
Comparison of Genetic Profiling between Primary Tumor and Circulating Tumor Cells Captured by Microfluidics in Epithelial Ovarian Cancer: Tumor Heterogeneity or Allele Dropout?

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Clinical Information
2.2. Tumor Tissue and Germline Control Correction
2.3. CTCs Capture and Recovery
2.4. Whole Genome Amplification (WGA)
2.5. Whole Exome Sequencing
2.6. Exome Variation Analysis
3. Results
3.1. CTCs Capture and Recovery
3.2. Capture Efficiency and Uniformity of WES
3.3. Germline Variations in PBMCs
3.4. Somatic Mutations in Tumors
3.5. Somatic Mutations in CTCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kroeger, P.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, S.; Zhang, K.; Qing, Y.; Xu, T. A comprehensive overview of exosomes in ovarian cancer: Emerging biomarkers and therapeutic strategies. J. Ovarian Res. 2017, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Kamps, R.; Brandão, R.D.; Van Den Bosch, B.J.; Aimee, D.C.; Paulussen, A.D.C.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-generation sequencing in oncology: Genetic diagnosis, risk prediction and cancer classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019, 110, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Calabuig-Fariñas, S.; Jantus-Lewintre, E.; Herreros-Pomares, A.; Camps, C. Circulating tumor cells versus circulating tumor DNA in lung cancer-which one will win? Transl. Lung Cancer Res. 2016, 5, 466–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Balaj, L.; Stott, S.L.; Nahed, B.; Carter, B.S. Liquid biopsy for brain tumors. Expert Rev. Mol. Diagn. 2017, 17, 943–947. [Google Scholar] [CrossRef]
- Huang, H.M.; Li, H.X. Tumor heterogeneity and the potential role of liquid biopsy in bladder cancer. Cancer Commun. 2021, 41, 91–108. [Google Scholar] [CrossRef]
- Giannopoulou, L.; Kasimir-Bauer, S.; Lianidou, E.S. Liquid Biopsy in Ovarian Cancer: Recent Advances on Circulating Tumor Cells and Circulating Tumor DNA. Clin. Chem. Lab. Med. 2018, 56, 186–197. [Google Scholar] [CrossRef]
- Asante, D.B.; Calapre, L.; Ziman, M.; Meniawy, T.M.; Gray, E.S. Liquid biopsy in ovarian cancer using circulating tumor DNA and cells: Ready for prime time? Cancer Lett. 2020, 468, 59–71. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Ilié, M.; Szafer-Glusman, E.; Hofman, V.; Chamorey, E.; Lalvée, S.; Selva, E.; Leroy, S.; Marquette, C.H.; Kowanetz, M.; Hedge, P.; et al. Detection of PD-L1 in circulating tumor cells and white blood cells from patients with advanced non-small-cell lung cancer. Ann. Oncol. 