
Novel Decision Tool for More Severe α-Thalassemia Genotypes Screening with Functional Loss of Two or More α-Globin Genes: A Diagnostic Test Study
 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analytical Methods
2.2. Binomial Logistic Regression (BLR) Model Building
3. Results
3.1. Univariate Analysis
3.2. Development of BLR Model
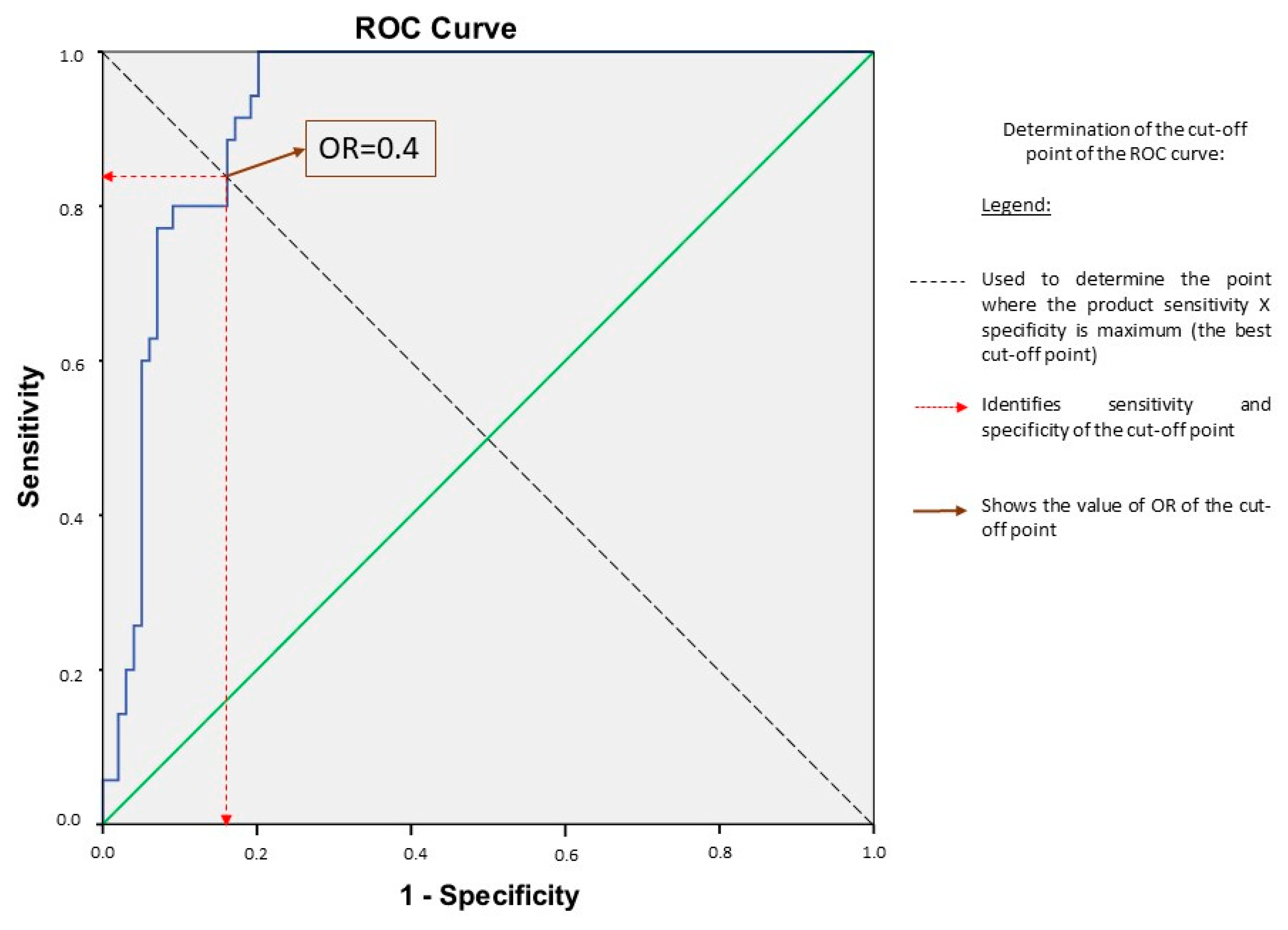
3.3. Determination of the Best Cut-Off Point and Its Performance
3.4. Validation Process
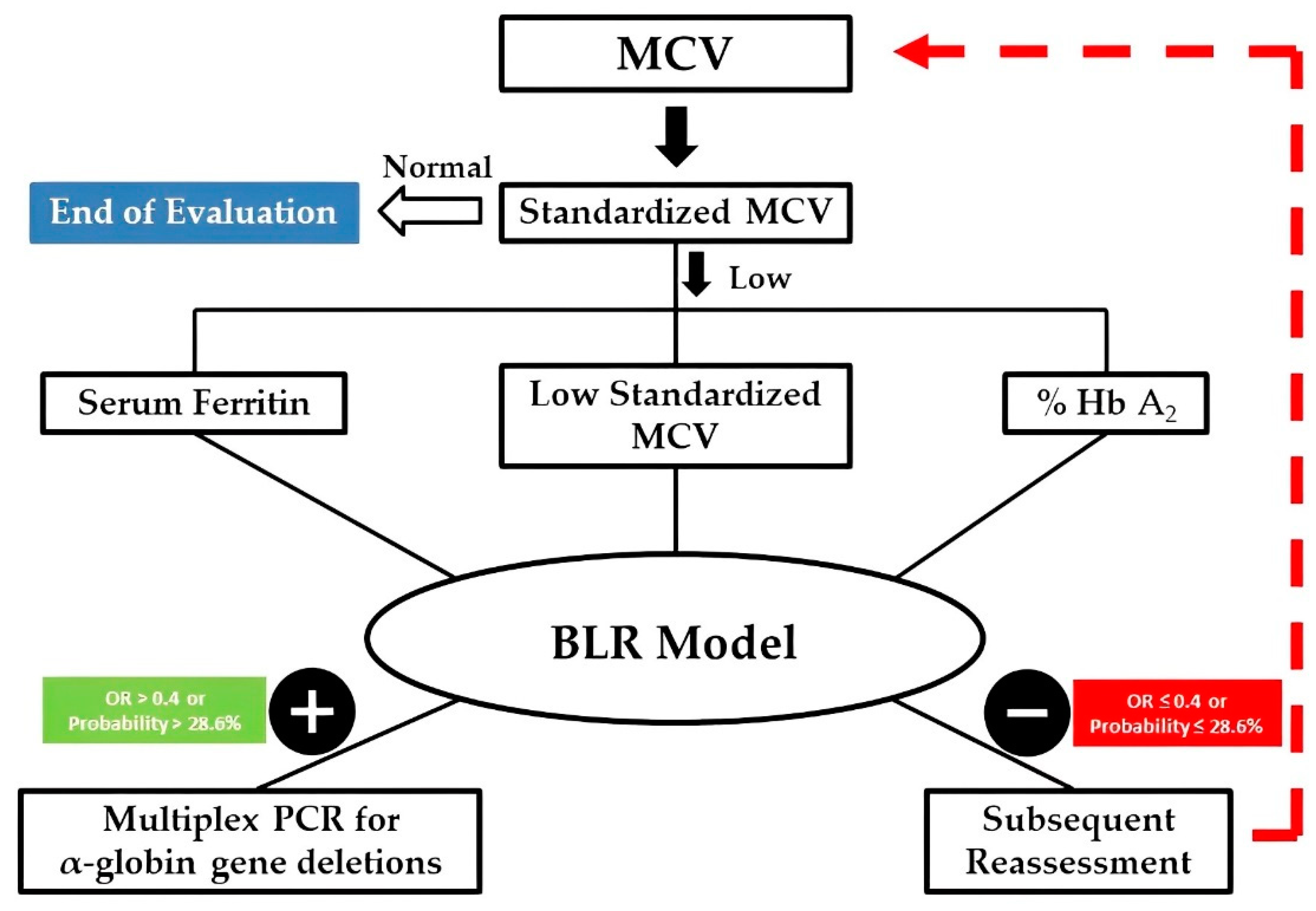
3.5. Improving the Decision-Making Process
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muncie, H.L.; Campbell, J. Alpha and Beta Thalassemia. Am. Fam. Physician 2009, 80, 339–344. [Google Scholar] [PubMed]
- Harteveld, C.L.; Higgs, D.R. Alpha-Thalassaemia. Orphanet J. Rare Dis. 2010, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, S.S.; Boehm, C.D.; Higgs, D.R.; Cutting, G.R. Single-Tube Multiplex-Pcr Screen for Common Deletional Determinants of Alpha-Thalassemia. Blood 2000, 95, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Modell, B.; Darlison, M. Global Epidemiology of Haemoglobin Disorders And Derived Service Indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef]
- Sonati, M.F.; Farah, S.B.; Ramalho, A.S.; Costa, F.F. High Prevalence of Alpha-Thalassemia in A Black Population Of Brazil. Hemoglobin 1991, 15, 309–311. [Google Scholar] [CrossRef]
- Adorno, E.V.; Couto, F.D.; Moura Neto, J.P.; Menezes, J.F.; Rêgo, M.; Reis, M.G.; Gonçalves, M.S. Hemoglobinopathies in Newborns from Salvador, Bahia, Northeast Brazil. Cad. Saude Publica 2005, 21, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Souza, A.E.; Cardoso, G.L.; Takanashi, S.Y.; Guerreiro, J.F. Alpha Thalassemia (3.7 Kb Deletion) In A Population from The Brazilian Amazon Region: Santarém, Pará State. Genet. Mol. Res. 2009, 8, 477–481. [Google Scholar] [CrossRef]
- Couto, F.D.; De Albuquerque, A.B.; Adorno, E.V.; De Moura Neto, J.P.; De Freitas Abbehusen, L.; De Oliveira, J.L.; Dos Reis, M.G.; De Souza Gonçalves, M. Alpha-Thalassemia 2, 3.7 Kb Deletion and Hemoglobin Ac Heterozygosity In Pregnancy: A Molecular And Hematological Analysis. Clin. Lab. Haematol. 2003, 25, 29–34. [Google Scholar] [CrossRef]
