
Multifocal Neuroblastoma and Central Hypoventilation in An Infant with Germline ALK F1174I Mutation
 , , , , , and
, , , , , and
Abstract
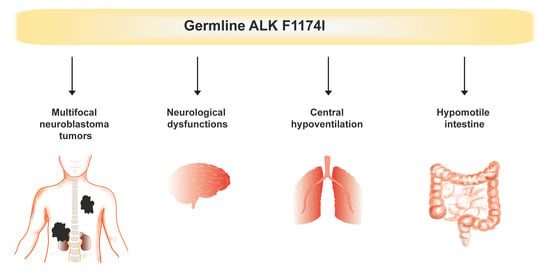
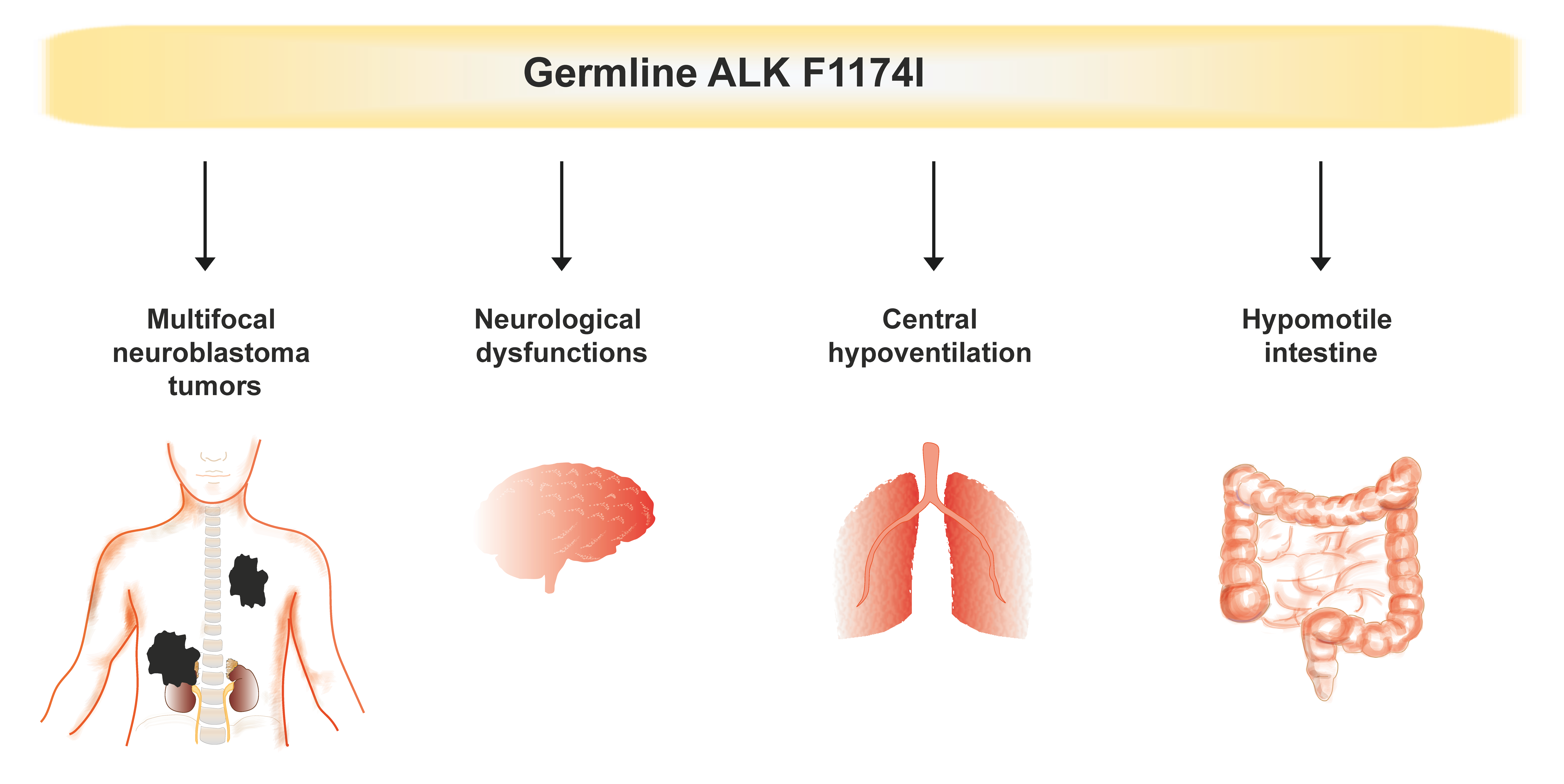
:
1. Introduction
2. Materials and Methods
2.1. Patient
2.2. ALK and PHOX2B Mutation Analysis
2.3. High Resolution SNP Array Analysis
2.4. Whole Genome Sequencing
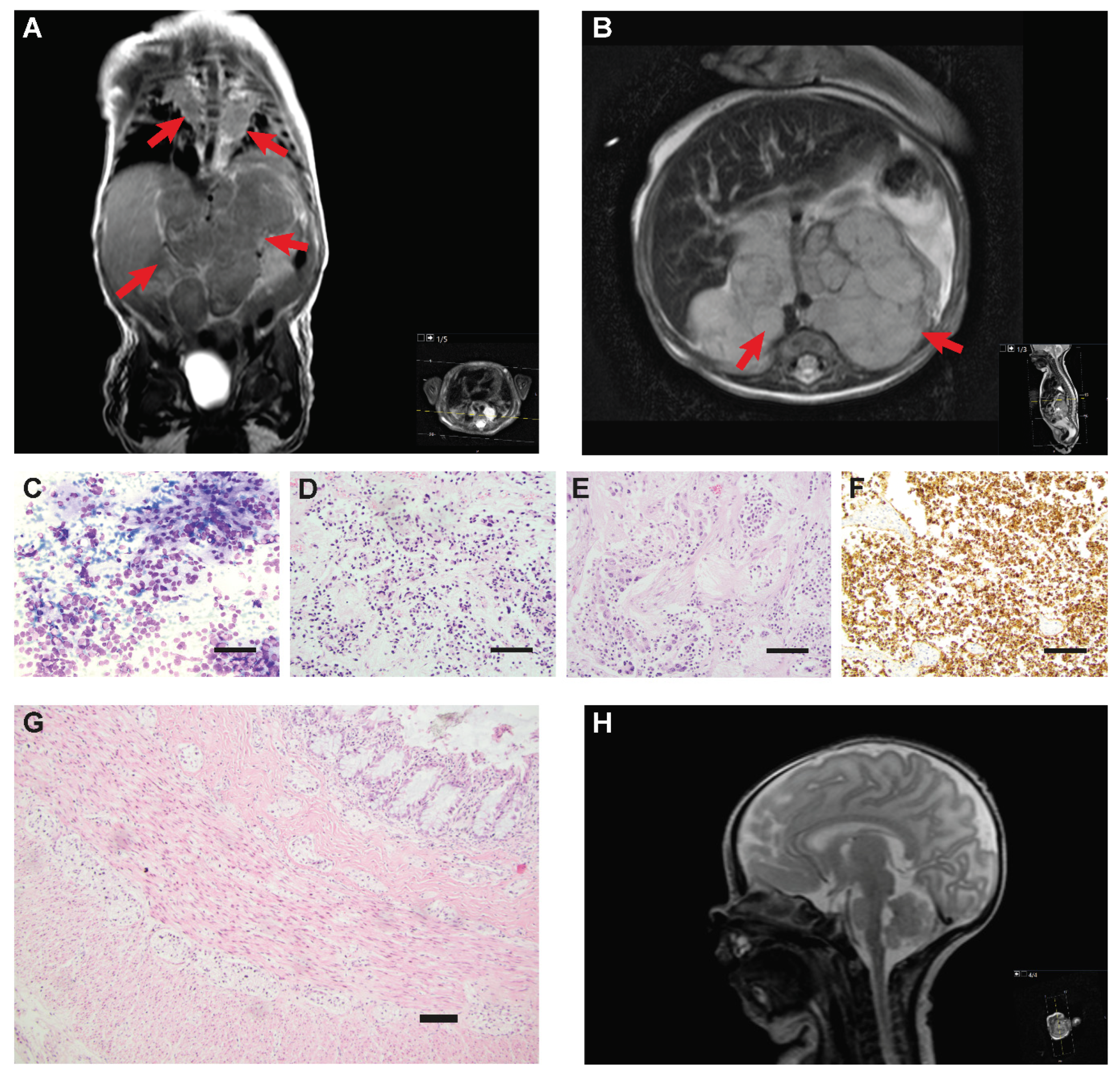
3. Results
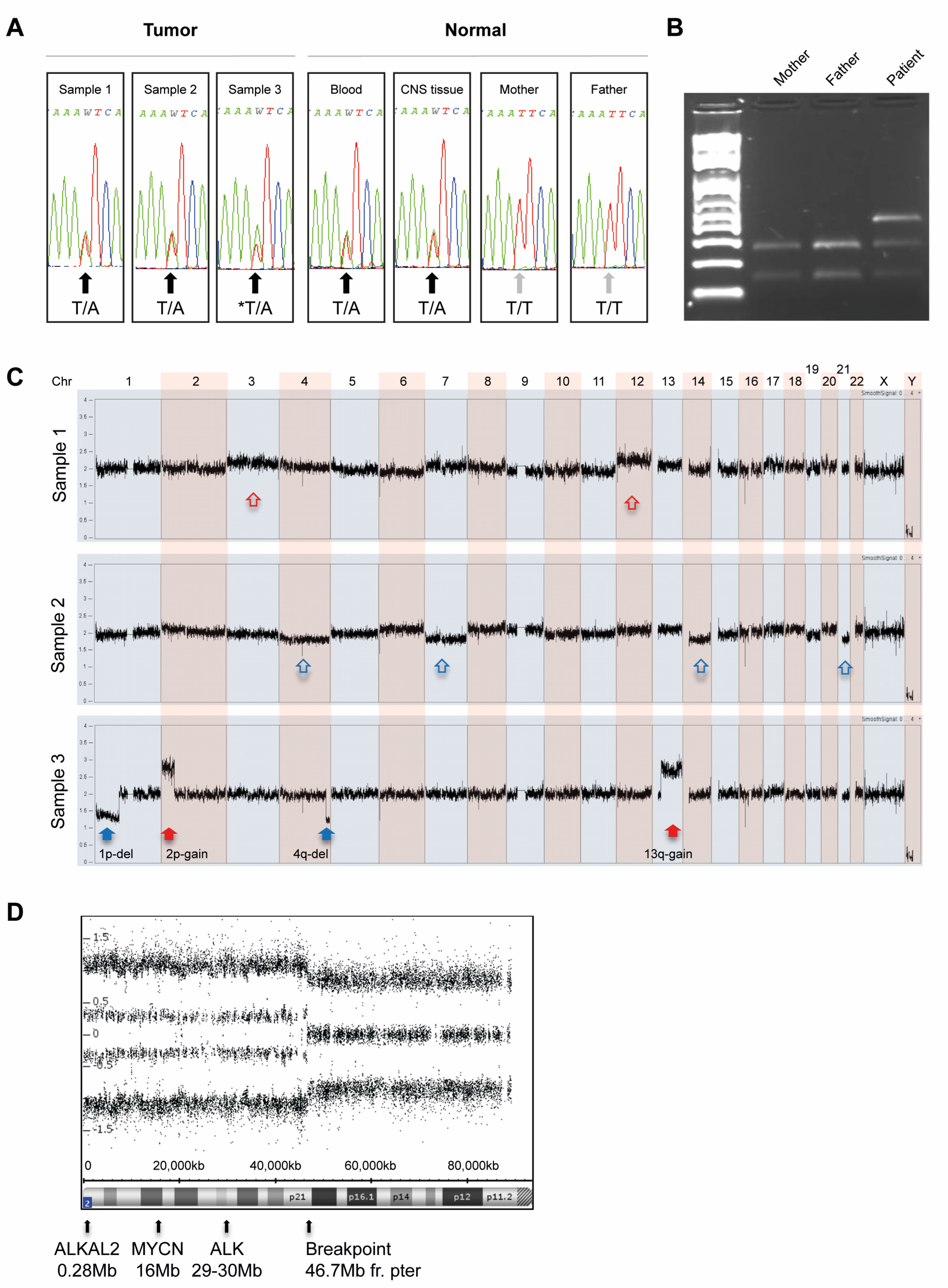
3.1. Analysis of ALK and PHOX2B
3.2. Copy Number Analysis with SNP Microarrays of Tumor Specimens
3.3. Whole Genome Sequencing on Constitutional and Tumor DNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vega-Lopez, G.A.; Cerrizuela, S.; Tribulo, C.; Aybar, M.J. Neurocristopathies: New insights 150 years after the neural crest discovery. Dev. Biol. 2018, 444 (Suppl. S1), S110–S143. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Caren, H.; Kryh, H.; Nethander, M.; Sjoberg, R.M.; Trager, C.; Nilsson, S.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc. Natl. Acad. Sci. USA 2010, 107, 4323–4328. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Sausen, M.; Leary, R.J.; Jones, S.; Wu, J.; Reynolds, C.P.; Liu, X.; Blackford, A.; Parmigiani, G.; Diaz, L.A., Jr.; Papadopoulos, N.; et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 2013, 45, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.K.; Mitra, S.; Subhash, S.; Hertwig, F.; Kanduri, M.; Mishra, K.; Fransson, S.; Ganeshram, A.; Mondal, T.; Bandaru, S.; et al. The risk-associated long noncoding RNA NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell 2014, 26, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr.; Strong, L.C. Mutation and cancer: Neuroblastoma and pheochromocytoma. Am. J. Hum. Genet. 