
Head and Neck Paragangliomas in the Czech Republic: Management at the Otorhinolaryngology Department
_(OMFS)_(Lond)_MD_PhD.jpg)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Subject Selection Criteria
2.2. Management Protocol
2.2.1. Characterization of Tumors Based on Clinicoradiological Investigations
- Biochemical examination: Plasma metanephrine and normetaphrine levels were measured to ascertain secretory status of tumors. Chromogranin A, a tumor marker for pheochromocytomas/paragangliomas, was also determined for all patients.
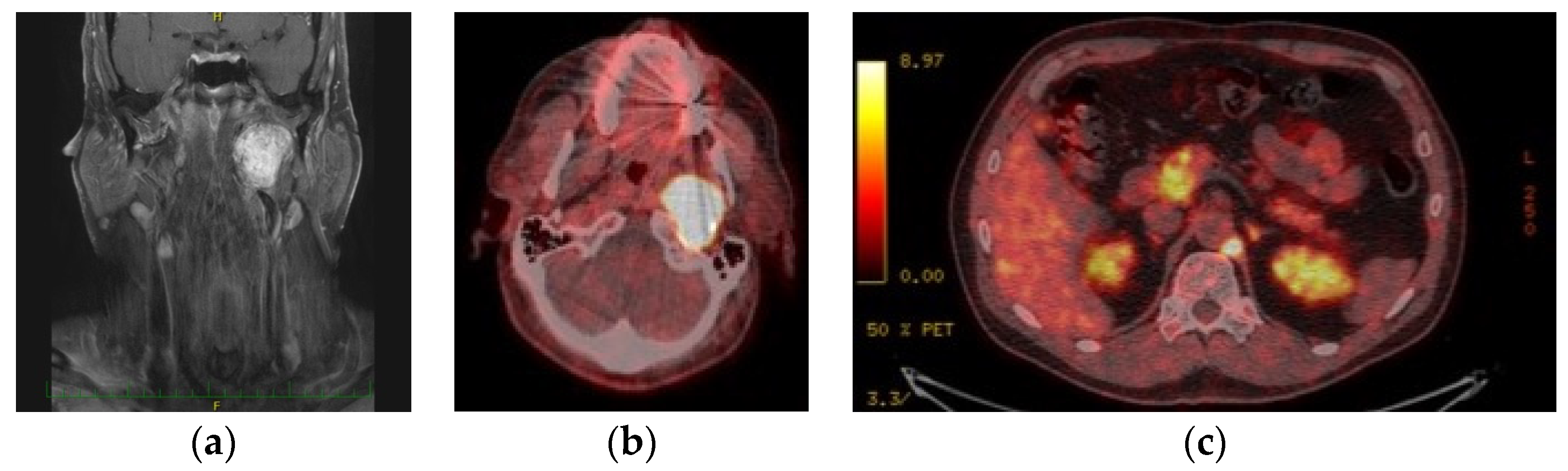
- Analysis using imaging techniques: Following ultrasound of the neck, CT or MRI including CT- or MR-angiography according to the localization, Shamblin’s classification was employed to classify the extent of CBPGLs, whilst Fisch’s classification was used for vagal and jugular PGLs. Furthermore, 18F-FDOPA PET/CT scan was done for all patients with multiple tumors, suspected metastases, and/or progression of disease on follow-up.
- Germline genetic testing: According to our protocol, all patients who consented underwent genetic examination. Peripheral blood samples were taken for DNA extraction and analysis. Firstly, preliminary germline genetic testing using Polymerase Chain Reaction (PCR) Sanger sequencing was done using the Mutation Surveyor® (Carolina Biosystems, Czech Republic) to exclude SDHD followed by SDHB mutation; subsequently, patients were referred for genetic counselling to complete Next Generation Sequencing (NGS) examination. NGS utilizing NextSeq 500 (Illumina®, San Diego, CA, USA) was used to analyze 123 standard panel genes for pheochromocytoma/paraganglioma. In certain cases, NGS examination was already completed before referral to us. Relevant family members of index patients with positive genetic mutation were also invited for genetic carrier testing.