- Anselmo, F.C.; Ferreira, N.S.; da Mota, A.J.; Gonçalves, M.S.; Albuquerque, S.R.L.; Fraiji, N.A.; Ferreira, A.C.D.; de Moura Neto, J.P. Deletional Alpha-Thalassemia Alleles In Amazon Blood Donors. Adv. Hematol. 2020, 2020, 4170259. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, F.C.; Soumanou, A.G.; de Aguiar Ferreira, C.; Sobrinha, F.M.V.; Castro, A.C.S.; Brito, R.O.; da Mota, A.J.; de Souza Gonçalves, M.; de Moura Neto, J.P. Detection Of Common Deletional Of A-Thalassemia 3.7 Kb From Metropolitan Region Of Manaus, Amazonas, Brazil. Mediterr. J. Hematol Infect. Dis. 2021, 13, E2021001. [Google Scholar] [CrossRef]
- Silva Filho, I.; Gonçalves, M.S.; Adôrno, E.V.; Campos, D.P.; Fleury, M.K. Triagem De Hemoglobinopatias E Avaliação Da Degeneração Oxidativa Da Hemoglobina Em Trabalhadores Portadores Do Traço Falciforme (Hbas), Expostos A Riscos Ocupacionais. Rev. Bras. Hematol. Hemoter. 2005, 27, 183–187. [Google Scholar] [CrossRef]
- Borges, E.; Wenning, M.R.; Kimura, E.M.; Gervásio, S.A.; Costa, F.F.; Sonati, M.F. High Prevalence Of Alpha-Thalassemia Among Individuals With Microcytosis And Hypochromia Without Anemia. Braz. J. Med. Biol. Res. 2001, 34, 759–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siqueira, P. Nova Ferramenta De Suporte Para O Rastreio De A-Talassemia Em Pacientes Com Microcitose. Master’s Thesis, Federal University of Rio De Janeiro, Rio De Janeiro, Brazil, 2015. [Google Scholar]
- Clark, B.E.; Thein, S.L. Molecular Diagnosis of Haemoglobin Disorders. Clin. Lab. Haematol. 2004, 26, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.N.; Saminathan, R.; Choolani, M. Thalassaemia Screening and Confirmation of Carriers in Parents. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 39, 27–40. [Google Scholar] [CrossRef]
- Van Vranken, M. Evaluation of Microcytosis. Am. Fam. Physician 2010, 82, 1117–1122. [Google Scholar]
- Deloughery, T.G. Microcytic Anemia. N. Engl. J. Med. 2014, 371, 2537. [Google Scholar] [CrossRef]
- Langlois, S.; Ford, J.C.; Chitayat, D. Carrier Screening For Thalassemia and Hemoglobinopathies In Canada. J. Obstet. Gynaecol. Can. 2008, 30, 950–959. [Google Scholar] [CrossRef]