1972, 24, 514–532. [Google Scholar]
- Kushner, B.H.; Gilbert, F.; Helson, L. Familial neuroblastoma. Case reports, literature review, and etiologic considerations. Cancer 1986, 57, 1887–1893. [Google Scholar] [CrossRef]
- Bourdeaut, F.; Ferrand, S.; Brugieres, L.; Hilbert, M.; Ribeiro, A.; Lacroix, L.; Benard, J.; Combaret, V.; Michon, J.; Valteau-Couanet, D.; et al. ALK germline mutations in patients with neuroblastoma: A rare and weakly penetrant syndrome. Eur. J. Hum. Genet. 2012, 20, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Kyemba, S.M.; Rebbeck, T.R.; White, P.S.; Sulman, E.P.; Jensen, S.J.; Allen, C.; Biegel, J.A.; Brodeur, G.M. Molecular genetic analysis of familial neuroblastoma. Eur. J. Cancer 1997, 33, 1923–1928. [Google Scholar] [CrossRef]
- Cohen, M.D.; Auringer, S.T.; Grosfeld, J.L.; Galliani, C.A.; Heerema, N.A. Multifocal primary neuroblastoma. Pediatr. Radiol. 1993, 23, 463–466. [Google Scholar] [CrossRef]
- Robertson, C.M.; Tyrrell, J.C.; Pritchard, J. Familial neural crest tumours. Eur. J. Pediatr. 1991, 150, 789–792. [Google Scholar] [CrossRef]
- Trochet, D.; Bourdeaut, F.; Janoueix-Lerosey, I.; Deville, A.; de Pontual, L.; Schleiermacher, G.; Coze, C.; Philip, N.; Frebourg, T.; Munnich, A.; et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am. J. Hum. Genet. 2004, 74, 761–764. