
Pulmonary Hypertension Associated Genetic Variants in Sarcoidosis Associated Pulmonary Hypertension

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
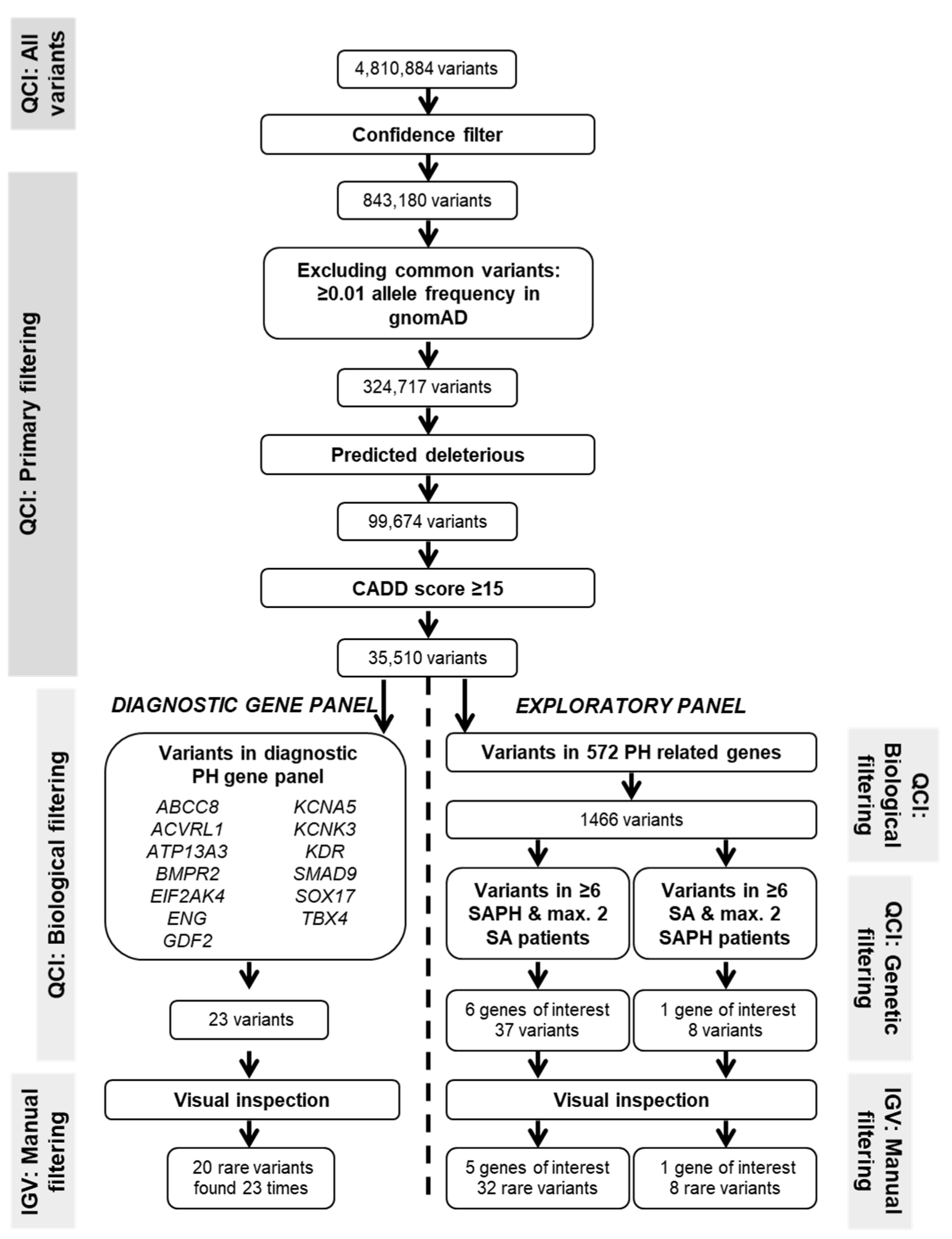
2.2. Whole Exome Sequencing and Variant Filtering
2.3. Diagnostic PH Gene Panel
2.4. Exploratory Analysis in Genes Biologically Involved in PH Pathways
2.5. Statistical Analysis
3. Results
3.1. Patient Cohorts
3.2. Diagnostic Gene Panel
3.3. Genes of Biological Relevance in PH
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baughman, R.P.; Culver, D.A.; Judson, M.A. A Concise Review of Pulmonary Sarcoidosis. Am. J. Respir. Crit. Care Med. 2011, 183, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef]
- Huitema, M.P.; Bakker, A.L.M.; Mager, J.J.; Rensing, B.J.W.M.; Smits, F.; Snijder, R.J.; Grutters, J.C.; Post, M.C. Prevalence of Pulmonary Hypertension in Pulmonary Sarcoidosis: The First Large European Prospective Study. Eur. Respir. J. 2019, 54, 1900897. [Google Scholar] [CrossRef]
- Shorr, A.F.; Helman, D.L.; Davies, D.B.; Nathan, S.D. Pulmonary Hypertension in Advanced Sarcoidosis: Epidemiology and Clinical Characteristics. Eur. Respir. J. 2005, 25, 783–788. [Google Scholar] [CrossRef] [Green Version]
- Nunes, H.; Humbert, M.; Capron, F.; Brauner, M.; Sitbon, O.; Battesti, J.-P.; Simonneau, G.; Valeyre, D. Pulmonary Hypertension Associated with Sarcoidosis: Mechanisms, Haemodynamics and Prognosis. Thorax 2006, 61, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathijssen, H.; Huitema, M.P.; Bakker, A.L.M.; Smits, F.; Mager, J.J.; Snijder, R.J.; Grutters, J.C.; Post, M.C. Clinical Phenotypes of Sarcoidosis-Associated Pulmonary Hypertension. Heart Lung Circ. 2021, 30, 1502–1508. [Google Scholar] [CrossRef]