- Mosca, A.; Paleari, R.; Ivaldi, G.; Galanello, R.; Giordano, P.C. The Role Of Haemoglobin A(2) Testing In The Diagnosis Of Thalassaemias And Related Haemoglobinopathies. J. Clin. Pathol. 2009, 62, 13–17. [Google Scholar] [CrossRef]
- Demir, A.; Yarali, N.; Fisgin, T.; Duru, F.; Kara, A. Most Reliable Indices In Differentiation Between Thalassemia Trait And Iron Deficiency Anemia. Pediatr. Int. 2002, 44, 612–616. [Google Scholar] [CrossRef]
- Nalbantoğlu, B.; Güzel, S.; Büyükyalçın, V.; Donma, M.M.; Güzel, E.Ç.; Nalbantoğlu, A.; Karasu, E.; Özdilek, B. Indices Used In Differentiation Of Thalassemia Trait From Iron Deficiency Anemia In Pediatric Population: Are They Reliable? Pediatr. Hematol. Oncol. 2012, 29, 472–478. [Google Scholar] [CrossRef]
- Mentzer, W.C. Differentiation of Iron Deficiency from Thalassaemia Trait. Lancet 1973, 1, 882. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.C.; Bevington, J.M. Iron Deficiency and-or Thalassaemia Trait. Lancet 1973, 1, 832. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.F.; Dusse, L.M.; Borges, K.B.; de Castro, R.L.; Coura-Vital, W.; Carvalho, M. A New Index To Discriminate Between Iron Deficiency Anemia And Thalassemia Trait. Rev. Bras. Hematol. Hemoter. 2016, 38, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janel, A.; Roszyk, L.; Rapatel, C.; Mareynat, G.; Berger, M.G.; Serre-Sapin, A.F. Proposal Of A Score Combining Red Blood Cell Indices For Early Differentiation Of Beta-Thalassemia Minor From Iron Deficiency Anemia. Hematology 2011, 16, 123–127. [Google Scholar] [CrossRef]
- Ehsani, M.A.; Shahgholi, E.; Rahiminejad, M.S.; Seighali, F.; Rashidi, A. A New Index for Discrimination Between Iron Deficiency Anemia And Beta-Thalassemia Minor: Results In 284 Patients. Pak. J. Biol. Sci. 2009, 12, 473–475. [Google Scholar] [CrossRef] [Green Version]
- Urrechaga, E.; Hoffmann, J.J.M.L. Critical Appraisal of Discriminant Formulas for Distinguishing Thalassemia From Iron Deficiency In Patients With Microcytic Anemia. Clin. Chem. Lab. Med. 2017, 55, 1582–1591. [Google Scholar] [CrossRef]
- Huang, T.C.; Wu, Y.Y.; Chen, Y.G.; Lai, S.W.; Wu, S.C.; Ye, R.H.; Lu, C.S.; Chen, J.H. Discrimination Index Of Microcytic. Anemia In Young Soldiers: A Single Institutional Analysis. PLoS ONE 2015, 10, E0114061. [Google Scholar] [CrossRef]