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Khazi, D.; Carlisle, A.J.; Winter, C.L.; Rappaport, E.; Maris, J.M. Germline PHOX2B mutation in hereditary neuroblastoma. Am. J. Hum. Genet. 2004, 75, 727–730. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Caren, H.; Abel, F.; Kogner, P.; Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 2008, 416, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Bachetti, T.; Di Paolo, D.; Di Lascio, S.; Mirisola, V.; Brignole, C.; Bellotti, M.; Caffa, I.; Ferraris, C.; Fiore, M.; Fornasari, D.; et al. PHOX2B-mediated regulation of ALK expression: In vitro identification of a functional relationship between two genes involved in neuroblastoma. PLoS ONE 2010, 5, e13108. [Google Scholar] [CrossRef]
- Stovroff, M.; Dykes, F.; Teague, W.G. The complete spectrum of neurocristopathy in an infant with congenital hypoventilation, Hirschsprung’s disease, and neuroblastoma. J. Pediatr. Surg. 1995, 30, 1218–1221. [Google Scholar] [CrossRef]
- Hiyama, E.; Yokoyama, T.; Hiyama, K.; Yamaoka, H.; Matsuura, Y.; Nishimura, S.; Ueda, K. Multifocal neuroblastoma: Biologic behavior and surgical aspects. Cancer 2000, 88, 1955–1963. [Google Scholar] [CrossRef]
- Rohrer, T.; Trachsel, D.; Engelcke, G.; Hammer, J. Congenital central hypoventilation syndrome associated with Hirschsprung’s disease and neuroblastoma: Case of multiple neurocristopathies. Pediatr. Pulmonol. 2002, 33, 71–76. [Google Scholar] [CrossRef]
