
Epithelial–Mesenchymal Transition by Synergy between Transforming Growth Factor-β and Growth Factors in Cancer Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Dual Roles of TGF-β in Pathophysiological Conditions
2. Collective or Single Cancer Cell Migration by EMT
3. EMT Transcription Factors (EMT-TFs)
4. Synergy between TGF-β and Growth Factors in EMT
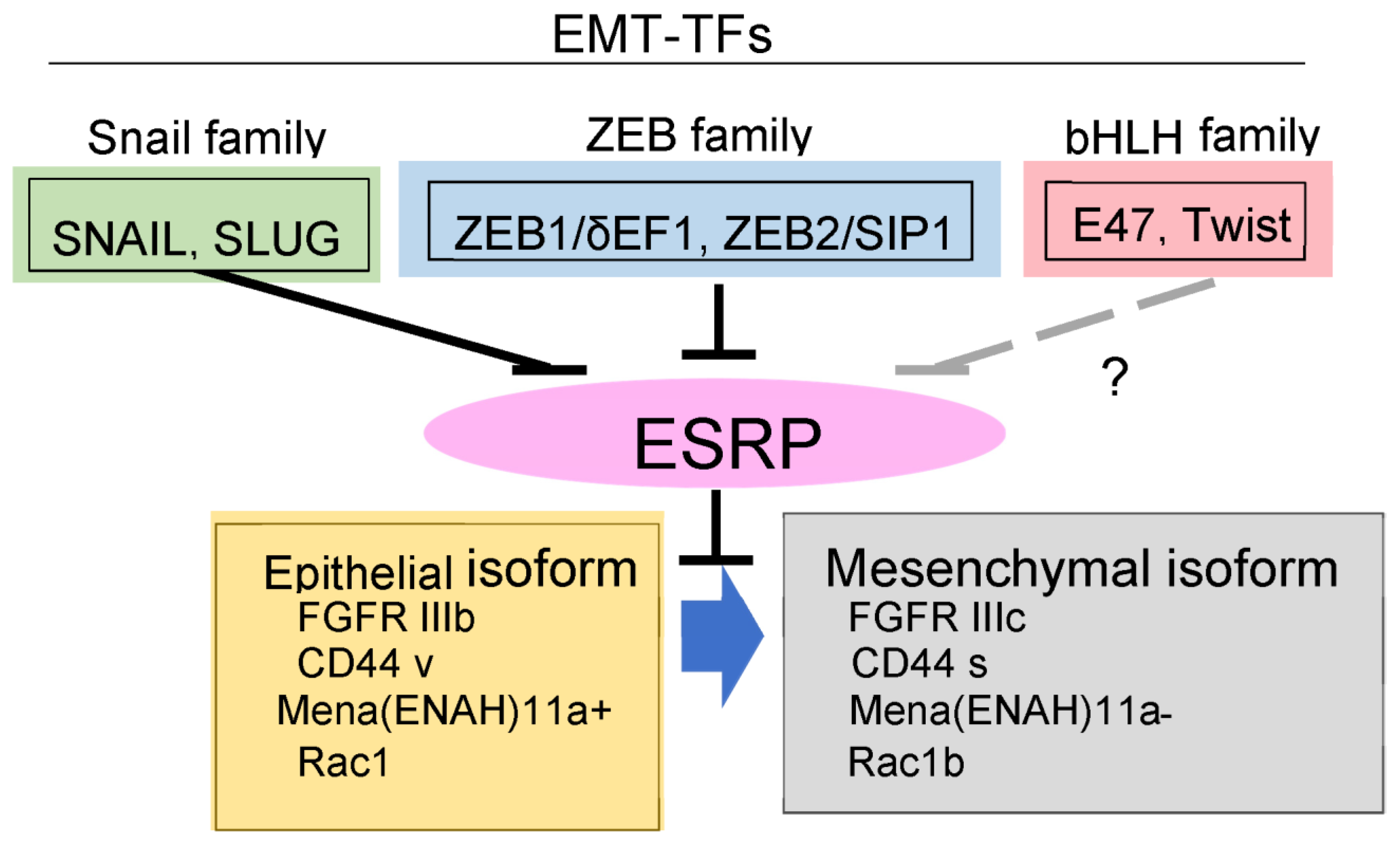
5. Unique Synergy between TGF-β and FGF through Regulation of Alternative Splicing during EMT
6. EMT Transcription Factors (EMT-TFs) in Breast Cancer and Head and Neck Squamous Carcinoma (HNSCC) Cells
7. Pivotal Roles of Ets Family Proteins to Regulate SNAIL and ZEB in Cancer Cells
8. ZEB Family Proteins Are Sustained by Dual Positive Feedback Loops
9. Molecules That Regulate EMT as Potential Diagnostic Markers
10. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- ten Dijke, P.; Miyazono, K.; Heldin, C.H. Signaling via hetero-oligomeric complexes of type I and type II serine/threonine kinase receptors. Curr. Opin. Cell Biol. 1996, 8, 139–145. [Google Scholar] [CrossRef]
- Massague, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Derynck, R. Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-β family. Dev. Cell 2021, 56, 726–746. [Google Scholar] [CrossRef]
- Massague, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Cells Tissues Organs 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef]
- Zeisberg, M.; Kalluri, R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J. Mol. Med. 2004, 82, 175–181. [Google Scholar] [CrossRef]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef]
- Gaggioli, C. Collective invasion of carcinoma cells: When the fibroblasts take the lead. Cell Adh. Migr. 2008, 2, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Labernadie, A.; Kato, T.; Brugues, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; Gonzalez-Tarrago, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef]
- Qin, L.; Yang, D.; Yi, W.; Cao, H.; Xiao, G. Roles of leader and follower cells in collective cell migration. Mol. Biol. Cell 2021, 32, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGFβ/Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Firulli, B.A.; Krawchuk, D.; Centonze, V.E.; Vargesson, N.; Virshup, D.M.; Conway, S.J.; Cserjesi, P.; Laufer, E.; Firulli, A.B. Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat. Genet. 2005, 37, 373–381. [Google Scholar] [CrossRef]
- Firulli, A.B.; Conway, S.J. Phosphoregulation of Twist1 provides a mechanism of cell fate control. Curr. Med. Chem. 2008, 15, 2641–2647. [Google Scholar] [CrossRef]
- Gajula, R.P.; Chettiar, S.T.; Williams, R.D.; Thiyagarajan, S.; Kato, Y.; Aziz, K.; Wang, R.; Gandhi, N.; Wild, A.T.; Vesuna, F.; et al. The twist box domain is required for Twist1-induced prostate cancer metastasis. Mol. Cancer Res. 2013, 11, 1387–1400. [Google Scholar] [CrossRef]
- Kang, E.; Seo, J.; Yoon, H.; Cho, S. The Post-Translational Regulation of Epithelial-Mesenchymal Transition-Inducing Transcription Factors in Cancer Metastasis. Int. J. Mol. Sci. 2021, 22, 3591. [Google Scholar] [CrossRef]
- Ren, J.; Crowley, S.D. Twist1: A Double-Edged Sword in Kidney Diseases. Kidney Dis. 2020, 6, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990. [Google Scholar] [CrossRef]