- Fu, Y.K.; Liu, H.M.; Lee, L.H.; Chen, Y.J.; Chien, S.H.; Lin, J.S.; Chen, W.C.; Cheng, M.H.; Lin, P.H.; Lai, J.Y.; et al. The Tvgh-Nycu Thal-Classifier: Development of A Machine-Learning Classifier For Differentiating Thalassemia And Non-Thalassemia Patients. Diagnostics 2021, 11, 1725. [Google Scholar] [CrossRef]
- Huber, A.R.; Ottiger, C.; Risch, L.; Regenass, S.; Hergersberg, M.; Herklotz, R. Syndromes Thalassémiques: Clinique Et Diagnostic. Swiss. Med. Forum 2004, 4, 947–952. [Google Scholar]
- Zhou, Y.; Zhang, J.; Wang, C.; Zhou, L.; Zhou, L.; Ou, D.; Peng, D. Application Of Hba. Int. J. Lab. Hematol. 2020, 42, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Ambayya, A.; Sahibon, S.; Yang, T.W.; Zhang, Q.Y.; Hassan, R.; Sathar, J. A Novel Algorithm Using Cell Population Data (Vcs Parameters) As A Screening Discriminant Between Alpha And Beta Thalassemia Traits. Diagnostics 2021, 11, 2163. [Google Scholar] [CrossRef] [PubMed]
- de Medeiros Alcoforado, G.H.; Bezerra, C.M.; Araújo Moura Lemos, T.M.; de Oliveira, D.M.; Kimura, E.M.; Ferreira Costa, F.; de Fátima Sonati, M.; de Medeiros, T.M. Prevalence Of A-Thalassemia 3.7 Kb Deletion In The Adult Population Of Rio Grande Do Norte, Brazil. Genet. Mol. Biol. 2012, 35, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.C.; de Castro, S.M.; Gonzalez, T.P.; Santin, A.P.; Filippon, L.; Zaleski, C.F.; Azevedo, L.A.; Amorin, B.; Callegari-Jacques, S.M.; Hutz, M.H. Prevalence Of Common A-Thalassemia Determinants In South Brazil: Importance For The Diagnosis Of Microcytic Anemia. Genet. Mol. Biol. 2010, 33, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, G.L.; Takanashi, S.Y.; Guerreiro, J.F. Inherited Hemoglobin Disorders In An Afro-Amazonian Community: Saracura. Genet. Mol. Biol. 2012, 35, 553–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucharoen, S.; Viprakasit, V. Hb H Disease: Clinical Course and Disease Modifiers. Hematol. Am. Soc. Hematol. Educ Program. 2009, 1, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Giambona, A.; Passarello, C.; Renda, D.; Maggio, A. The Significance Of The Hemoglobin A(2) Value In Screening For Hemoglobinopathies. Clin. Biochem. 