2.2.2. Institutional Ethics Board Statement
2.2.3. Treatment Plan
Therapeutic Approaches
Protocol for Surgery
- Preoperative embolization
- Surgical techniques
- Postoperative care
3. Results
3.1. Analysis of Audiometric Examination
3.2. Characteristics of PGLs and Genetic Results
3.2.1. Localization of Head and Neck Tumors
- Unilateral PGLs
Carotid Body
Jugular
Tympanic
Vagal
- Multiple or Bilateral PGLs
3.2.2. Localization of Paragangliomas below the Neck
3.2.3. Biochemical Activity of Tumors
3.3. Treatment Modality and Outcome
3.3.1. Surgery
Preoperative Embolization
Surgical Approaches
Histopathology Results
3.3.2. ‘Wait and Scan’ Approach
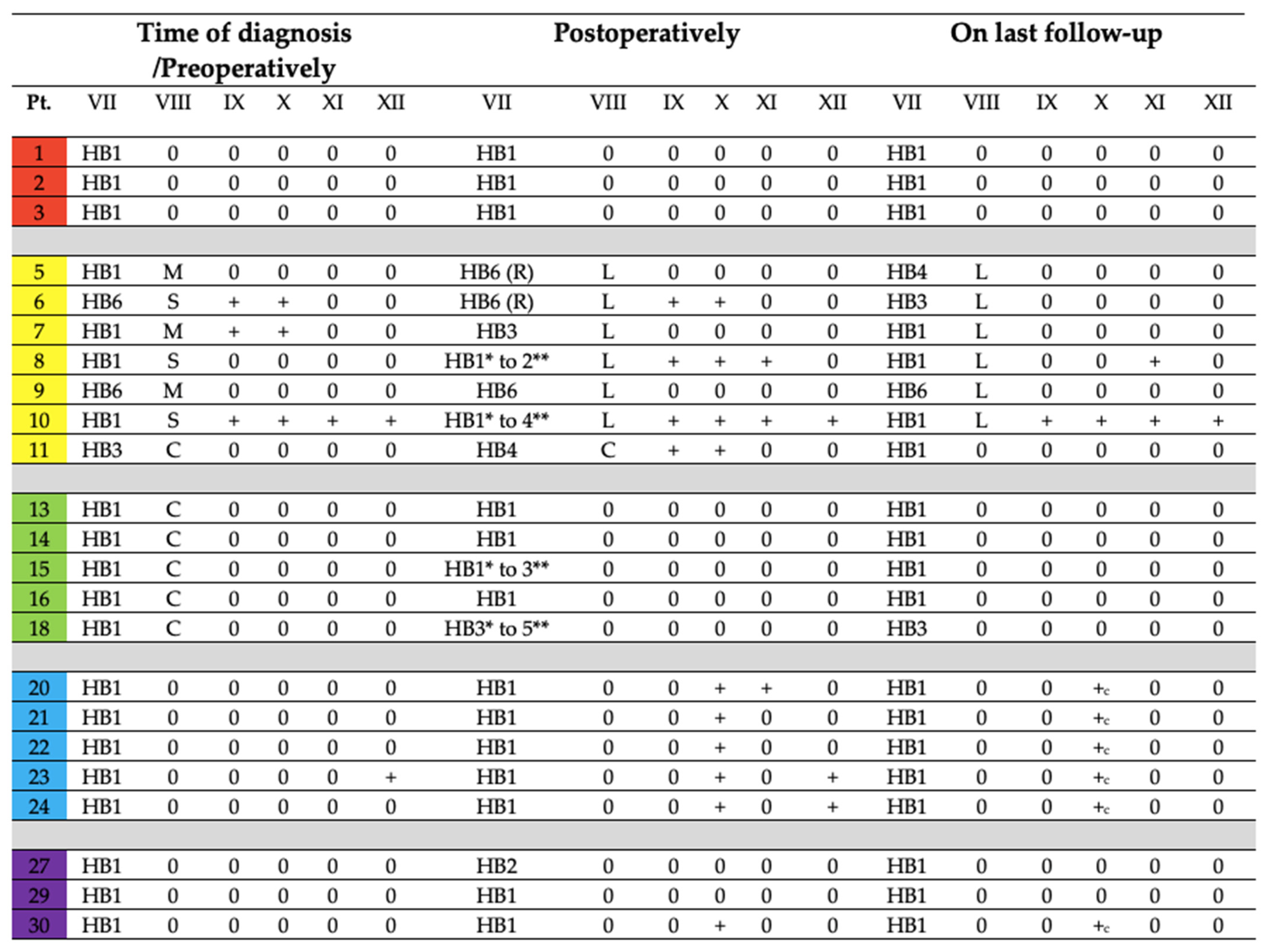
3.3.3. Disease Control and Cranial Nerve Function Status on Last Follow-Up
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Whalen, R.K.; Althausen, A.F.; Daniels, G.H. Extra-Adrenal Pheochromocytoma. J. Urol. 1992, 147, 1–10. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Osamura, R.Y.; Kloppel, G.; Rosai, J. (Eds.) Head and neck paragangliomas. In WHO Classification of Tumours of Endocrine Organs, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 190–192. [Google Scholar]
- Taïeb, D.; Kaliski, A.; Boedeker, C.C.; Martucci, V.; Fojo, T.; Adler, J.R., Jr.; Pacak, K. Current Approaches and Recent Developments in the Management of Head and Neck Paragangliomas. Endocr. Rev. 2014, 35, 795–819. [Google Scholar] [CrossRef]
- Wasserman, P.G.; Savargaonkar, P. Paragangliomas. Otolaryngol. Clin. N. Am. 2001, 34, 845–862. [Google Scholar] [CrossRef]
- Offergeld, C.; Brase, C.; Yaremchuk, S.; Mader, I.; Rischke, H.C.; Gläsker, S.; Schmid, K.W.; Wiech, T.; Preuss, S.F.; Suárez, C.; et al. Head and Neck Paragangliomas: Clinical and Molecular Genetic Classification. Clinics 2012, 67 (Suppl. 1), 19–28. [Google Scholar] [CrossRef]
- Guha, A.; Musil, Z.; Vicha, A.; Zelinka, T.; Pacak, K.; Astl, J.; Chovanec, M. A Systematic Review on the Genetic Analysis of Paragangliomas: Primarily Focused on Head and Neck Paragangliomas. Neoplasma 2019, 66, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Boedeker, C.C.; Hensen, E.F.; Neumann, H.P.H.; Maier, W.; van Nederveen, F.H.; Suárez, C.; Kunst, H.P.; Rodrigo, J.P.; Takes, R.P.; Pellitteri, P.K.; et al. Genetics of Hereditary Head and Neck Paragangliomas: Hereditary Head and Neck Paragangliomas. Head Neck 2014, 36, 907–916. [Google Scholar] [CrossRef]
- Neumann, H.P.H.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-Line Mutations in Nonsyndromic Pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boedeker, C.C. Paragangliomas and Paraganglioma Syndromes. GMS Curr. Top. Otorhinolaryngol. Head Neck Surg. 2011, 10, Doc03. [Google Scholar]