2018, 29, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Konczalla, L.; Wöstemeier, A.; Kemper, M.; Karstens, K.F.; Izbicki, J.; Reeh, M. Clinical significance of circulating tumor cells in gastrointestinal carcinomas. Diagnostics 2020, 10, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsköld, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Kularatne, S.A.; Kalli, K.R.; Prendergast, F.G.; Amato, R.J.; Klee, G.G.; Hartmann, L.C.; Low, P.S. Quantitation of circulating tumor cells in blood samples from ovarian and prostate cancer patients using tumor-specific fluorescent ligands. Int. J. Cancer 2008, 123, 1968–1973. [Google Scholar] [CrossRef] [Green Version]
- Kolostova, K.; Spicka, J.; Matkowski, R.; Bobek, V. Isolation, primary culture, morphological and molecular characterization of circulating tumor cells in gynecological cancers. Am. J. Trans. Res. 2015, 7, 1203–1213. [Google Scholar]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef]
- Fernández-Lázaro, D.; Hernández, J.; García, A.C.; Castillo, A.; Hueso, M.V.; Cruz-Hernández, J.J. Clinical Perspective and Translational Oncology of Liquid Biopsy. Diagnostics 2020, 10, 443. [Google Scholar] [CrossRef]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Torga, G.; Pienta, K.J. Patient-Paired Sample Congruence between 2 Commercial Liquid Biopsy Tests. JAMA Oncol. 2018, 4, 868–870. [Google Scholar] [CrossRef]
- Lyu, M.; Zhou, J.; Ning, K.; Ying, B. The diagnostic value of circulating tumor cells and ctDNA for gene mutations in lung cancer. Onco. Targets Ther. 2019, 12, 2539–2552. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Yamada, T.; Takahashi, G.; Iwai, T.; Ueda, K.; Kuriyama, S.; Koizumi, M.; Matsuda, A.; Shinji, S.; Ohta, R.; et al. Analysis of colorectal cancer-related mutations by liquid biopsy: Utility of circulating cell-free DNA and circulating tumor cells. Cancer Sci. 2019, 110, 3497–3509. [Google Scholar] [CrossRef]
- Huang, C.E.; Ma, G.C.; Jou, H.J.; Lin, W.H.; Lee, D.J.; Lin, Y.S.; Ginsberg, N.; Chen, H.F.; Chang, M.C.; Chen, M. Noninvasive prenatal diagnosis of fetal aneuploidy by circulating fetal nucleated red blood cells and extravillous trophoblasts using silicon-based nanostructured microfluidics. Mol. Cytogenet. 