2009, 42, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Higgs, D.R. The Pathophysiology and Clinical Features Of Alpha-Thalassemia. In Disorders of Hemoglobin: Genetics, Pathophysiology, Clinical Management, 2nd ed.; Steinberg, M.H., Forget, B.G., Eds.; Cambridge University Press: New York, NY, USA, 2009; pp. 266–295. [Google Scholar]
- Steinberg, M.H.; Nagel, R.L. Hemoglobin of the Embryo, Fetus, and Adult. In Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management, 2nd ed.; Steinberg, M., Forget, B., Eds.; Cambridge University Press: New York, NY, USA, 2009; pp. 119–138. [Google Scholar]
- Van Delft, P.; Lenters, E.; Bakker-Verweij, M.; de Korte, M.; Baylan, U.; Harteveld, C.L.; Giordano, P.C. Evaluating Five Dedicated Automatic Devices For Haemoglobinopathy Diagnostics In Multi-Ethnic Populations. Int. J. Lab. Hematol. 2009, 31, 484–495. [Google Scholar] [CrossRef]
- Chen, F.E.; Ooi, C.; Ha, S.Y.; Cheung, B.M.; Todd, D.; Liang, R.; Chan, T.K.; Chan, V. Genetic And Clinical Features Of Hemoglobin H Disease In Chinese Patients. N. Engl. J. Med. 2000, 343, 544–550. [Google Scholar] [CrossRef] [Green Version]
- Chui, D.H.; Fucharoen, S.; Chan, V. Hemoglobin H Disease: Not Necessarily A Benign Disorder. Blood 2003, 101, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Singer, S.T. Variable Clinical Phenotypes of Alpha-Thalassemia Syndromes. Sci. World J. 2009, 9, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Higgs, D.R.; Weatherall, D.J. The Alpha Thalassaemias. Cell Mol. Life Sci. 2009, 66, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E. Complexity of Alpha Thalassemia: Growing Health Problem with New Approaches To Screening, Diagnosis, And Therapy. Ann. N. Y. Acad. Sci. 2010, 1202, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Wang, Y.; Ye, Y.; Yang, C.; Wu, X.; Wu, X.; Zhang, X.; Li, T.; Xiao, J.; Zhuang, L.; et al. Differential Diagnosis Of Thalassemia And Iron Deficiency Anemia In Pregnant Women Using New Formulas From Multidimensional Analysis Of Red Blood Cells. Ann. Transl. Med. 2021, 9, 141. [Google Scholar] [CrossRef] [PubMed]