- Patetsios, P.; Gable, D.R.; Garrett, W.V.; Lamont, J.P.; Kuhn, J.A.; Shutze, W.P.; Kourlis, H.; Grimsley, B.; Pearl, G.J.; Smith, B.L.; et al. Management of Carotid Body Paragangliomas and Review of a 30-Year Experience. Ann. Vasc. Surg. 2002, 16, 331–338. [Google Scholar] [CrossRef]
- Suárez, C.; Rodrigo, J.P.; Ferlito, A.; Cabanillas, R.; Shaha, A.R.; Rinaldo, A. Tumours of Familial Origin in the Head and Neck. Oral Oncol. 2006, 42, 965–978. [Google Scholar] [CrossRef]
- Erickson, D.; Kudva, Y.C.; Ebersold, M.J.; Thompson, G.B.; Grant, C.S.; van Heerden, J.A.; Young, W.F., Jr. Benign Paragangliomas: Clinical Presentation and Treatment Outcomes in 236 Patients. J. Clin. Endocrinol. Metab. 2001, 86, 5210–5216. [Google Scholar] [CrossRef] [PubMed]
- Robertson, V.; Poli, F.; Hobson, B.; Saratzis, A.; Ross Naylor, A. A Systematic Review and Meta-Analysis of the Presentation and Surgical Management of Patients with Carotid Body Tumours. Eur. J. Vasc. Endovasc. Surg. 2019, 57, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamblin, W.R.; ReMine, W.H.; Sheps, S.G.; Harrison, E.G., Jr. Carotid Body Tumor (Chemodectoma). Am. J. Surg. 1971, 122, 732–739. [Google Scholar] [CrossRef]
- Smith, J.D.; Harvey, R.N.; Darr, O.A.; Prince, M.E.; Bradford, C.R.; Wolf, G.T.; Else, T.; Basura, G.J. Head and Neck Paragangliomas: A Two-Decade Institutional Experience and Algorithm for Management. Laryngoscope Investig. Otolaryngol. 2017, 2, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, A.; Myssiorek, D.; Devaney, K.O.; Ferlito, A. Which Paragangliomas of the Head and Neck Have a Higher Rate of Malignancy? Oral Oncol. 2004, 40, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Biller, H.F.; Lawson, W.; Som, P.; Rosenfeld, R. Glomus Vagale Tumors. Ann. Otol. Rhinol. Laryngol. 1989, 98 Pt 1, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Hamersley, E.R.S.; Barrows, A.; Perez, A.; Schroeder, A.; Castle, J.T. Malignant Vagal Paraganglioma. Head Neck Pathol. 2016, 10, 201–205. [Google Scholar] [CrossRef]
- Granato, L.; Próspero, J.D.; Filho, D.M. Nasal Paraganglioma: A Case Report and Literature Review. Int. Arch. Otorhinolaryngol. 2013, 17, 92–95. [Google Scholar]
- Barnes, L. Paraganglioma of the Larynx. A Critical Review of the Literature. ORL J. Otorhinolaryngol. Relat. Spec. 1991, 53, 220–234. [Google Scholar] [CrossRef]
- Connor, S.E.J.; Gleeson, M.J.; Odell, E. Extracranial Glomus Faciale Tumour. J. Laryngol. Otol. 2008, 122, 986–989. [Google Scholar] [CrossRef]