- Sulica, R.; Teirstein, A.S.; Kakarla, S.; Nemani, N.; Behnegar, A.; Padilla, M.L. Distinctive Clinical, Radiographic, and Functional Characteristics of Patients with Sarcoidosis-Related Pulmonary Hypertension. Chest 2005, 128, 1483–1489. [Google Scholar] [CrossRef] [Green Version]
- Bourbonnais, J.M.; Samavati, L. Clinical Predictors of Pulmonary Hypertension in Sarcoidosis. Eur. Respir. J. 2008, 32, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Huitema, M.P.; Grutters, J.C.; Rensing, B.J.W.M.; Reesink, H.J.; Post, M.C. Pulmonary Hypertension Complicating Pulmonary Sarcoidosis. Neth. Heart J. 2016, 24, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Eyries, M.; Montani, D.; Nadaud, S.; Girerd, B.; Levy, M.; Bourdin, A.; Trésorier, R.; Chaouat, A.; Cottin, V.; Sanfiorenzo, C.; et al. Widening the Landscape of Heritable Pulmonary Hypertension Mutations in Paediatric and Adult Cases. Eur. Respir. J. 2019, 53, 1801371. [Google Scholar] [CrossRef]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of Rare Sequence Variation Underlying Heritable Pulmonary Arterial Hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, E.D.; Phillips, J.A.; Loyd, J.E. Heritable Pulmonary Arterial Hypertension Overview. GeneReviews®; 1993. Available online: https://pubmed.ncbi.nlm.nih.gov/20301658/ (accessed on 2 September 2022).
