
Dose Dependence Effect in Biallelic WNT10A Variant-Associated Tooth Agenesis Phenotype

 and
and
Abstract
:1. Introduction
2. Methods
2.1. Study Subjects
2.2. Variant Detection
2.3. Sanger Sequencing and Clone Sequencing
2.4. Bioinformatics Analysis
3. Results
3.1. Clinical Findings
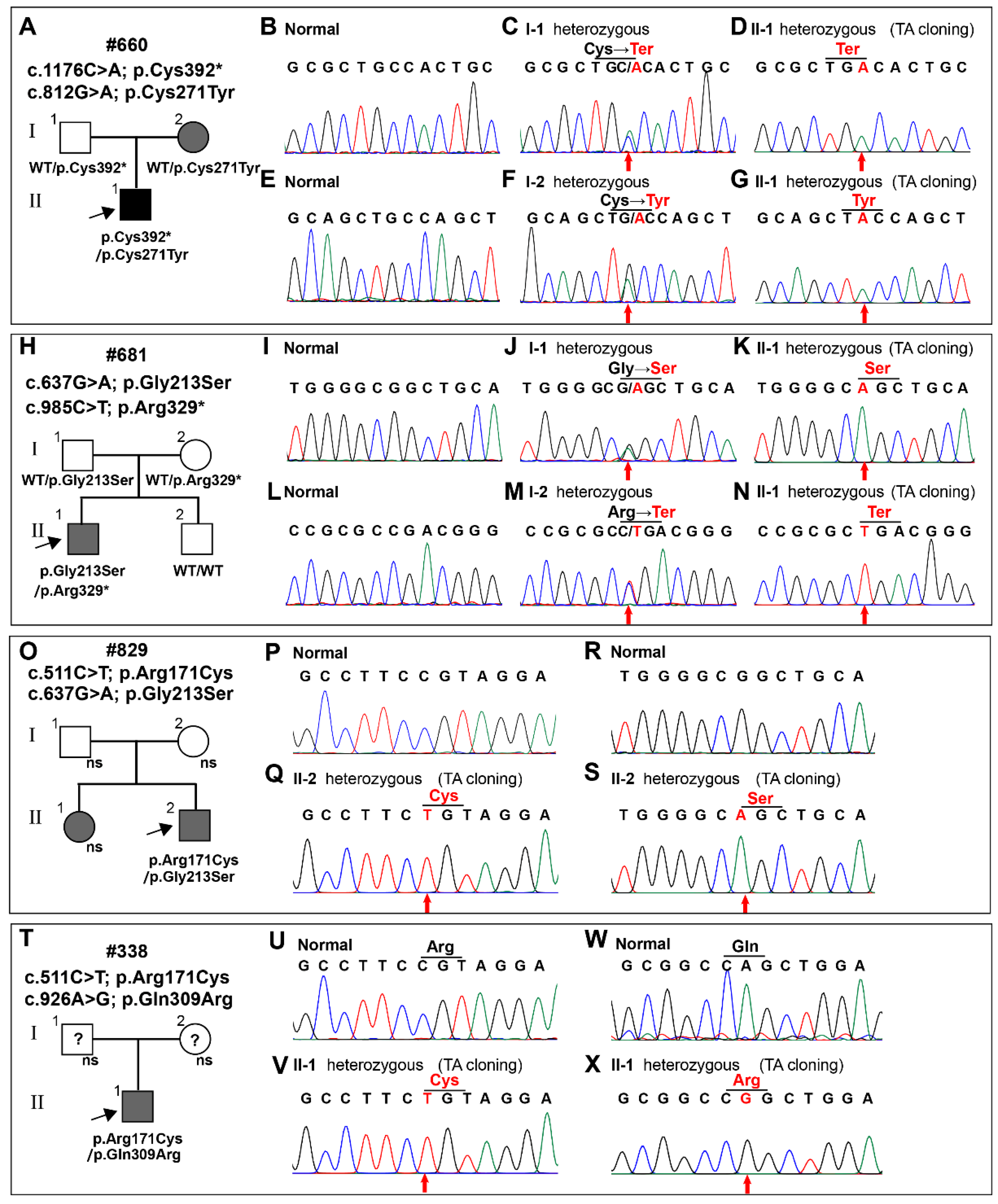
3.2. The Identification of WNT10A Variants
3.3. Bioinformatics Analysis of the WNT10A Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nieminen, P. Genetic basis of tooth agenesis. J. Exp. Zool. Part B: Mol. Dev. Evol. 2009, 312B, 320–342. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Maeda, T. Prevalence and genetic basis of tooth agenesis. Jpn. Dent. Sci. Rev. 2009, 45, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Polder, B.J.; Hof, M.A.V.; Van Der Linden, F.P.G.M.; Kuijpers-Jagtman, A.M. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dent. Oral Epidemiol. 2004, 32, 217–226. [Google Scholar] [CrossRef]
- Rølling, S.; Poulsen, S. Oligodontia in Danish schoolchildren. Acta Odontol. Scand. 2001, 59, 111–112. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.C.; Lyu, X.; Shen, G.H.; Deng, X.X.; Li, W.R.; Zhang, X.X.; Feng, H.L. Prevalence of tooth agenesis in adolescent Chinese populations with or without orthodontics. Chin. J. Dent. Res. 2015, 18, 59–65. [Google Scholar]
- Liu, H.; Liu, H.; Su, L.; Zheng, J.; Feng, H.; Liu, Y.; Yu, M.; Han, D. Four Novel PAX9 Variants and the PAX9-Related Non-Syndromic Tooth Agenesis Patterns. Int. J. Mol. Sci. 2022, 23, 8142. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, N.; Goswami, M.; Chhabra, A. Genetic basis of dental agenesis—Molecular genetics patterning clinical dentistry. Med Oral 2014, 19, e112–e119. [Google Scholar] [CrossRef]
- Yu, M.; Wong, S.-W.; Han, D.; Cai, T. Genetic analysis: Wnt and other pathways in nonsyndromic tooth agenesis. Oral Dis. 2019, 25, 646–651. [Google Scholar] [CrossRef] [Green Version]
- Doolan, B.J.; Onoufriadis, A.; Kantaputra, P.; McGrath, J.A. WNT10A, dermatology and dentistry. Br. J. Dermatol. 2021, 185, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Adaimy, L.; Chouery, E.; Mégarbané, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Mégarbané, A. Mutation in WNT10A Is Associated with an Autosomal Recessive Ectodermal Dysplasia: The Odonto-onycho-dermal Dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A Mutations Are a Frequent Cause of a Broad Spectrum of Ectodermal Dysplasias with Sex-Biased Manifestation Pattern in Heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef]
- Song, S.; Zhao, R.; He, H.; Zhang, J.; Feng, H.; Lin, L. WNT10A variants are associated with non-syndromic tooth agenesis in the general population. Hum. Genet. 2014, 133, 117–124. [Google Scholar] [CrossRef]
- Arzoo, P.S.; Klar, J.; Bergendal, B.; Norderyd, J.; Dahl, N. WNT10A mutations account for ¼ of population-based isolated oligodontia and show phenotypic correlations. Am. J. Med Genet. Part A 2013, 164, 353–359. [Google Scholar] [CrossRef]