- Langerman, A.; Athavale, S.M.; Rangarajan, S.V.; Sinard, R.J.; Netterville, J.L. Natural History of Cervical Paragangliomas: Outcomes of Observation of 43 Patients. Arch. Otolaryngol. Head Neck Surg. 2012, 138, 341–345. [Google Scholar]
- Von Dobschuetz, E.; Leijon, H.; Schalin-Jäntti, C.; Schiavi, F.; Brauckhoff, M.; Peczkowska, M.; Spiazzi, G.; Demattè, S.; Cecchini, M.E.; Sartorato, P.; et al. A Registry-Based Study of Thyroid Paraganglioma: Histological and Genetic Characteristics. Endocr. Relat. Cancer 2015, 22, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.T.; Braun, J.T.; Pennant, M.; Thompson, L.D.R. Primary Paraganglioma of the Parathyroid: A Case Report and Clinicopathologic Review. Head Neck Pathol. 2010, 4, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harries, K.; Nunn, T.; Shah, V.; Richards, D.; Manson, J.M. First Reported Case of Esophageal Paraganglioma. A Review of the Literature of Gastrointestinal Tract Paraganglioma Including Gangliocytic Paraganglioma. Dis. Esophagus 2004, 17, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Guha, A.; Vicha, A.; Zelinka, T.; Musil, Z.; Chovanec, M. Genetic Variants in Patients with Multiple Head and Neck Paragangliomas: Dilemma in Management. Biomedicines 2021, 9, 626. [Google Scholar] [CrossRef] [PubMed]
- Papaspyrou, K.; Mewes, T.; Rossmann, H.; Fottner, C.; Schneider-Raetzke, B.; Bartsch, O.; Schreckenberger, M.; Lackner, K.J.; Amedee, R.G.; Mann, W.J. Head and Neck Paragangliomas: Report of 175 Patients (1989–2010). Head Neck 2012, 34, 632–637. [Google Scholar] [CrossRef]
- Piccini, V.; Rapizzi, E.; Bacca, A.; Di Trapani, G.; Pulli, R.; Giachè, V.; Zampetti, B.; Lucci-Cordisco, E.; Canu, L.; Corsini, E.; et al. Head and Neck Paragangliomas: Genetic Spectrum and Clinical Variability in 79 Consecutive Patients. Endocr. Relat. Cancer 2012, 19, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Nölting, S.; Ullrich, M.; Pietzsch, J.; Ziegler, C.G.; Eisenhofer, G.; Grossman, A.; Pacak, K. Current Management of Pheochromocytoma/Paraganglioma: A Guide for the Practicing Clinician in the Era of Precision Medicine. Cancers 2019, 11, 1505. [Google Scholar] [CrossRef] [Green Version]
- Crona, J.; Taïeb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef]
- Santhanam, P.; Taïeb, D. Role of (18) F-FDOPA PET/CT Imaging in Endocrinology. Clin. Endocrinol. 2014, 81, 789–798. [Google Scholar] [CrossRef]