2017, 10, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.C.; Lin, W.H.; Huang, C.E.; Chang, T.Y.; Liu, J.Y.; Yang, Y.J.; Lee, M.H.; Wu, W.J.; Chang, Y.S.; Chen, M. A Silicon-based coral-like nanostructured microfluidics to isolate rare cells in human circulation: Validation by SK-BR-3 cancer cell line and its utility in circulating fetal nucleated red blood cells. Micromachines 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jou, H.J.; Chou, L.Y.; Chang, W.C.; Ho, H.C.; Zhang, W.T.; Ling, P.Y.; Tsai, K.H.; Chen, T.H.; Chen, S.H.; Lo, P.H.; et al. A novel automatic platform based on nanostructured microfluidic chip for isolating and identification of circulating tumor cells. Micromachines 2021, 12, 473. [Google Scholar] [CrossRef]
- Po, J.W.; Roohullah, A.; Lynch, D.; DeFazio, A.; Harrison, M.; Harnett, P.R.; Kennedy, C.; de Souza, P.; Becker, T.M. Improved ovarian cancer EMT-CTC isolation by immunomagnetic targeting of epithelial EpCAM and mesenchymal N-cadherin. J. Circ. Biomark. 2018, 7, 1849454418782617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szulwach, K.E.; Chen, P.; Wang, X.; Wang, J.; Weaver, L.S.; Gonzales, M.L.; Sun, G.; Unger, M.A.; Ramakrishnan, R. Single-cell genetic analysis using automated microfluidics to resolve somatic mosaicism. PLoS ONE 2015, 10, e0135007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and analytical validation of foundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE 2020, 15, e0237802. [Google Scholar] [CrossRef]
- Chen, H.F.; Chang, S.P.; Wu, S.H.; Lin, W.H.; Lee, Y.C.; Ni, Y.H.; Chen, C.A.; Ma, G.C.; Ginsberg, N.A.; You, E.M.; et al. Validating a rapid, real-time, PCR-based direct mutation detection assay for preimplantation genetic diagnosis. Gene 2014, 548, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Chang, S.P.; Chen, H.F.; Ma, G.C.; Lin, W.H.; Lin, C.F.; Tsai, F.P.; Wu, C.H.; Tsai, H.D.; Lee, T.H.; et al. Preimplantation genetic screening of blastocysts by multiplex qPCR followed by fresh embryo transfer: Validation and verification. Mol. Cytogenet. 2015, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.C.; Chen, H.F.; Yang, Y.S.; Lin, W.H.; Tsai, F.P.; Lin, C.F.; Chiu, C.; Chen, M. A pilot proof-of-principle study to compare fresh and vitrified cycle preimplantation genetic screening by chromosome microarray and next generation sequencing. Mol. Cytogenet. 2016, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.F.; Chen, S.U.; Ma, G.C.; Hsieh, S.T.; Tsai, H.D.; Yang, Y.S.; Chen, M. Preimplantation genetic diagnosis and screening: Current status and future challenges. J. Formos. Med. Assoc. 