- Dhamo, B.; Fennis, W.; Créton, M.; Vucic, S.; Cune, M.; van Amstel, H.K.P.; Wolvius, E.B.; Boogaard, M.-J.V.D.; Ongkosuwito, E.M. The association between WNT10A variants and dental development in patients with isolated oligodontia. Eur. J. Hum. Genet. 2016, 25, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Liu, Y.; Liu, H.; Wong, S.-W.; He, H.; Zhang, X.; Wang, Y.; Han, D.; Feng, H. Distinct impacts of bi-allelic WNT10A mutations on the permanent and primary dentitions in odonto-onycho-dermal dysplasia. Am. J. Med Genet. Part A 2019, 179, 57–64. [Google Scholar] [CrossRef]
- Kantaputra, P.; Jatooratthawichot, P.; Tantachamroon, O.; Nanekrungsan, K.; Intachai, W.; Olsen, B.; Tongsima, S.; Ngamphiw, C.; Cairns, J.R.K. Novel Dental Anomaly–associated Mutations in WNT10A Protein Binding Sites. Int. Dent. J. 2022. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, H.; Camhi, H.; Seymen, F.; Koruyucu, M.; Kasimoglu, Y.; Kim, J.-W.; Kim-Berman, H.; Yuson, N.M.R.; Benke, P.J.; et al. Analyses of oligodontia phenotypes and genetic etiologies. Int. J. Oral Sci. 2021, 13, 32. [Google Scholar] [CrossRef]
- Krøigård, A.B.; Clemmensen, O.; Gjørup, H.; Hertz, J.M.; Bygum, A. Odonto-onycho-dermal dysplasia in a patient homozygous for a WNT10A nonsense mutation and mild manifestations of ectodermal dysplasia in carriers of the mutation. BMC Dermatol. 2016, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, S.; Klar, J.; Wajid, M.; Aslam, M.; Tariq, M.; Schuster, J.; Baig, S.M.; Dahl, N. WNT10A missense mutation associated with a complete Odonto-Onycho-Dermal Dysplasia syndrome. Eur. J. Hum. Genet. 2009, 17, 1600–1605. [Google Scholar] [CrossRef] [Green Version]
- Mallaiah, U.; Dickinson, J. Photo Essay: Bilateral Multiple Eyelid Apocrine Hidrocystomas and Ectodermal Dysplasia. Arch. Ophthalmol. 2001, 119, 1866–1867. [Google Scholar] [CrossRef]
- Tziotzios, C.; Petrof, G.; Liu, L.; Verma, A.; Wedgeworth, E.; Mellerio, J.; McGrath, J. Clinical features and WNT 10A mutations in seven unrelated cases of Schöpf–Schulz–Passarge syndrome. Br. J. Dermatol. 2014, 171, 1211–1214. [Google Scholar] [CrossRef]
- Kantaputra, P.; Sripathomsawat, W. WNT10A and isolated hypodontia. Am. J. Med Genet. Part A 2011, 155, 1119–1122. [Google Scholar] [CrossRef]
- van den Boogaard, M.-J.; Créton, M.; Bronkhorst, Y.; van der Hout, A.; Hennekam, E.; Lindhout, D.; Cune, M.; Ploos van Amstel, H.K. Mutations inWNT10Aare present in more than half of isolated hypodontia cases. J. Med. Genet. 2012, 49, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Prasad, M.K.; Geoffroy, V.; Vicaire, S.; Jost, B.; Dumas, M.; Le Gras, S.; Switala, M.; Gasse, B.; Laugel-Haushalter, V.; Paschaki, M.; et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J. Med Genet. 2016, 53, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Yu, M.; Yeh, I.; Zhang, L.; Liu, H.; Cai, T.; Feng, H.; Liu, Y.; Han, D. Functional study of novel PAX9 variants: The paired domain and non-syndromic oligodontia. Colorectal Dis. 2021, 27, 1468–1477. [Google Scholar] [CrossRef]