- Van Berkel, A.; Vriens, D.; Visser, E.P.; Janssen, M.J.R.; Gotthardt, M.; Hermus, A.R.M.M.; de Geus-Oei, L.-F.; Timmers, H.J.L.M. Metabolic Subtyping of Pheochromocytoma and Paraganglioma by 18F-FDG Pharmacokinetics Using Dynamic PET/CT Scanning. J. Nucl. Med. 2019, 60, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Lenders, J.W.M.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr.; Endocrine Society. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Parameswaran, V.; Burgess, J.R. Clinical Utility of Chromogranin A for the Surveillance of Succinate Dehydrogenase B- and Succinate Dehydrogenase D-Related Paraganglioma. Ann. Clin. Biochem. 2019, 56, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, R.J.; Neumann, H.P.; Parmer, R.J.; Barbosa, J.A.; O’Connor, D.T. Chromogranin A in Familial Pheochromocytoma: Diagnostic Screening Value, Prediction of Tumor Mass, and Post-Resection Kinetics Indicating Two-Compartment Distribution. Am. J. Med. 1990, 88, 607–613. [Google Scholar] [CrossRef]
- Jansen, J.C.; van den Berg, R.; Kuiper, A.; van der Mey, A.G.; Zwinderman, A.H.; Cornelisse, C.J. Estimation of Growth Rate in Patients with Head and Neck Paragangliomas Influences the Treatment Proposal. Cancer 2000, 88, 2811–2816. [Google Scholar] [CrossRef]
- Wieneke, J.A.; Smith, A. Carotid Body Paraganglioma. In Diagnostic Imaging: Head and Neck; Elsevier: Amsterdam, The Netherlands, 2017; pp. 140–143. [Google Scholar]
- Chaloupka, J.C.; Mangla, S.; Huddle, D.C.; Roth, T.C.; Mitra, S.; Ross, D.A.; Sasaki, C.T. Evolving Experience with Direct Puncture Therapeutic Embolization for Adjunctive and Palliative Management of Head and Neck Hypervascular Neoplasms. Laryngoscope 1999, 109, 1864–1872. [Google Scholar] [CrossRef]
- Sanna, M.; Piazza, P.; Shin, S.-H.; Flanagan, S.; Mancini, F. (Eds.) Embolization of paragangliomas. In Microsurgery of Skull Base Paragangliomas, 1st ed.; Thieme Medical: New York, NY, USA, 2013. [Google Scholar]
- Gemmete, J.J.; Chaudhary, N.; Pandey, A.; Gandhi, D.; Sullivan, S.E.; Marentette, L.J.; Chepeha, D.B.; Ansari, S.A. Usefulness of Percutaneously Injected Ethylene-Vinyl Alcohol Copolymer in Conjunction with Standard Endovascular Embolization Techniques for Preoperative Devascularization of Hypervascular Head and Neck Tumors: Technique, Initial Experience, and Correlation with Surgical Observations. AJNR Am. J. Neuroradiol. 2010, 31, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Pedicelli, A.; Lozupone, E.; Valente, I.; Snider, F.; Rigante, M.; D’Argento, F.; Alexandre, A.; Garignano, G.; Chiumarulo, L.; Paludetti, G.; et al. Pre-Operative Direct Puncture Embolization of Head and Neck Hypervascular Tumors Using SQUID 12. Interv. Neuroradiol. 2020, 26, 346–353. [Google Scholar] [CrossRef]