- Baloira Villar, A.; Pousada Fernández, G.; Núñez Fernández, M.; Valverde Pérez, D. Estudio Clínico y Molecular de 4 Casos de Hipertensión Pulmonar Asociada a Sarcoidosis. Arch. Bronconeumol. 2015, 51, e19–e21. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O.; Leong, I.U.S.; Smith, K.R.; Gerasimenko, O.; Haraldsdottir, E.; et al. PanelApp Crowdsources Expert Knowledge to Establish Consensus Diagnostic Gene Panels. Nat. Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef] [PubMed]
- Costabel, U.; Hunninghake, G.W.; on behalf of the Sarcoidosis Statement Committee. ATS/ERS/WASOG Statement on Sarcoidosis. Eur. Respir. J. 1999, 14, 735. [Google Scholar] [CrossRef] [Green Version]
- Scadding, J.G. Prognosis of Intrathoracic Sarcoidosis in England. BMJ 1961, 2, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Pousada, G.; Baloira, A.; Fontán, D.; Núñez, M.; Valverde, D. Mutational and Clinical Analysis of the ENG Gene in Patients with Pulmonary Arterial Hypertension. BMC Genet. 2016, 17, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Letteboer, T.G.W.; Zewald, R.A.; Kamping, E.J.; de Haas, G.; Mager, J.J.; Snijder, R.J.; Lindhout, D.; Hennekam, F.A.M.; Westermann, C.J.J.; Ploos van Amstel, J.K. Hereditary Hemorrhagic Telangiectasia: ENG and ALK-1 Mutations in Dutch Patients. Hum. Genet. 2005, 116, 8–16. [Google Scholar] [CrossRef]
- Christophersen, I.E.; Olesen, M.S.; Liang, B.; Andersen, M.N.; Larsen, A.P.; Nielsen, J.B.; Haunsø, S.; Olesen, S.-P.; Tveit, A.; Svendsen, J.H.; et al. Genetic Variation in KCNA5: Impact on the Atrial-Specific Potassium Current IKur in Patients with Lone Atrial Fibrillation. Eur. Heart J. 2013, 34, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Mann, S.A.; Otway, R.; Guo, G.; Soka, M.; Karlsdotter, L.; Trivedi, G.; Ohanian, M.; Zodgekar, P.; Smith, R.A.; Wouters, M.A.; et al. Epistatic Effects of Potassium Channel Variation on Cardiac Repolarization and Atrial Fibrillation Risk. J. Am. Coll. Cardiol. 2012, 59, 1017–1025. [Google Scholar] [CrossRef]
- Simard, C.; Drolet, B.; Yang, P.; Kim, R.B.; Roden, D.M. Polymorphism Screening in the Cardiac K+ Channel Gene KCNA5*. Clin. Pharmacol. Ther. 2005, 77, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Mallet, C.; Lamribet, K.; Giraud, S.; Dupuis-Girod, S.; Feige, J.J.; Bailly, S.; Tillet, E. Functional Analysis of Endoglin Mutations from Hereditary Hemorrhagic Telangiectasia Type 1 Patients Reveals Different Mechanisms for Endoglin Loss of Function. Hum. Mol. Genet. 2015, 24, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Richards-Yutz, J.; Grant, K.; Chao, E.C.; Walther, S.E.; Ganguly, A. Update on Molecular Diagnosis of Hereditary Hemorrhagic Telangiectasia. Hum. Genet. 