- Trochet, D.; O’Brien, L.M.; Gozal, D.; Trang, H.; Nordenskjold, A.; Laudier, B.; Svensson, P.J.; Uhrig, S.; Cole, T.; Niemann, S.; et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am. J. Hum. Genet. 2005, 76, 421–426. [Google Scholar] [CrossRef]
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef]
- Pulford, K.; Lamant, L.; Morris, S.W.; Butler, L.H.; Wood, K.M.; Stroud, D.; Delsol, G.; Mason, D.Y. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997, 89, 1394–1404. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Javanmardi, N.; Bernard, V.; Leroy, Q.; Cappo, J.; Rio Frio, T.; Pierron, G.; Lapouble, E.; Combaret, V.; Speleman, F.; et al. Emergence of new ALK mutations at relapse of neuroblastoma. J. Clin. Oncol. 2014, 32, 2727–2734. [Google Scholar] [CrossRef]
- Bellini, A.; Potschger, U.; Bernard, V.; Lapouble, E.; Baulande, S.; Ambros, P.F.; Auger, N.; Beiske, K.; Bernkopf, M.; Betts, D.R.; et al. Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J. Clin. Oncol. 2021, 39, 3377–3390. [Google Scholar] [CrossRef]
- de Pontual, L.; Kettaneh, D.; Gordon, C.T.; Oufadem, M.; Boddaert, N.; Lees, M.; Balu, L.; Lachassinne, E.; Petros, A.; Mollet, J.; et al. Germline gain-of-function mutations of ALK disrupt central nervous system development. Hum. Mutat. 2011, 32, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Krona, C.; Caren, H.; Sjoberg, R.M.; Sandstedt, B.; Laureys, G.; Kogner, P.; Martinsson, T. Analysis of neuroblastoma tumour progression; loss of PHOX2B on 4p13 and 17q gain are early events in neuroblastoma tumourigenesis. Int. J. Oncol. 2008, 32, 575–583. [Google Scholar] [CrossRef]