- Suárez, C.; Rodrigo, J.P.; Bödeker, C.C.; Llorente, J.L.; Silver, C.E.; Jansen, J.C.; Takes, R.P.; Strojan, P.; Pellitteri, P.K.; Rinaldo, A.; et al. Jugular and Vagal Paragangliomas: Systematic Study of Management with Surgery and Radiotherapy. Head Neck 2013, 35, 1195–1204. [Google Scholar] [CrossRef]
- Kollert, M.; Minovi, A.; Mangold, R.; Hendus, J.; Draf, W.; Bockmühl, U. Paragangliome des Kopf-Hals-Bereiches—Tumorkontrolle, funktionelle Ergebnisse und Lebensqualität nach Operation. Laryngorhinootologie. 2006, 85, 649–656. [Google Scholar] [CrossRef]
- Hinerman, R.W.; Mendenhall, W.M.; Amdur, R.J.; Stringer, S.P.; Antonelli, P.J.; Cassisi, N.J. Definitive Radiotherapy in the Management of Chemodectomas Arising in the Temporal Bone, Carotid Body, and Glomus Vagale. Head Neck 2001, 23, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Krych, A.J.; Foote, R.L.; Brown, P.D.; Garces, Y.I.; Link, M.J. Long-Term Results of Irradiation for Paraganglioma. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 1063–1066. [Google Scholar] [CrossRef]
- Ibrahim, R.; Ammori, M.B.; Yianni, J.; Grainger, A.; Rowe, J.; Radatz, M. Gamma Knife Radiosurgery for Glomus Jugulare Tumors: A Single-Center Series of 75 Cases. J. Neurosurg. 2017, 126, 1488–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netterville, J.L.; Civantos, F.J. Rehabilitation of Cranial Nerve Deficits after Neurotologic Skull Base Surgery. Laryngoscope 1993, 103, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.; Hague, K.; Kacchara, R.; Jenkins, A.; Das, S.; Catalano, P. Jugular Foramen: Microscopic Anatomic Features and Implications for Neural Preservation with Reference to Glomus Tumors Involving the Temporal Bone. Neurosurgery 2001, 48, 838–847. [Google Scholar]
- Gottfried, O.N.; Liu, J.K.; Couldwell, W.T. Comparison of Radiosurgery and Conventional Surgery for the Treatment of Glomus Jugulare Tumors. Neurosurg. Focus 2004, 17, 22–30. [Google Scholar] [CrossRef]
- Lalwani, A.K.; Jackler, R.K.; Gutin, P.H. Lethal Fibrosarcoma Complicating Radiation Therapy for Benign Glomus Jugulare Tumor. Am. J. Otol. 1993, 14, 398–402. [Google Scholar]
- Preissig, S.H.; Bohmfalk, G.L.; Reichel, G.W.; Smith, M.T. Anaplastic Astrocytoma Following Radiation for a Glomus Jugular Tumor. Cancer 1979, 43, 2243–2247. [Google Scholar] [CrossRef]
- Lekovic, G.P.; Mehta, G.U.; Maxwell, A.K.; Peng, K.A.; Brackmann, D.E. Radiation-Induced Malignant Peripheral Nerve Sheath Tumor of the Vagus Nerve Following Radiation Treatment of Cervical Paraganglioma. J. Neurol. Surg. Rep. 2020, 81, e66–e70. [Google Scholar] [CrossRef]
