|---|---|---|---|
| Bone marrow trephine biopsy under 20× |  |  |  |
| Bone marrow trephine biopsy under 40× |  |  |  |
| Description | Hypercellularity, panmyelosis with a notable predominance of megakaryocytic and erythroid lineages, and megakaryocytes increase in size and with frequent hyperlobated forms, minimal or absence of reticulin fibrosis. | Normal or mildly hypercellularity, a normal proliferation of erythroid and granulocytic lineages, enlarged megakaryocytes (mature with hyperlobulated nuclei), delicate reticulin fibres. | Hypercellularity with clustered and dysplastic large size megakaryocytes, presence of hyperlobulated and hyperchromatic megakaryocytes, often naked megakaryocytes, increase reticulin around clustered megakaryocytes. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chia, Y.C.; Siti Asmaa, M.J.; Ramli, M.; Woon, P.Y.; Johan, M.F.; Hassan, R.; Islam, M.A. Molecular Genetics of Thrombotic Myeloproliferative Neoplasms: Implications in Precision Oncology. Diagnostics 2023, 13, 163. https://doi.org/10.3390/diagnostics13010163
Chia YC, Siti Asmaa MJ, Ramli M, Woon PY, Johan MF, Hassan R, Islam MA. Molecular Genetics of Thrombotic Myeloproliferative Neoplasms: Implications in Precision Oncology. Diagnostics. 2023; 13(1):163. https://doi.org/10.3390/diagnostics13010163
Chicago/Turabian StyleChia, Yuh Cai, Mat Jusoh Siti Asmaa, Marini Ramli, Peng Yeong Woon, Muhammad Farid Johan, Rosline Hassan, and Md Asiful Islam. 2023. "Molecular Genetics of Thrombotic Myeloproliferative Neoplasms: Implications in Precision Oncology" Diagnostics 13, no. 1: 163. https://doi.org/10.3390/diagnostics13010163