- Sun, T.; Fu, J.; Shen, T.; Lin, X.; Liao, L.; Feng, X.H.; Xu, J. The Small C-terminal Domain Phosphatase 1 Inhibits Cancer Cell Migration and Invasion by Dephosphorylating Ser(P)68-Twist1 to Accelerate Twist1 Protein Degradation. J. Biol. Chem. 2016, 291, 11518–11528. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Restuccia, D.F.; Lan, Q.; Hynx, D.; Dirnhofer, S.; Hess, D.; Ruegg, C.; Hemmings, B.A. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov. 2012, 2, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Xia, W.; Lim, S.O.; Hsu, J.L.; Huo, L.; Wu, Y.; Li, L.Y.; Lai, C.C.; Chang, S.S.; Hsu, Y.H.; et al. AKT1 Inhibits Epithelial-to-Mesenchymal Transition in Breast Cancer through Phosphorylation-Dependent Twist1 Degradation. Cancer Res. 2016, 76, 1451–1462. [Google Scholar] [CrossRef]
- Su, Y.W.; Xie, T.X.; Sano, D.; Myers, J.N. IL-6 stabilizes Twist and enhances tumor cell motility in head and neck cancer cells through activation of casein kinase 2. PLoS ONE 2011, 6, e19412. [Google Scholar] [CrossRef]
- Tedja, R.; Roberts, C.M.; Alvero, A.B.; Cardenas, C.; Yang-Hartwich, Y.; Spadinger, S.; Pitruzzello, M.; Yin, G.; Glackin, C.A.; Mor, G. Protein kinase Calpha-mediated phosphorylation of Twist1 at Ser-144 prevents Twist1 ubiquitination and stabilizes it. J. Biol. Chem. 2019, 294, 5082–5093. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef]
- Mingot, J.-M.; Vega, S.; Maestro, B.; Sanz, J.M.; Nieto, M.A. Characterization of Snail nuclear import pathways as representatives of C2H2 zinc finger transcription factors. J. Cell Sci. 2009, 122, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Manzanares, M.; Locascio, A.; Nieto, M.A. The increasing complexity of the Snail gene superfamily in metazoan evolution. Trends Genet. 2001, 17, 178–181. [Google Scholar] [CrossRef]
- Xu, Y.; Lee, S.; Kim, H.; Kim, N.; Piao, S.; Park, S.; Jung, Y.; Yook, J.; Park, B.; Ha, N. Role of CK1 in GSK3β-mediated phosphorylation and degradation of snail. Oncogene 2010, 29, 3124–3133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Gururaj, A.E.; Barnes, C.J. p21-activated kinases in cancer. Nat. Rev. Cancer 2006, 6, 459–471. [Google Scholar] [CrossRef]
- Yang, Z.; Rayala, S.; Nguyen, D.; Vadlamudi, R.K.; Chen, S.; Kumar, R. Pak1 phosphorylation of snail, a master regulator of epithelial-to-mesenchyme transition, modulates snail’s subcellular localization and functions. Cancer Res. 2005, 65, 3179–3184. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, Y.M.; Yang, C.H.; Cho, S.K.; Lee, J.W.; Cho, M. Functional regulation of Slug/Snail2 is dependent on GSK-3β-mediated phosphorylation. The FEBS J. 2012, 279, 2929–2939. [Google Scholar] [CrossRef]
- Virtakoivu, R.; Mai, A.; Mattila, E.; De Franceschi, N.; Imanishi, S.Y.; Corthals, G.; Kaukonen, R.; Saari, M.; Cheng, F.; Torvaldson, E. Vimentin–ERK Signaling Uncouples Slug Gene Regulatory FunctionSlug Phosphorylation Induces Vimentin. Cancer Res. 2015, 75, 2349–2362. [Google Scholar] [CrossRef]
- Park, J.-J.; Park, M.-H.; Oh, E.H.; Soung, N.-K.; Lee, S.J.; Jung, J.-K.; Lee, O.-J.; Yun, S.J.; Kim, W.-J.; Shin, E.-Y. The p21-activated kinase 4-Slug transcription factor axis promotes epithelial—mesenchymal transition and worsens prognosis in prostate cancer. Oncogene 2018, 37, 5147–5159. [Google Scholar] [CrossRef]