2010, 128, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Sadick, H.; Hage, J.; Goessler, U.; Stern-Straeter, J.; Riedel, F.; Hoermann, K.; Bugert, P. Mutation Analysis of “Endoglin” and “Activin Receptor-like Kinase” Genes in German Patients with Hereditary Hemorrhagic Telangiectasia and the Value of Rapid Genotyping Using an Allele-Specific PCR-Technique. BMC Med. Genet. 2009, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Gedge, F.; McDonald, J.; Phansalkar, A.; Chou, L.S.; Calderon, F.; Mao, R.; Lyon, E.; Bayrak-Toydemir, P. Clinical and Analytical Sensitivities in Hereditary Hemorrhagic Telangiectasia Testing and a Report of de Novo Mutations. J. Mol. Diagn. 2007, 9, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Ali, B.R.; Ben-Rebeh, I.; John, A.; Akawi, N.A.; Milhem, R.M.; Al-Shehhi, N.A.; Al-Ameri, M.M.; Al-Shamisi, S.A.; Al-Gazali, L. Endoplasmic Reticulum Quality Control Is Involved in the Mechanism of Endoglin-Mediated Hereditary Haemorrhagic Telangiectasia. PLoS ONE 2011, 6, e26206. [Google Scholar] [CrossRef] [Green Version]
- Prigoda, N.L.; Savas, S.; Abdalla, S.A.; Piovesan, B.; Rushlow, D.; Vandezande, K.; Zhang, E.; Ozcelik, H.; Gallie, B.L.; Letarte, M. Hereditary Haemorrhagic Telangiectasia: Mutation Detection, Test Sensitivity and Novel Mutations. J. Med. Genet. 2006, 43, 722–728. [Google Scholar] [CrossRef] [Green Version]
- Ricard, N.; Bidart, M.; Mallet, C.; Lesca, G.; Giraud, S.; Prudent, R.; Feige, J.-J.; Bailly, S. Functional Analysis of the BMP9 Response of ALK1 Mutants from HHT2 Patients: A Diagnostic Tool for Novel ACVRL1 Mutations. Blood 2010, 116, 1604–1612. [Google Scholar] [CrossRef] [Green Version]
- Lesca, G.; Plauchu, H.; Coulet, F.; Lefebvre, S.; Plessis, G.; Odent, S.; Rivière, S.; Leheup, B.; Goizet, C.; Carette, M.F.; et al. Molecular Screening of ALK1/ACVRL1 and ENG Genes in Hereditary Hemorrhagic Telangiectasia in France. Hum. Mutat. 2004, 23, 289–299. [Google Scholar] [CrossRef]
- McDonald, J.; Gedge, F.; Burdette, A.; Carlisle, J.; Bukjiok, C.J.; Fox, M.; Bayrak-Toydemir, P. Multiple Sequence Variants in Hereditary Hemorrhagic Telangiectasia Cases: Illustration of Complexity in Molecular Diagnostic Interpretation. J. Mol. Diagn. 2009, 11, 569–575. [Google Scholar] [CrossRef]
- Mattassi, R.; Manara, E.; Colombo, P.G.; Manara, S.; Porcella, A.; Bruno, G.; Bruson, A.; Bertelli, M. Variant Discovery in Patients with Mendelian Vascular Anomalies by Next-Generation Sequencing and Their Use in Patient Clinical Management. J. Vasc. Surg. 2018, 67, 922–932.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez, J.; Reguero, J.R.; Alvarez, C.; Junquera, M.R.; Arango, A.; Morís, C.; Coto, E. A Semiconductor Chip-Based Next Generation Sequencing Procedure for the Main Pulmonary Hypertension Genes. Lung 2015, 193, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.H.F.; Roofthooft, M.T.R.