- Caren, H.; Erichsen, J.; Olsson, L.; Enerback, C.; Sjoberg, R.M.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-resolution array copy number analyses for detection of deletion, gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: Four cases of homozygous deletions of the CDKN2A gene. BMC Genom. 2008, 9, 353. [Google Scholar] [CrossRef]
- Nannya, Y.; Sanada, M.; Nakazaki, K.; Hosoya, N.; Wang, L.; Hangaishi, A.; Kurokawa, M.; Chiba, S.; Bailey, D.K.; Kennedy, G.C.; et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005, 65, 6071–6079. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Roller, E.; Ivakhno, S.; Lee, S.; Royce, T.; Tanner, S. Canvas: Versatile and scalable detection of copy number variants. Bioinformatics 2016, 32, 2375–2377. [Google Scholar] [CrossRef]
- Weese-Mayer, D.E.; Rand, C.M.; Khaytin, I.; Slattery, S.M.; Yap, K.L.; Marazita, M.L.; Berry-Kravis, E.M. Congenital Central Hypoventilation Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Trang, H.; Samuels, M.; Ceccherini, I.; Frerick, M.; Garcia-Teresa, M.A.; Peters, J.; Schoeber, J.; Migdal, M.; Markstrom, A.; Ottonello, G.; et al. Guidelines for diagnosis and management of congenital central hypoventilation syndrome. Orphanet J. Rare Dis. 2020, 15, 252. [Google Scholar] [CrossRef]
- Hernandez-Miranda, L.R.; Ibrahim, D.M.; Ruffault, P.L.; Larrosa, M.; Balueva, K.; Muller, T.; Weerd, W.; Stolte-Dijkstra, I.; Hostra, R.M.W.; Brunet, J.F.; et al. Mutation in LBX1/Lbx1 precludes transcription factor cooperativity and causes congenital hypoventilation in humans and mice. Proc. Natl. Acad. Sci. USA 2018, 115, 13021–13026. [Google Scholar] [CrossRef]