- Na, A.F.; Lai, L.T.; Kaye, A.H. Radiation Induced Brainstem Glioblastoma in a Patient Treated for Glomus Jugulare Tumour. J. Clin. Neurosci. 2015, 22, 219–221. [Google Scholar] [CrossRef]
- Springate, S.C.; Weichselbaum, R.R. Radiation or Surgery for Chemodectoma of the Temporal Bone: A Review of Local Control and Complications. Head Neck 1990, 12, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Muracciole, X.; Régis, J. Radiosurgery and Carcinogenesis Risk. Prog. Neurol. Surg. 2008, 21, 207–213. [Google Scholar] [PubMed] [Green Version]
- Suárez, C.; Rodrigo, J.P.; Mendenhall, W.M.; Hamoir, M.; Silver, C.E.; Grégoire, V.; Strojan, P.; Neumann, H.P.H.; Obholzer, R.; Offergeld, C.; et al. Carotid Body Paragangliomas: A Systematic Study on Management with Surgery and Radiotherapy. Eur. Arch. Otorhinolaryngol. 2014, 271, 23–34. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Pt. | Age (Years) | Gender | Medical History | F/H of PPGLs | Tumor(s) | Type of Treatment | |||
|---|---|---|---|---|---|---|---|---|---|
| Laterality | Classification | CBPGL | |||||||
| 1 | 47 | M | Asthma | - | R | Shamblin II | Surgery | JPGL | |
| 2 | 62 | M | Hypertension | - | L | Shamblin II | Surgery | TPGL | |
| 3 | 64 | M | Pangastritis | - | R | Shamblin II | Surgery | VPGL | |
| 4 | 76 | F | - | + | R | Shamblin I | W + S | ||
| 5 | 37 | F | Asthma, hypertension, renal angiomyolipoma | - | L | Fisch C3 Di2 | Surgery | ||
| 6 | 37 | F | - | - | L | Fisch C3 | Surgery | ||
| 7 | 43 | F | Hypertension, hydrocephalus | - | L | Fisch C3 Di3 | Surgery | ||
| 8 | 54 | F | Hypertension, diabetes mellitus | - | R | Fisch C3Di1 | Surgery | ||
| 9 | 60 | F | Hypertension | - | L | Fisch C3 De2 | Surgery | ||
| 10 | 64 | M | Diabetes mellitus | - | L | Fisch C3 | Surgery | ||
| 11 | 69 | F | Diabetes mellitus, Leiden mutation | - | R | Fisch C1 | Surgery | ||
| 12 | 80 | F | Hypertension | - | L | Fisch C3 Di3 | W + S | ||
| 13 | 42 | M | Hypertension, diabetes mellitus | - | L | Fisch B3 | Surgery | ||
| 14 | 48 | F | Hypertension | - | L | Fisch A1 | Surgery | ||
| 15 | 48 | F | - | - | R | Fisch B2 | Surgery | ||
| 16 | 55 | F | Hypothyroidism, duodenal ulcer | - | L | Fisch B2 | Surgery | ||
| 17 | 57 | M | - | - | L | Fisch C2 Di1 | W + S | ||
| 18 | 67 | F | - | - | R | Fisch C1 | Surgery | ||
| 19 | 71 | F | - | - | L | Fisch C1 | W + S | ||
| 20 | 39 | F | Asthma | - | L | Fisch A | Surgery | ||