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 Mutations (Small Patella Syndrome) Are Associated with Childhood-Onset Pulmonary Arterial Hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A.; Hamid, R.; Loyd, J.E. Longitudinal Analysis Casts Doubt on the Presence of Genetic Anticipation in Heritable Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tew, M.B.; Arnett, F.C.; Reveille, J.D.; Tan, F.K. Mutations of Bone Morphogenetic Protein Receptor Type II Are Not Found in Patients with Pulmonary Hypertension and Underlying Connective Tissue Diseases. Arthritis Rheum. 2002, 46, 2829–2830. [Google Scholar] [CrossRef]
- Morse, J.; Barst, R.; Horn, E.; Cuervo, N.; Deng, Z.; Knowles, J. Pulmonary Hypertension in Scleroderma Spectrum of Disease: Lack of Bone Morphogenetic Protein Receptor 2 Mutations. J. Rheumatol. 2002, 29, 2379–2381. [Google Scholar]
- Chida, A.; Shintani, M.; Matsushita, Y.; Sato, H.; Eitoku, T.; Nakayama, T.; Furutani, Y.; Hayama, E.; Kawamura, Y.; Inai, K.; et al. Mutations of Notch3 in Childhood Pulmonary Arterial Hypertension. Mol. Genet. Genom. Med. 2014, 2, 229–239. [Google Scholar] [CrossRef]
- Baeten, J.T.; Lilly, B. Notch Signaling in Vascular Smooth Muscle Cells. Adv. Pharmacol. 2017, 78, 351–382. [Google Scholar]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.-J.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 Signaling Promotes the Development of Pulmonary Arterial Hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Boucly, A.; Cottin, V.; Nunes, H.; Jaïs, X.; Tazi, A.; Prévôt, G.; Reynaud-Gaubert, M.; Dromer, C.; Viacroze, C.; Horeau-Langlard, D.; et al. Management and Long-Term Outcomes of Sarcoidosis-Associated Pulmonary Hypertension. Eur. Respir. J. 2017, 50, 1700465. [Google Scholar] [CrossRef]
- Shlobin, O.A.; Kouranos, V.; Barnett, S.D.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Cordova, F.C.; Carmona, E.M.; Scholand, M.B.; Wijsenbeek, M.; et al. Physiological Predictors of Survival in Patients with Sarcoidosis-Associated Pulmonary Hypertension: Results from an International Registry. Eur. Respir. J. 2020, 55, 1901747. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 2, ISBN 0471142905. [Google Scholar]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving Genome-Wide Variant Effect Prediction Using Deep Learning-Derived Splice Scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]


{kind=link}
| SAPH (n = 39) | SA (n = 39) | p-Value | ||
|---|---|---|---|---|
| Male, n (%) | 19 (48.7) | 19 (48.7) | 1.00 | |
| Age mean (SD), y | 59.2 (11.3) | 60.9 (10.9) | 0.50 | |
| Ancestry | White, n (%) | 27 (69.2) | 27 (69.2) | 1.00 |
| Black, n (%) | 9 (23.1) | 9 (23.1) | ||