- Spielmann, M.; Hernandez-Miranda, L.R.; Ceccherini, I.; Weese-Mayer, D.E.; Kragesteen, B.K.; Harabula, I.; Krawitz, P.; Birchmeier, C.; Leonard, N.; Mundlos, S. Mutations in MYO1H cause a recessive form of central hypoventilation with autonomic dysfunction. J. Med. Genet. 2017, 54, 754–761. [Google Scholar] [CrossRef]
- Chand, D.; Yamazaki, Y.; Ruuth, K.; Schonherr, C.; Martinsson, T.; Kogner, P.; Attiyeh, E.F.; Maris, J.; Morozova, O.; Marra, M.A.; et al. Cell culture and Drosophila model systems define three classes of anaplastic lymphoma kinase mutations in neuroblastoma. Dis. Models Mech. 2013, 6, 373–382. [Google Scholar] [CrossRef]
- Siaw, J.T.; Wan, H.; Pfeifer, K.; Rivera, V.M.; Guan, J.; Palmer, R.H.; Hallberg, B. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells, Drosophila and mice. Oncotarget 2016, 7, 29011–29022. [Google Scholar] [CrossRef]
- Vivancos Stalin, L.; Gualandi, M.; Schulte, J.H.; Renella, R.; Shakhova, O.; Muhlethaler-Mottet, A. Expression of the Neuroblastoma-Associated ALK-F1174L Activating Mutation During Embryogenesis Impairs the Differentiation of Neural Crest Progenitors in Sympathetic Ganglia. Front. Oncol. 2019, 9, 275. [Google Scholar] [CrossRef]
- Lopez-Delisle, L.; Pierre-Eugene, C.; Bloch-Gallego, E.; Birling, M.C.; Duband, J.L.; Durand, E.; Bourgeois, T.; Matrot, B.; Sorg, T.; Huerre, M.; et al. Hyperactivation of Alk induces neonatal lethality in knock-in AlkF1178L mice. Oncotarget 2014, 5, 2703–2713. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