2018, 117, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.H.; Chang, M.Y.; Ma, G.C.; Chang, S.P.; Lin, C.F.; Lin, W.H.; Chen, H.F.; Chen, S.U.; Lee, Y.C.; Chao, C.C.; et al. Preimplantation genetic diagnosis of neurodegenerative diseases: Review of methodologies and report of our experience as a regional reference laboratory. Diagnostics 2019, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Chang, C.J.; Guan, Y.; Szafer-Glusman, E.; Punnoose, E.; Do, A.; Suttmann, B.; Gagnon, R.; Rodriguez, A.; Landers, M.; et al. Genomic analysis of circulating tumor cells at the single-cell level. J. Mol. Diagn. 2020, 22, 770–781. [Google Scholar] [CrossRef]
- Unrau, P.; Deugau, K.V. Non-cloning amplification of specific DNA fragments from whole genomic DNA digests using DNA ‘Indexers’. Gene 1994, 145, 163–169. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
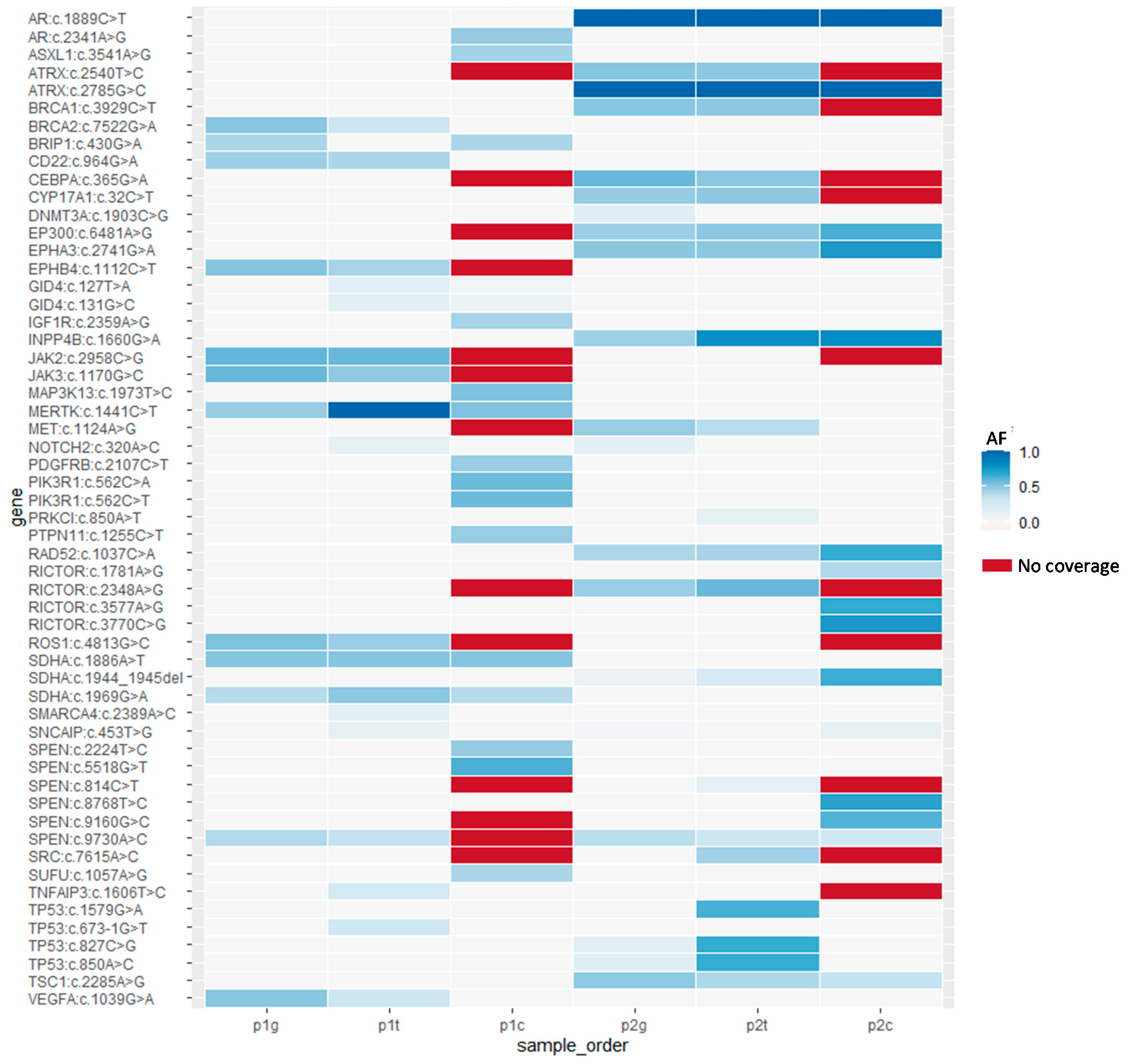
| Gene | Variation | Patient | PBMCs | Tumor | CTCs |
|---|---|---|---|---|---|
| Category Ig | |||||
| AR | c.1889C>T | p2 | Homo | Homo | Homo |
| ATRX | c.2785G>C | p2 | Homo | Homo | Homo |
| CD22 | c.964G>A | p1 | Het | Het | Het |
| EP300 | c.6481A>G | p2 | Het | Het | Het |
| EPHA3 | c.2741G>A | p2 | Het | Het | Het |
| INPP4B | c.1660G>A | p2 | Het | Het | Het |
| RAD52 | c.1037C>A | p2 | Het | Het | Het |
| SDHA | c.1886A>T | p1 | Het | Het | Het |
| SDHA | c.1969G>A | p1 | Het | Het | Het |
| SDHA | c.1944_1945del | p2 | Het | Het | Het |
| SPEN | c.9730A>C | p1 and p2 | Het | Het | Het |
| TSC1 | c.2285A>G | p2 | Het | Het | Het |
| Category IIg | |||||
| ATRX | c.2540T>C | p2 | Het | Het | NC |
| BRCA1 | c.3929C>T | p2 | Het | Het | NC |
| CEBPA | c.365G>A | p2 | Het | Het | NC |
| CYP17A1 | c.32C>T | p2 | Het | Het | NC |
| EPHB4 | c.1112C>T | p1 | Het | Het | NC |
| JAK2 | c.2958C>G | p1 | Het | Het | NC |
| JAK3 | c.1170G>C | p1 | Het | Het | NC |
| RICTOR | c.2348A>G | p2 | Het | Het | NC |
| ROS1 | c.4813G>C | p1 | Het | Het | NC |
| Category IIIg | |||||
| BRCA2 | c.7522G>A | p1 | Het | Het | Wt |
| MET | c.1124A>G | p2 | Het | Het | Wt |
| TP53 | c.850A>C | p2 | Het | Het | Wt |
| VEGFA | c.1039G>A | p1 | Het | Het | Wt |
| Category IVg | |||||
| BRIP1 | c.430G>A | p1 | Het | Wt | Het |
| DNMT3A | c.1903C>G | p2 | Het | Wt | Wt |
| MERTK | c.1441C>T | p1 | Het | Homo | Het |
| Gene | Variation | Patient | Tumor AF | CTCs AF | Pathway |
|---|---|---|---|---|---|
| Category It | |||||
| GID4 | c.127T>A | p1 | 0.16 | 0.06 | NA |
| GID4 | c.131G>C | p1 | 0.15 | 0.086 | |