| Hindustan, n (%) | 3 (7.7) | 3 (7.7) | ||
| Scadding stage (n = 38; n = 39) | 0, n (%) | 2 (5.3) | 8 (20.5) | 0.016 |
| 1, n (%) | 4 (10.5) | 3 (7.7) | ||
| 2, n (%) | 4 (10.5) | 9 (23.1) | ||
| 3, n (%) | 2 (5.3) | 6 (15.4) | ||
| 4, n (%) | 26 (68.4) | 13 (33.3) | ||
| Lung fibrosis score (n = 38; n = 39) | <5%, n (%) | 9 (23.7) | 22 (56.4) | <0.001 |
| 5–20%, n (%) | 1 (2.6) | 7 (17.9) | ||
| >20%, n (%) | 28 (73.7) | 10 (25.6) | ||
| FVC % predicted, mean (SD) (n = 37; n = 36) | 66.6 (19.4) | 94.2 (23.1) | <0.001 | |
| DLCO SB% predicted, mean, mean (SD) (n = 32; n = 32) | 47.8 (18.5) | 68.4 (15.2) | <0.001 | |
| Mean PAP mmHg, mean (SD) | 37.4 (10.9) | ND | ||
| PVR Wood units, mean (SD) (n = 36; ND) | 5.6 (3.1) | ND | ||
| Cardiac output L·min-1, mean (SD) (n = 38; ND) | 5.7 (1.8) | ND | ||
| PCWP mmHg, mean (SD) (n = 37; ND) | 9.7 (3.6) | ND |
| Gene | Transcript | Nucleotide Change | Amino Acid Change | dbSNP | gnomAD Frequency | CADD | SIFT a | PPh2 b | Mutation Taster c | Mutation Assessor d | Variant Classification e | SAPH Carrier ID(s) | SA Carrier ID(s) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KCNK3 | NM_002246.3 | c.418C>A | p.(L140M) | rs755486166 | 0.0000 | 25.5 | D | PrD | D | M | VUS | - | SA67 |
| BMPR2 | NM_001204.7 | c.86A>G | p.(N29S) | rs112862820 | 0.0007 | 16.2 | T | B | B | N | LB | SAPH5 | - |
| KDR | NM_002253.4 | c.3762+8C>A | NA | rs78801899 | 0.0003 | 17.0 | NA | NA | B | NA | VUS | - | SA71 |
| KDR | NM_002253.4 | c.2854G>A | p.(V952I) | rs13129474 | 0.0000 | 20.0 | T | B | B | N | VUS | - | SA41 |
| KDR | NM_002253.4 | c.2837G>A | p.(R946H) | rs140041720 | 0.0002 | 28.7 | D | PrD | B | N | VUS | SAPH1 | - |
| KDR | NM_002253.4 | c.406G>A | p.(V136M) | rs35636987 | 0.0007 | 24.1 | D | PrD | B | M | VUS | SAPH6 | - |
| KDR | NM_002253.4 | c.170G>C | p.(R57T) | rs139047809 | 0.0012 | 19.3 | T | PoD | B | M | VUS | - | SA41 |
| ENG | NM_000118.3 | c.1844C>T | p.(S615L) | rs148002300 | 0.0015 | 21.1 | T | B | B | M | LB | SAPH10 | - |
| ENG | NM_000118.3 | c.583G>A | p.(E195K) | rs1255912441 | 0.0000 | 17.6 | T | B | B | M | VUS | SAPH15 | - |
| ENG | NM_000118.3 | c.572G>A | p.(G191D) | rs41322046 | 0.0091 | 24.2 | D | PrD | B | M | B | SAPH11 | - |
| KCNA5 | NM_002234.4 | c.188G>T | p.(G63V) | rs768062067 | 0.0000 | 20.1 | - | B | B | N | VUS | SAPH34 | - |
| KCNA5 | NM_002234.4 | c.929C>T | p.(P310L) | rs17215402 | 0.0040 | 18.2 | T | B | B | L | LB | SAPH12 | - |
| KCNA5 | NM_002234.4 | c.1733G>A | p.(R578K) | rs12720445 | 0.0054 | 17.2 | T | B | B | M | LB | SAPH24; SAPH13; SAPH11 | - |
| ACVRL1 | NM_001077401.2 | c.-159C>T | NA | rs542225698 | 0.0009 | 17.3 | NA | NA | B | NA | VUS | SAPH23; SAPH8 | - |
| ACVRL1 | NM_000020.3 | c.1445C>T | p.(A482V) | rs139142865 | 0.0016 | 23.2 | D | PrD | B | M | LB | SAPH26 | - |