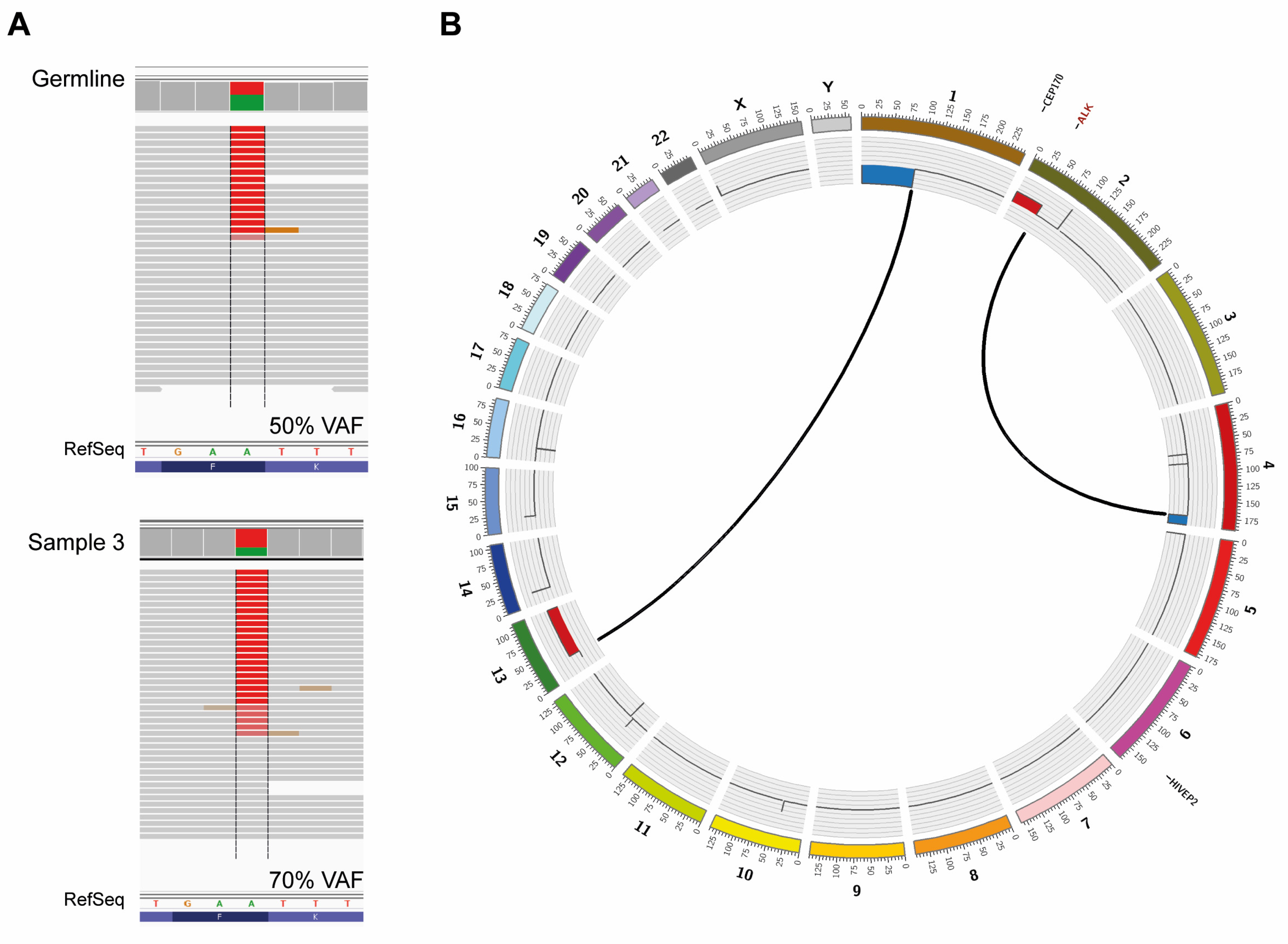{kind=link}
| INRGSS Stage | Age at NB Diagnosis | INRG Risk Group | Histopathological Diagnosis | Genetic Profile | Ploidy |
|---|---|---|---|---|---|
| Undifferentiated neuroblastoma in thorax | Sample 1: numerical only. No MNA or 11q-deletion. | 4n | |||
| L2 | 4 weeks | Low | Ganglioneuroblastoma in abdomen | Sample 2: numerical only. No MNA or 11q-deletion. | 4n |
| Ganglioneuroblastoma in abdomen | Sample 3: other segmental aberrations. No MNA or 11q-deletion. | 2n |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djos, A.; Treis, D.; Fransson, S.; Gordon Murkes, L.; Wessman, S.; Ásmundsson, J.; Markström, A.; Kogner, P.; Martinsson, T. Multifocal Neuroblastoma and Central Hypoventilation in An Infant with Germline ALK F1174I Mutation. Diagnostics 2022, 12, 2260. https://doi.org/10.3390/diagnostics12092260
Djos A, Treis D, Fransson S, Gordon Murkes L, Wessman S, Ásmundsson J, Markström A, Kogner P, Martinsson T. Multifocal Neuroblastoma and Central Hypoventilation in An Infant with Germline ALK F1174I Mutation. Diagnostics. 2022; 12(9):2260. https://doi.org/10.3390/diagnostics12092260
Chicago/Turabian StyleDjos, Anna, Diana Treis, Susanne Fransson, Lena Gordon Murkes, Sandra Wessman, Jurate Ásmundsson, Agneta Markström, Per Kogner, and Tommy Martinsson. 2022. "Multifocal Neuroblastoma and Central Hypoventilation in An Infant with Germline ALK F1174I Mutation" Diagnostics 12, no. 9: 2260. https://doi.org/10.3390/diagnostics12092260