- Tongsong, T.; Srisupundit, K.; Luewan, S. Outcomes of Pregnancies Affected By Hemoglobin H Disease. Int. J. Gynaecol. Obstet. 2009, 104, 206–208. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variables | OR | 95% Confidence Interval for OR | p-Value | |
|---|---|---|---|---|
| Lower Bound | Upper Bound | |||
| RCC | 8.813 | 3.365 | 23.087 | <0.001 |
| Hemoglobin | 0.807 | 0.598 | 1.089 | 0.161 |
| Hematocrit | 0.986 | 0.889 | 1.095 | 0.797 |
| MCV | 0.860 | 0.804 | 0.919 | <0.001 |
| MCV standardized by age | 0.814 | 0.751 | 0.883 | <0.001 |
| MCH | 0.618 | 0.508 | 0.752 | <0.001 |
| MCHC | 0.596 | 0.444 | 0.800 | 0.001 |
| RDW | 1.040 | 0.909 | 1.189 | 0.567 |
| Reticulocyte Count | 1.005 | 0.995 | 1.015 | 0.343 |
| % Hb A1 | 1.041 | 0.989 | 1.097 | 0.124 |
| % Hb A2 | 0.393 | 0.176 | 0.878 | 0.023 |
| % Hb F | 0.754 | 0.471 | 1.207 | 0.240 |
| Serum Iron | 1.019 | 1.006 | 1.032 | 0.003 |
| TIBC | 0.996 | 0.990 | 1.003 | 0.264 |
| Serum Ferritin | 1.007 | 0.999 | 1.014 | 0.074 |
| Transferrin Saturation | 1.002 | 0.992 | 1.012 | 0.688 |
| LDH | 0.997 | 0.993 | 1.000 | 0.085 |
| Indirect Bilirubin | 1.737 | 0.300 | 10.043 | 0.538 |
| Two or More α-Globin Gene Deletions | B | p-Value | OR (Exp(B)) | IC95 |
|---|---|---|---|---|
| Intercept | 21.905 | <0.001 | ||
| MCV standardized by age | −0.284 | <0.001 | 0.753 | 0.676–0.839 |
| Serum Ferritin | 0.024 | <0.001 | 1.024 | 1.011–1.037 |
| % Hb A2 | −1.142 | 0.006 | 0.319 | 0.142–0.715 |
| Prediction Equation | |
|---|---|
| OR | =exp [21.905 − 0.284 (MCV standardized by age(fL)) + 0.024 (serum ferritin(ng/mL)) − 1.142 (Hb A2(%))] |
| Probability (P α-thal with 2 or more α-globin genes deletions) | =OR/(1 + OR) |
| Training Set Frequency (%)/Median (IQR)/Mean (SE) n = 134 | Validation Set Frequency (%)/Median (IQR)/Mean (SE) n = 160 | p-Value | |
|---|---|---|---|
| Age (years) | 9.5 (10.3) | 8.5 (10.1) | 0.551 * |
| Gender | |||
| Male | 67 (50.0%) | 86 (53.8%) | 0.559 *** |
| Female | 67 (50.0%) | 74 (46.2%) | |
| The Genotype of the α-Globin Gene | |||
| αα/αα | 53 (39.6%) | 69 (43.1%) | 0.801 *** |
| -α3.7/αα | 46 (34.3%) | 50 (31.3%) | |
| -α3.7/-α3.7 | 33 (24.6%) | 41 (25.6%) | |
| -α3.7/- SEA | 1 (0.7%) | — | |
| αα/- SEA | 1 (0.7%) | — | |