- Zhuge, X.; Kataoka, H.; Tanaka, M.; Murayama, T.; Kawamoto, T.; Sano, H.; Togi, K.; Yamauchi, R.; Ueda, Y.; Xu, Y.; et al. Expression of the novel Snai-related zinc-finger transcription factor gene Smuc during mouse development. Int. J. Mol. Med. 2005, 15, 945–948. [Google Scholar] [CrossRef]
- Gheldof, A.; Hulpiau, P.; van Roy, F.; De Craene, B.; Berx, G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol. Life Sci. 2012, 69, 2527–2541. [Google Scholar] [CrossRef] [PubMed]
- Llorens, M.C.; Lorenzatti, G.; Cavallo, N.L.; Vaglienti, M.V.; Perrone, A.P.; Carenbauer, A.L.; Darling, D.S.; Cabanillas, A.M. Phosphorylation regulates functions of ZEB1 transcription factor. J. Cell. Physiol. 2016, 231, 2205–2217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Babaei-Jadidi, R.; Lorenzi, F.; Spencer-Dene, B.; Clarke, P.; Domingo, E.; Tulchinsky, E.; Vries, R.G.; Kerr, D.; Pan, Y. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis 2019, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, K.; Shirakihara, T.; Nakano, A.; Imamura, T.; Miyazono, K.; Saitoh, M. Role of Ras signaling in the induction of snail by transforming growth factor-β. J. Biol. Chem. 2009, 284, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Morgani, S.M.; David, C.J.; Wang, Q.; Er, E.E.; Huang, Y.H.; Basnet, H.; Zou, Y.; Shu, W.; Soni, R.K.; et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020, 577, 566–571. [Google Scholar] [CrossRef]
- Saitoh, M.; Endo, K.; Furuya, S.; Minami, M.; Fukasawa, A.; Imamura, T.; Miyazawa, K. STAT3 integrates cooperative Ras and TGF-β signals that induce Snail expression. Oncogene 2016, 35, 1049–1057. [Google Scholar] [CrossRef]
- Coumoul, X.; Deng, C.X. Roles of FGF receptors in mammalian development and congenital diseases. Birth Defects Res. Part C Embryo Today Rev. 2003, 69, 286–304. [Google Scholar] [CrossRef]
- Shirakihara, T.; Horiguchi, T.; Miyazawa, M.; Ehata, S.; Shibata, T.; Morita, I.; Miyazono, K.; Saitoh, M. TGF-β regulates isoform switching of FGF receptors and epithelial-mesenchymal transition. EMBO J. 2011, 30, 783–795. [Google Scholar] [CrossRef]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef] [PubMed]
- Manchado, E.; Weissmueller, S.; Morris, J.P.t.; Chen, C.C.; Wullenkord, R.; Lujambio, A.; de Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016, 534, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Sakamoto, K.; Endo, K.; Sakamoto, K.; Kayamori, K.; Ehata, S.; Ichikawa, J.; Ando, T.; Nakamura, R.; Kimura, Y.; Yoshizawa, K.; et al. EHF suppresses cancer progression by inhibiting ETS1-mediated ZEB expression. Oncogenesis 2021, 10, 26. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Monville, F.; Finetti, P.; Adelaide, J.; Cervera, N.; Fekairi, S.; Xerri, L.; Jacquemier, J.; Birnbaum, D.; et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006, 25, 2273–2284. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef]
