
Copy Number Variant Detection with Low-Coverage Whole-Genome Sequencing Represents a Viable Alternative to the Conventional Array-CGH

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Theoretical Minimal Read Count Estimation
2.3. Variant Identification
- Mapping and binning
- Mapping reads using Bowtie 2 [20];
- Binning reads into same-size 20 kb bins;
- Normalizing bin counts.
- Normalization (similar to the one published previously by [21])
- GC bias correction by LOESS smoothing method [22];
- Principal component analysis (PCA) normalization to remove higher-order population artifacts on autosomal chromosomes;
- Subtracting per-bin mean bin count to obtain data normalized around zero.
- Filtration of unusable bins
- Unmappable or poorly mappable regions (zero or low mean of bin count);
- Repetitive regions or areas with certain systematically increased mappability (high mean of bin count);
- Highly variable regions (high variance of bin count).
- Segment identification and reporting
2.3.1. Mapping and Binning
2.3.2. Normalization
2.3.3. Filtration of Unusable Bins
2.3.4. Segment Identification and Reporting
2.4. In-Silico Analysis
3. Results
3.1. Theoretical Minimal Read Count
3.2. Detection Accuracy for Variable CNV Lengths and Read Count (In Silico)
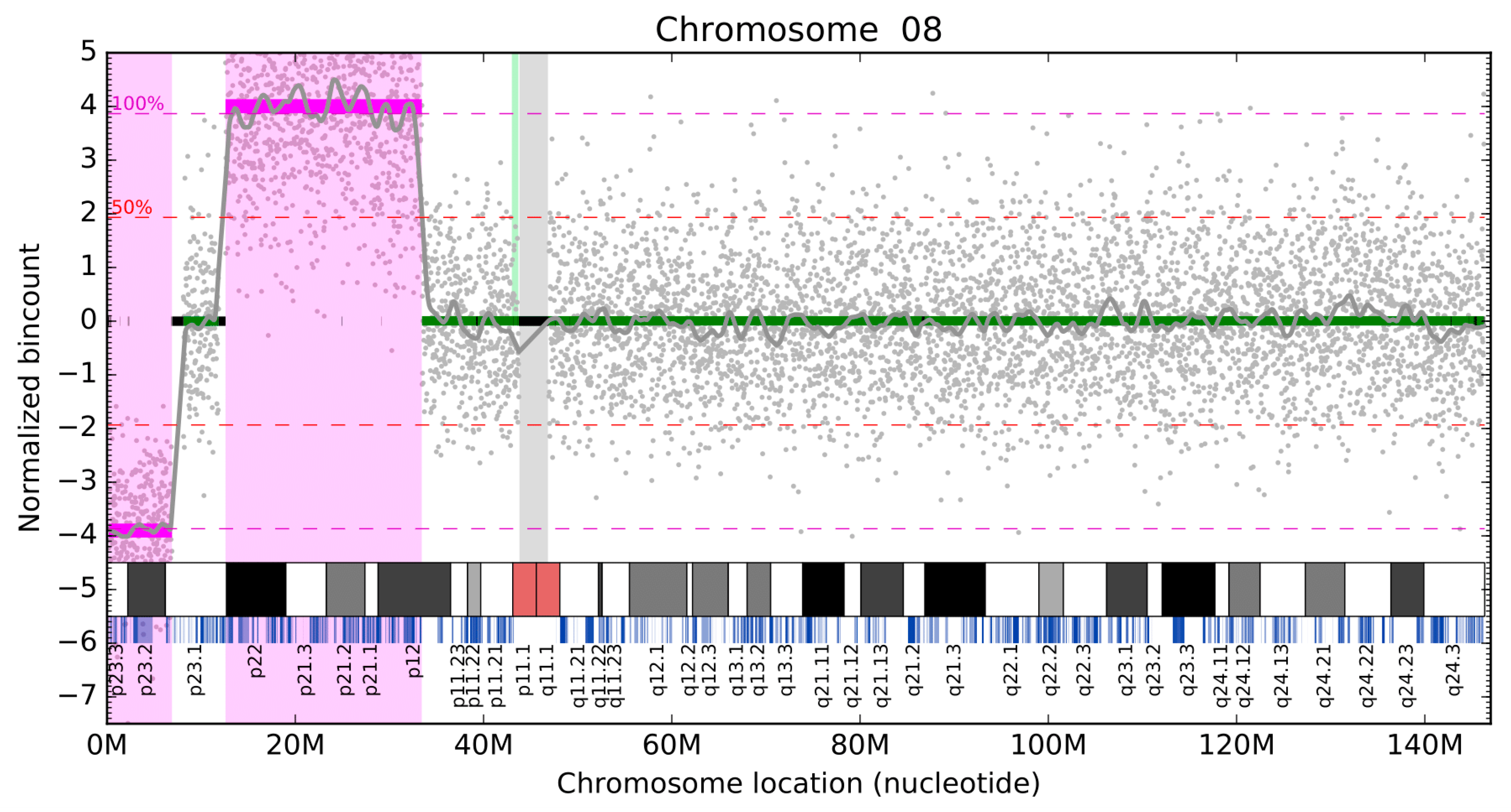
3.3. Validation of Clinical Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aCGH | array-based comparative genomic hybridization |
| CBS | circular binary segmentation |
| CNV | copy number variant |
| NGS | next-generation sequencing |
| NIPT | non-invasive prenatal testing |
| WGS | whole-genome sequencing |
References
- Pös, O.; Budis, J.; Kubiritova, Z.; Kucharik, M.; Duris, F.; Radvanszky, J.; Szemes, T. Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing. Int. J. Mol. Sci. 2019, 20, 4403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.; Xuan, Z.; Makarov, V.; Ye, K.; Sebat, J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009, 19, 1586–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartha, Á.; Győrffy, B. Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology. Cancers 2019, 11, 1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, C.D.; Di Giacomo, G.; Cignini, P.; Padula, F.; Mangiafico, L.; Mesoraca, A.; D’Emidio, L.; McCluskey, M.R.; Paganelli, A.; Giorlandino, C. Comparative study of aCGH and Next Generation Sequencing (NGS) for chromosomal microdeletion and microduplication screening. J. Prenat Med. 2014, 8, 57–69. [Google Scholar]
- Wang, H.; Nettleton, D.; Ying, K. Copy number variation detection using next generation sequencing read counts. BMC Bioinform. 2014, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Fromer, M.; Moran, J.L.; Chambert, K.; Banks, E.; Bergen, S.E.; Ruderfer, D.M.; Handsaker, R.E.; McCarroll, S.A.; O’Donovan, M.C.; Owen, M.J.; et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012, 91, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E.; Project, N.E.S. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Oldridge, D.A.; Diskin, S.J.; Zhang, N.R. CODEX: A normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 2015, 43, e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, J.S.; Maxwell, E.K.; O’Dushlaine, C.; Lopez, A.E.; Dewey, F.E.; Chernomorsky, R.; Baras, A.; Overton, J.D.; Habegger, L.; Reid, J.G. CLAMMS: A scalable algorithm for calling common and rare copy number variants from exome sequencing data. Bioinformatics 2016, 32, 133–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, A.; Mahamdallie, S.; Ruark, E.; Seal, S.; Ramsay, E.; Clarke, M.; Uddin, I.; Wylie, H.; Strydom, A.; Lunter, G.; et al. Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 2016, 1, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, L.F.; van Dijk, F.; de Boer, E.N.; van Dijk-Bos, K.K.; Jongbloed, J.D.H.; van der Hout, A.H.; Westers, H.; Sinke, R.J.; Swertz, M.A.; Sijimons, R.H.; et al. CoNVaDING: Single Exon Variation Detection in Targeted NGS Data. Hum. Mutat. 2016, 37, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Murrell, J.R.; Luo, M.; Conlin, L.K. A highly sensitive and specific workflow for detecting rare copy-number variants from exome sequencing data. Genome Med. 2020, 12, 14. [Google Scholar] [CrossRef] [Green Version]
- Raman, L.; Dheedene, A.; De Smet, M.; Van Dorpe, J.; Menten, B. WisecondorX: Improved copy number detection for routine shallow whole-genome sequencing. Nucleic Acids Res. 2019, 47, 1605–1614. [Google Scholar] [CrossRef]
- Straver, R.; Sistermans, E.A.; Holstege, H.; Visser, A.; Oudejans, C.B.M.; Reinders, M.J.T. WISECONDOR: Detection of fetal aberrations from shallow sequencing maternal plasma based on a within-sample comparison scheme. Nucleic Acids Res. 2014, 42, e31. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharanipragada, P.; Vogeti, S.; Parekh, N. iCopyDAV: Integrated platform for copy number variations—Detection, annotation and visualization. PLoS ONE 2018, 13, e0195334. [Google Scholar] [CrossRef] [Green Version]
- Hyblova, M.; Harsanyova, M.; Nikulenkov-Grochova, D.; Kadlecova, J.; Kucharik, M.; Budis, J.; Minarik, G. Validation of Copy Number Variants Detection from Pregnant Plasma Using Low-Pass Whole-Genome Sequencing in Noninvasive Prenatal Testing-Like Settings. Diagnostics 2020, 10, 569. [Google Scholar] [CrossRef]
- Kucharik, M.; Gnip, A.; Hyblova, M.; Budis, J.; Strieskova, L.; Harsanyova, M.; Pös, O.; Kubiritova, Z.; Radvanszky, J.; Minarik, G.; et al. Non-invasive prenatal testing (NIPT) by low coverage genomic sequencing: Detection limits of screened chromosomal microdeletions. PLoS ONE 2020, 15, e0238245. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Tynan, J.; Ehrich, M.; Hannum, G.; McCullough, R.; Saldivar, J.-S.; Oeth, P.; Boom, D.V.D.; Deciu, C. Detection of fetal subchromosomal abnormalities by sequencing circulating cell-free DNA from maternal plasma. Clin. Chem. 2015, 61, 608–616. [Google Scholar] [CrossRef]