| Variables of the BLR model | |||
| MCV standardized by age (fL) | 76.3 (± 0.71) | 75.6 (± 0.63) | 0.470 ** |
| Serum Ferritin (ng/mL) | 32.1 (35.7) | 31.2 (36.2) | 0.726 * |
| Hb A2 (%) | 2.8 (0.4) | 2.8 (0.4) | 0.514 * |
| Training Set | Validation Set | Total Sample | ||||
|---|---|---|---|---|---|---|
| (n = 134) | (n = 160) | (n = 294) | ||||
| C.I. (95%) | C.I. (95%) | C.I. (95%) | ||||
| Sensitivity | 88.6% | 73.3–96.8% | 95.1% | 83.5–99.4% | 92.1% | 83.6–97.1% |
| Specificity | 83.8% | 75.1–90.5% | 82.4% | 74.3–88.7% | 83.0% | 77.4–87.8% |
| Positive Likelihood Ratio | 5.48 | 3.45 -8.72 | 5.39 | 3.63–8.00 | 5.43 | 4.02–7.33 |
| Negative Likelihood Ratio | 0.14 | 0.05–0.34 | 0.06 | 0.02–0.23 | 0.10 | 0.04–0.21 |
| Disease Prevalence | 26.1% | 18.9–34.4% | 25.6% | 19.1–33.1% | 25.9% | 20.9–31.3% |
| Positive Predictive Value | 66.0% | 54.9–75.5% | 65.0% | 55.6–73.4% | 65.4% | 58.3–71.9% |
| Negative Predictive Value | 95.4% | 89.2–98.1% | 98.0% | 92.7–99.5% | 96.8% | 93.3–98.5% |
| Cut-off Point Accuracy | 85.1% | 77.9–90.6% | 85.6% | 79.2–90.7% | 85.4% | 80.8–89.2% |
| Global Accuracy (AUC) | 0.927 | 0.883–0.970 | 0.902 | 0.853–0.950 | 0.914 | 0.881–0.947 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siqueira, P.F.R.; Fleury, M.K.; Pontes, R.M.; Silva, R.S.P.; Costa, E.S.; Land, M.G.P. Novel Decision Tool for More Severe α-Thalassemia Genotypes Screening with Functional Loss of Two or More α-Globin Genes: A Diagnostic Test Study. Diagnostics 2022, 12, 3008. https://doi.org/10.3390/diagnostics12123008
Siqueira PFR, Fleury MK, Pontes RM, Silva RSP, Costa ES, Land MGP. Novel Decision Tool for More Severe α-Thalassemia Genotypes Screening with Functional Loss of Two or More α-Globin Genes: A Diagnostic Test Study. Diagnostics. 2022; 12(12):3008. https://doi.org/10.3390/diagnostics12123008
Chicago/Turabian StyleSiqueira, Patricia F. R., Marcos K. Fleury, Robéria M. Pontes, Renata S. P. Silva, Elaine S. Costa, and Marcelo G. P. Land. 2022. "Novel Decision Tool for More Severe α-Thalassemia Genotypes Screening with Functional Loss of Two or More α-Globin Genes: A Diagnostic Test Study" Diagnostics 12, no. 12: 3008. https://doi.org/10.3390/diagnostics12123008