- Zheng, J.; Yu, M.; Liu, H.; Cai, T.; Feng, H.; Liu, Y.; Han, D. Novel MSX1 variants identified in families with nonsyndromic oligodontia. Int. J. Oral Sci. 2021, 13, 2. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Ye, X.; Attaie, A.B. Genetic Basis of Nonsyndromic and Syndromic Tooth Agenesis. J. Pediatr. Genet. 2016, 05, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.-F.; Chai, Y. Molecular regulatory mechanism of tooth root development. Int. J. Oral Sci. 2012, 4, 177–181. [Google Scholar] [CrossRef] [Green Version]
- Thesleff, I.; Sharpe, P. Signalling networks regulating dental development. Mech. Dev. 1997, 67, 111–123. [Google Scholar] [CrossRef]
- Lammi, L.; Arte, S.; Somer, M.; Järvinen, H.; Lahermo, P.; Thesleff, I.; Pirinen, S.; Nieminen, P. Mutations in AXIN2 Cause Familial Tooth Agenesis and Predispose to Colorectal Cancer. Am. J. Hum. Genet. 2004, 74, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Han, D.; Feng, H.; Qu, H.; Song, S.; Bai, B.; Zhang, Z. Involvement of and Interaction between WNT10A and EDA Mutations in Tooth Agenesis Cases in the Chinese Population. PLoS ONE 2013, 8, e80393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanchanasevee, C.; Sriwattanapong, K.; Theerapanon, T.; Thaweesapphithak, S.; Chetruengchai, W.; Porntaveetus, T.; Shotelersuk, V. Phenotypic and Genotypic Features of Thai Patients with Nonsyndromic Tooth Agenesis and WNT10A Variants. Front. Physiol. 2020, 11, 573214. [Google Scholar] [CrossRef]
- Machida, J.; Goto, H.; Tatematsu, T.; Shibata, A.; Miyachi, H.; Takahashi, K.; Izumi, H.; Nakayama, A.; Shimozato, K.; Tokita, Y. WNT10A variants isolated from Japanese patients with congenital tooth agenesis. Hum. Genome Var. 2017, 4, hgv201747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Han, D.; Song, S.; Wang, Y.; Zhao, H.; Pan, S.; Bai, B.; Feng, H. Correlation between the phenotypes and genotypes of X-linked hypohidrotic ectodermal dysplasia and non-syndromic hypodontia caused by ectodysplasin-A mutations. Eur. J. Med Genet. 2011, 54, e377–e382. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Song, J.-S.; Shin, T.J.; Hyun, H.-K.; Kim, Y.-J.; Kim, J.-W. WNT10A mutations causing oligodontia. Arch. Oral Biol. 2019, 103, 8–11. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Variant | Proband | Domain | Mutation Taster | Fathmm | PolyPhen-2 | gnomAD, dbSNP, 1000G | ACMG Classification (Evidence of Pathogenicity) |
|---|---|---|---|---|---|---|---|
| c.1176C > A/ p.Cys392* | #660 II-1 | Wnt | Disease causing | - | - | rs1553623389 | Pathogenic PVS1 + PP1 + PP5 |
| c.812G > A/ p.Cys271Tyr | #660 II-1 | Wnt | Disease causing | DAMAGING (−2.18) | Probably damaging (1.000) | Not found | Likely pathogenic PM2 + PM3 + PP1 + PP2 + PP3 |
| c.511C > T/ p.Arg171Cys | #829 II-2 #338 II-1 | Wnt | Disease causing | TOLERATED (−1.04) | Probably damaging (0.999) | rs116998555 | Uncertain significance BP6 |
| c.637G > A/ p.Gly213Ser | #829 II-2 #681 II-1 | Wnt | Disease causing | DAMAGING (−1.66) | Probably damaging (1.000) | rs147680216 | Uncertain significance PP1 + PP3 |
| c.985C > T/ p.Arg329* | #681 II-1 | Wnt | Disease causing | - | - | Not found | Pathogenic PVS1 + PM2 + PP1 + PP2 |
| c.926A > G/ p.Gln309Arg | #338 II-1 | Wnt | Disease causing | TOLERATED (−0.92) | Benign (0.006) | rs1461989045 | Uncertain significance PP3 + PM1 + BS4 + BP2 |