| Category IIt | |||||
| NOTCH2 | c.320A>C | p1 | 0.13 | Wt | Thyroid hormone signaling |
| PRKCI | c.850A>T | p2 | 0.13 | Wt | Tight junction |
| SMARCA4 | c.2389A>C | p1 | 0.15 | Wt | NA |
| SNCAIP | c.453T>G | p1 | 0.12 | Wt | NA |
| TNFAIP3 | c.1606T>C | p1 | 0.26 | Wt | NA |
| TP53 | c.673-1G>T | p1 | 0.3 | Wt | Thyroid hormone signaling |
| TP53 | c.827C>G | p2 | 0.66 | Wt | |
| TP53 | c.1579G>A | p2 | 0.64 | Wt | |
| Category IIIt | |||||
| SPEN | c.814C>T | p2 | 0.14 | NA | NA |
| SRC | c.7615A>C | p2 | 0.45 | NA |
|
| Gene | Variation | Patient | Tumor | CTCs AF | Pathway |
|---|---|---|---|---|---|
| AR | c.2341A>G | p1 | Wt | 0.48 | Pathways in cancer |
| ASXL1 | c.3541A>G | p1 | Wt | 0.44 | NA |
| IGF1R | c.2359A>G | p1 | Wt | 0.43 |
|
| MAP3K13 | c.1973T>C | p1 | Wt | 0.53 | NA |
| PDGFRB | c.2107C>T | p1 | Wt | 0.47 |
|
| PIK3R1 | c.562C>A | p1 | Wt | 0.45 |
|
| c.562C>T | p1 | Wt | 0.08 | ||
| PTPN11 | c.1255C>T | p1 | Wt | 0.47 |
|
| RICTOR | c.3770C>G | p1 | Wt | 0.75 | mTOR pathway |
| c.3577A>G | p2 | Wt | 0.67 | ||
| c.1781A>G | p2 | Wt | 0.41 | ||
| SNCAIP | c.453T>G | p2 | Wt | 0.12 | NA |
| SPEN | c.5518G>T | p1 | Wt | 0.63 | NA |
| c.2224T>C | p1 | Wt | 0.48 | ||
| c.8768T>C | p2 | Wt | 0.71 | ||
| c.9160G>C | p2 | Wt | 0.62 | ||
| SUFU | c.1057A>G | p1 | Wt | 0.42 | Pathways in cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, T.-Y.; Chen, S.-W.; Lin, W.-H.; Huang, C.-E.; Evans, M.I.; Chung, I.-F.; Wu, J.-W.; Ma, G.-C.; Chen, M. Comparison of Genetic Profiling between Primary Tumor and Circulating Tumor Cells Captured by Microfluidics in Epithelial Ovarian Cancer: Tumor Heterogeneity or Allele Dropout? Diagnostics 2021, 11, 1102. https://doi.org/10.3390/diagnostics11061102
Chang T-Y, Chen S-W, Lin W-H, Huang C-E, Evans MI, Chung I-F, Wu J-W, Ma G-C, Chen M. Comparison of Genetic Profiling between Primary Tumor and Circulating Tumor Cells Captured by Microfluidics in Epithelial Ovarian Cancer: Tumor Heterogeneity or Allele Dropout? Diagnostics. 2021; 11(6):1102. https://doi.org/10.3390/diagnostics11061102
Chicago/Turabian StyleChang, Ting-Yu, Sheng-Wen Chen, Wen-Hsiang Lin, Chung-Er Huang, Mark I. Evans, I-Fang Chung, Janne-Wha Wu, Gwo-Chin Ma, and Ming Chen. 2021. "Comparison of Genetic Profiling between Primary Tumor and Circulating Tumor Cells Captured by Microfluidics in Epithelial Ovarian Cancer: Tumor Heterogeneity or Allele Dropout?" Diagnostics 11, no. 6: 1102. https://doi.org/10.3390/diagnostics11061102