| SMAD9 | NM_005905.6 | c.677G>A | p.(R226Q) | rs78249575 | 0.0012 | 17.9 | A | B | B | N | VUS | - | SA77 |
| EIF2AK4 | NM_001013703.4 | c.13C>T | p.(R5C) | rs746773845 | 0.0000 | 23.3 | NA | B | B | L | VUS | SAPH10 | - |
| EIF2AK4 | NM_001013703.4 | c.4792G>A | p.(A1598T) | rs1045326564 | 21.0 | T | B | B | N | VUS | - | SA71 | |
| TBX4 | NM_018488.3 | c.104C>T | p.(A35V) | rs148424252 | 0.0089 | 16.5 | T | B | B | N | LB | - | SA58 |
| TBX4 | NM_018488.3 | c.1094A>G | p.(H365R) | rs1443274012 | 0.0000 | 25.5 | T | PrD | D | M | VUS | SAPH17 | - |
| Gene | Transcript | Nucleotide Change | Amino Acid Change | dbSNP | gnomAD Frequency | CADD | SIFT a | PPh2 b | Mutation Taster c | Mutation Assessor d | Variant Classification e | SAPH Carrier ID(s) | SA Carrier ID(s) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDE6B | NM_001145291.2 | c.35T>C | p.(L12P) | - | - | 24.1 | - | PrD | D | M | VUS | SAPH20 | - |
| PDE6B | NM_001145291.2 | c.143G>A | p.(R48Q) | rs113842820 | 0.0082 | 15.4 | - | B | B | L | B | SAPH1; SAPH38 | - |
| PDE6B | NM_001145291.2 | c.373C>G | p.(P125A) | rs28414606 | 0.0078 | 15.6 | - | B | B | N | B | SAPH38 | - |
| PDE6B | NM_001145291.2 | c.496G>A | p.(E166K) | rs115775983 | 0.0090 | 15.6 | - | B | B | N | LB | SAPH1 | SA68 |
| PDE6B | NM_001145291.2 | c.706G>A | p.(G236S) | rs75543439 | 0.0002 | 20.4 | - | B | B | N | VUS | SAPH14 | - |
| PDE6B | NM_001145291.2 | c.794G>A | p.(R265Q) | rs144562730 | 0.0011 | 26.1 | - | PrD | B | L | VUS | SAPH12 | - |
| PDE6B | NM_001145291.2 | c.1295C>T | p.(T432I) | rs775120495 | 0.0000 | 23.7 | - | PoD | B * | M | VUS | SAPH34 | - |
| PDE6B | NM_001145291.2 | c.2344G>A | p.(V782M) | rs145124626 | 0.0003 | 26.9 | - | PrD | D | M | VUS | - | SA41 |
| PDE6B | NM_001145291.2 | c.2524G>A | p.(G842S) | rs367709559 | 0.0000 | 22.3 | - | B | B | L | VUS | - | SA41 |
| COL5A1 | NM_001278074.1 | c.278C>T | p.(A93V) | rs41306397 | 0.0087 | 22.2 | D | B | B | L | LB | SAPH28; SAPH16 | SA61 |
| COL5A1 | NM_001278074.1 | c.2695G>A | p.(G899S) | rs149964491 | 0.0016 | 23.3 | T | PoD | B | N | LB | SAPH38 | - |
| COL5A1 | NM_001278074.1 | c.4906G>A | p.(A1636T) | rs113452150 | 0.0073 | 29.1 | D | PoD | D | H | VUS | SAPH40 | - |
| COL5A1 | NM_001278074.1 | c.5407G>A | p.(D1803N) | rs61729495 | 0.0061 | 19.0 | A | B | B | N | LB | SAPH30; SAPH5 | - |
| MMP21 | NM_147191.1 | c.1361C>T | p.(A454V) | rs28381319 | 0.0002 | 22.3 | T | B | B | M | B | SAPH31; SAPH35; SAPH39 | SA80 |
| MMP21 | NM_147191.1 | c.1046A>G | p.(E349G) | rs28381302 | 0.0021 | 23.2 | D | PoD | B | M | LB | SAPH21; SAPH12 | SA54 |
| MMP21 | NM_147191.1 | c.290C>T | p.(A97V) | rs554501102 | 0.0006 | 17.4 | D | B | B | M | VUS | SAPH27 | - |