- Sinh, N.D.; Endo, K.; Miyazawa, K.; Saitoh, M. Ets1 and ESE1 reciprocally regulate expression of ZEB1/ZEB2, dependent on ERK1/2 activity, in breast cancer cells. Cancer Sci. 2017, 108, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.K.; Endo, K.; Itoh, Y.; Osada, A.H.; Kimura, Y.; Ueki, K.; Yoshizawa, K.; Miyazawa, K.; Saitoh, M. Ets family proteins regulate the EMT transcription factors Snail and ZEB in cancer cells. FEBS Open Bio. 2022, 12, 1353–1364. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Ito, T.; Azuma, S.; Ito, E.; Honma, R.; Yanagisawa, Y.; Nishikawa, A.; Kawamura, M.; Imai, J.; Watanabe, S.; et al. Constitutive activation of nuclear factor-kappaB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 2009, 100, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Qiao, C.; Li, X.; Chen, Z.; Liu, H.; Wu, S.; Hu, S.; Qiu, Z.; Qian, M.; Tian, D.; et al. Forkhead box K2 promotes human colorectal cancer metastasis by upregulating ZEB1 and EGFR. Theranostics 2019, 9, 3879–3902. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, J. The biology of the Ets1 proto-oncogene. Mol. Cancer 2003, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, J. The role of the transcription factor Ets1 in carcinoma. Semin. Cancer Biol. 2015, 35, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Luk, I.Y.; Reehorst, C.M.; Mariadason, J.M. ELF3, ELF5, EHF and SPDEF Transcription Factors in Tissue Homeostasis and Cancer. Molecules 2018, 23, 2191. [Google Scholar] [CrossRef]
- Yachida, S.; Wood, L.D.; Suzuki, M.; Takai, E.; Totoki, Y.; Kato, M.; Luchini, C.; Arai, Y.; Nakamura, H.; Hama, N.; et al. Genomic Sequencing Identifies ELF3 as a Driver of Ampullary Carcinoma. Cancer Cell 2016, 29, 229–240. [Google Scholar] [CrossRef]
- Suzuki, M.; Saito-Adachi, M.; Arai, Y.; Fujiwara, Y.; Takai, E.; Shibata, S.; Seki, M.; Rokutan, H.; Maeda, D.; Horie, M.; et al. E74-Like Factor 3 Is a Key Regulator of Epithelial Integrity and Immune Response Genes in Biliary Tract Cancer. Cancer Res. 2021, 81, 489–500. [Google Scholar] [CrossRef]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef]
- Härkönen, K.; Oikari, S.; Kyykallio, H.; Capra, J.; Hakkola, S.; Ketola, K.; Thanigai Arasu, U.; Daaboul, G.; Malloy, A.; Oliveira, C.; et al. CD44s Assembles Hyaluronan Coat on Filopodia and Extracellular Vesicles and Induces Tumorigenicity of MKN74 Gastric Carcinoma Cells. Cells 2019, 8, 276. [Google Scholar] [CrossRef] [Green Version]
- Roy Burman, D.; Das, S.; Das, C.; Bhattacharya, R. Alternative splicing modulates cancer aggressiveness: Role in EMT/metastasis and chemoresistance. Mol. Biol. Rep. 2021, 48, 897–914. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Montoya, C.A.; Donnenberg, V.S.; Sant, S. Interplay between tumor microenvironment and partial EMT as the driver of tumor progression. iScience 2021, 24, 102113. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saitoh, M. Epithelial–Mesenchymal Transition by Synergy between Transforming Growth Factor-β and Growth Factors in Cancer Progression. Diagnostics 2022, 12, 2127. https://doi.org/10.3390/diagnostics12092127
Saitoh M. Epithelial–Mesenchymal Transition by Synergy between Transforming Growth Factor-β and Growth Factors in Cancer Progression. Diagnostics. 2022; 12(9):2127. https://doi.org/10.3390/diagnostics12092127
Chicago/Turabian StyleSaitoh, Masao. 2022. "Epithelial–Mesenchymal Transition by Synergy between Transforming Growth Factor-β and Growth Factors in Cancer Progression" Diagnostics 12, no. 9: 2127. https://doi.org/10.3390/diagnostics12092127