| 21 | 44 | F | Hypothyroidism | - | R | Fisch A | Surgery | ||
| 22 | 46 | M | - | - | R | Fisch A | Surgery | ||
| 23 | 51 | F | - | - | R | Fisch A | Surgery | ||
| 24 | 51 | F | Hypertension, Migraine | - | L | Fisch A | Surgery | ||
| 25 | 34 | M | Tetralogy Of Fallot | - | B | L: Shamblin III, R: Shamblin II | W + S | ||
| 26 | 36 | M | Paranoid schizophrenia | - | B | Shamblin III | Treatment declined Deceased | ||
| R | Fisch C | ||||||||
| R | Fisch C4 Di2 | ||||||||
| 27 | 43 | F | Spontaneous abortion | + | R | Shamblin II | Surgery | ||
| L | Fisch A | W + S | |||||||
| 28 | 47 | M | - | - | L | Fisch B | W + S | ||
| L | Fisch C1 | W + S | |||||||
| 29 | 51 | M | Hypertension | - | B | Shamblin II | Surgery | ||
| B | Fisch C1 | Surgery | |||||||
| L | Fisch A1 | W + S | |||||||
| 30 | 57 | F | Hypertension, Hyperlipoproteinemia | - | R | Fisch A | Surgery | ||
| L | Fisch C1 | W + S | |||||||
| Number of Patients [Total (N = 30)] n (%) | ||
|---|---|---|
| Demographic profile | ||
| Gender | Males | 11 (36.7%) |
| Females | 19 (63.3%) | |
| Age at presentation below 40 years | 5 (16.7%) | |
| Medical history of hypertension | 11 (36.7%) | |
| Patients with negative family history | 28 (93.3%) | |
| Clinical features | ||
| Asymptomatic | 2 (6.7%) | |
| Painless neck mass | 10 (33.3%) | |
| Pulsation in the neck | 1 (3.3%) | |
| Dysphonia/hoarseness of voice | 3 (10%) | |
| Dysphagia | 3 (10%) | |
| Dysarthria | 2 (6.7%) | |
| Facial nerve palsy | 2 (6.7%) | |
| Restrictive tongue movement Hearing difficulty Pulsatile tinnitus Pressure in the ear Otorrhagia | 1 (3.3%) | |
| 16 (53.3%) | ||
| 8 (26.7%) | ||
| 1 (3.3%) | ||
| 1 (3.3%) | ||
| Imaging studies | ||
| Presence of HNPGLs as incidentalomas | 5 (16.7%) | |
| HNPGLs | ||
| Solitary | 24 (80%) | |
| Multiple | 6 (20%) | |
| Presence of PGLs below the neck | 3 (10%) | |
| Presence of non-PGL tumors | 6 (20%) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guha, A.; Chovanec, M. Head and Neck Paragangliomas in the Czech Republic: Management at the Otorhinolaryngology Department. Diagnostics 2022, 12, 28. https://doi.org/10.3390/diagnostics12010028
Guha A, Chovanec M. Head and Neck Paragangliomas in the Czech Republic: Management at the Otorhinolaryngology Department. Diagnostics. 2022; 12(1):28. https://doi.org/10.3390/diagnostics12010028
Chicago/Turabian StyleGuha, Anasuya, and Martin Chovanec. 2022. "Head and Neck Paragangliomas in the Czech Republic: Management at the Otorhinolaryngology Department" Diagnostics 12, no. 1: 28. https://doi.org/10.3390/diagnostics12010028