| Origin | Variation | Variation Type | Disease | Right Quadrants | Left Quadrants | Permanent Tooth Missing Number | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Max | 7 | 6 | 5 | 4 | 3 | 2 | 1 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||||||
| Mand | 7 | 6 | 5 | 4 | 3 | 2 | 1 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||||||
| #660 II-1 | p.Cys392* p.Cys271Tyr | Biallelic | OODD |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 28 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #660 I-1 | WT p.Cys392* | Monoallelic | Normal |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 0 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #660 I-2 | WT p.Cys271Tyr | Monoallelic | NSTA |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 2 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #829 II-2 | p.Arg171Cys p.Gly213Ser | Biallelic | NSTA |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 2 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #829 II-1 | NA | NA | NSTA |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 1 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #829 I-1 | NA | NA | Normal |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 0 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #829 I-2 | NA | NA | Normal |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 0 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #681 II-1 | p.Gly213Ser p.Arg329* | Biallelic | NSTA |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 18 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #681 II-2 | WT WT | Normal | Normal |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 0 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #681 I-1 | WT p.Gly213Ser | Monoallelic | Normal |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 0 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #681 I-2 | WT p.Arg329* | Monoallelic | Normal |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 0 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #338 II-1 | p.Arg171Cys p.Gln309Arg | Biallelic | NATA |  |  |  |  |  |  |  |  |  |  |  |  |  |  | 5 | |||
 |  |  |  |  |  |  |  |  |  |  |  |  |  | ||||||||
| #338 I-1 | NA | NA | NA | NA | NA | ||||||||||||||||
| #681 II-2 | NA | NA | NA | NA | NA | ||||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Lin, B.; Liu, H.; Su, L.; Feng, H.; Liu, Y.; Yu, M.; Han, D. Dose Dependence Effect in Biallelic WNT10A Variant-Associated Tooth Agenesis Phenotype. Diagnostics 2022, 12, 3087. https://doi.org/10.3390/diagnostics12123087
Liu H, Lin B, Liu H, Su L, Feng H, Liu Y, Yu M, Han D. Dose Dependence Effect in Biallelic WNT10A Variant-Associated Tooth Agenesis Phenotype. Diagnostics. 2022; 12(12):3087. https://doi.org/10.3390/diagnostics12123087
Chicago/Turabian StyleLiu, Haochen, Bichen Lin, Hangbo Liu, Lanxin Su, Hailan Feng, Yang Liu, Miao Yu, and Dong Han. 2022. "Dose Dependence Effect in Biallelic WNT10A Variant-Associated Tooth Agenesis Phenotype" Diagnostics 12, no. 12: 3087. https://doi.org/10.3390/diagnostics12123087