- Alkan, C.; Kidd, J.M.; Marques-Bonet, T.; Aksay, G.; Antonacci, F.; Hormozdiari, F.; Kitzman, J.O.; Baker, C.; Malig, M.; Mutlu, O.; et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009, 41, 1061–1067. [Google Scholar] [CrossRef]
- Seshan, V.E.; Olshen, A. DNAcopy: DNA Copy Number Data Analysis. R Package Version 1.36.0. 2013. Available online: https://www.researchgate.net/publication/241191458_DNAcopy_A_Package_for_analyzing_DNA_copy_data (accessed on 14 April 2021).
- Minarik, G.; Repiska, G.; Hyblova, M.; Nagyova, E.; Soltys, K.; Budis, J.; Ďuriš, F.; Sysak, R.; Bujalkova, M.G.; Vlkova-Izrael, B.; et al. Utilization of Benchtop Next Generation Sequencing Platforms Ion Torrent PGM and MiSeq in Noninvasive Prenatal Testing for Chromosome 21 Trisomy and Testing of Impact of In Silico and Physical Size Selection on Its Analytical Performance. PLoS ONE 2015, 10, e0144811. [Google Scholar] [CrossRef]
- Sekelska, M.; Izsakova, A.; Kubosova, K.; Tilandyova, P.; Csekes, E.; Kuchova, Z.; Hyblova, M.; Harsanyova, M.; Kucharik, M.; Budis, J.; et al. Result of Prospective Validation of the Trisomy Test for the Detection of Chromosomal Trisomies. Diagnostics 2019, 9, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrananda, D.; Thorne, N.P.; Ganesamoorthy, D.; Bruno, D.L.; Benjamini, Y.; Speed, T.P.; Slater, H.R.; Bahlo, M. Investigating and correcting plasma DNA sequencing coverage bias to enhance aneuploidy discovery. PLoS ONE 2014, 9, e86993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazdarica, J.; Budis, J.; Duris, F.; Turna, J.; Szemes, T. Adaptable Model Parameters in Non-Invasive Prenatal Testing Lead to More Stable Predictions. Int. J. Mol. Sci. 2019, 20, 3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agilent Technologies, Inc. Human Genome Cgh Microarray Kit, 4 × 44 k. Available online: https://www.agilent.com/en/product/cgh-cgh-snp-microarray-platform/cgh-cgh-snp-microarrays/human-microarrays/human-genome-cgh-microarray-kit-4x44k-228410 (accessed on 5 August 2020).




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kucharík, M.; Budiš, J.; Hýblová, M.; Minárik, G.; Szemes, T. Copy Number Variant Detection with Low-Coverage Whole-Genome Sequencing Represents a Viable Alternative to the Conventional Array-CGH. Diagnostics 2021, 11, 708. https://doi.org/10.3390/diagnostics11040708
Kucharík M, Budiš J, Hýblová M, Minárik G, Szemes T. Copy Number Variant Detection with Low-Coverage Whole-Genome Sequencing Represents a Viable Alternative to the Conventional Array-CGH. Diagnostics. 2021; 11(4):708. https://doi.org/10.3390/diagnostics11040708
Chicago/Turabian StyleKucharík, Marcel, Jaroslav Budiš, Michaela Hýblová, Gabriel Minárik, and Tomáš Szemes. 2021. "Copy Number Variant Detection with Low-Coverage Whole-Genome Sequencing Represents a Viable Alternative to the Conventional Array-CGH" Diagnostics 11, no. 4: 708. https://doi.org/10.3390/diagnostics11040708