| NOTCH3 | NM_000435.3 | c.6221C>T | p.(P2074L) | rs114447350 | 0.0003 | 21.9 | T | B | B | N | B | SAPH33; SAPH30 | - |
| NOTCH3 | NM_000435.3 | c.5900T>C | p.(M1967T) | rs377589088 | 0.0012 | 26.9 | D | PrD | D | N | VUS | SAPH28 | - |
| NOTCH3 | NM_000435.3 | c.5854G>A | p.(V1952M) | rs115582213 | 0.0088 | 27.3 | D | PrD | B | L | B | - | SA48; SA68 |
| NOTCH3 | NM_000435.3 | c.5284G>A | p.(V1762M) | rs756495084 | 0.0030 | 15.5 | T | B | B | N | LB | SAPH27 | - |
| NOTCH3 | NM_000435.3 | c.4762A>C | p.(N1588H) | - | 0.0009 | 26.3 | D | PrD | B | M | VUS | SAPH11 | - |
| NOTCH3 | NM_000435.3 | c.4679G>C | p.(R1560P) | rs78501403 | 0.0097 | 24.7 | T | PrD | B | L | LB | SAPH35 | - |
| NOTCH3 | NM_000435.3 | c.3352A>T | p.(N1118Y) | rs376950447 | 0.0001 | 22.3 | D | PrD | B | L | VUS | SAPH24 | - |
| NOTCH3 | NM_000435.3 | c.3088G>A | p.(G1030R) | rs1179899018 | 0.0082 | 23.8 | D | PrD | D | H | VUS | SAPH21 | - |
| NOTCH3 | NM_000435.3 | c.2039G>A | p.(R680H) | rs10406745 | 0.0002 | 17.3 | T | B | B | L | LB | SAPH30 | - |
| NOTCH3 | NM_000435.3 | c.1490C>T | p.(S497L) | rs114207045 | - | 24.9 | T | B | B | L | B | SAPH9 | - |
| NOTCH3 | NM_000435.3 | c.182G>A | p.(R61Q) | rs1222763947 | 0.0015 | 18.0 | T | B | B | N | VUS | SAPH12 | - |
| GUCY2F | NM_001522.3 | c.2380G>A | p.(E794K) | rs35726803 | 0.0001 | 28.5 | D | PrD | B | M | VUS | SAPH24 | SA68 |
| GUCY2F | NM_001522.3 | c.1883G>A | p.(R628Q) | rs7883913 | 0.0000 | 24.1 | D | PrD | B | M | VUS | SAPH12 | - |
| GUCY2F | NM_001522.3 | c.1720A>C | p.(K574Q) | rs139586665 | 0.0048 | 25.2 | D | PrD | B | M | VUS | SAPH29 | - |
| GUCY2F | NM_001522.3 | c.1483A>C | p.(N495H) | rs148768857 | 0.0067 | 20.2 | T | B | B | L | LB | SAPH40; SAPH39 | - |
| GUCY2F | NM_001522.3 | c.688C>T | p.(R230W) | rs33973457 | 0.0000 | 22.7 | D | PoD | B | L | LB | SAPH33 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groen, K.; Huitema, M.P.; van der Vis, J.J.; Post, M.C.; Grutters, J.C.; Baughman, R.P.; van Moorsel, C.H.M. Pulmonary Hypertension Associated Genetic Variants in Sarcoidosis Associated Pulmonary Hypertension. Diagnostics 2022, 12, 2564. https://doi.org/10.3390/diagnostics12102564
Groen K, Huitema MP, van der Vis JJ, Post MC, Grutters JC, Baughman RP, van Moorsel CHM. Pulmonary Hypertension Associated Genetic Variants in Sarcoidosis Associated Pulmonary Hypertension. Diagnostics. 2022; 12(10):2564. https://doi.org/10.3390/diagnostics12102564
Chicago/Turabian StyleGroen, Karlijn, Marloes P. Huitema, Joanne J. van der Vis, Marco C. Post, Jan C. Grutters, Robert P. Baughman, and Coline H. M. van Moorsel. 2022. "Pulmonary Hypertension Associated Genetic Variants in Sarcoidosis Associated Pulmonary Hypertension" Diagnostics 12, no. 10: 2564. https://doi.org/10.3390/diagnostics12102564