
Distinct Somatic Alteration Features Identified by Gene Panel Sequencing in Korean Triple-Negative Breast Cancer with High Ki67 Expression


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Specimens
2.2. DNA Extraction and Purification
2.3. Library Preparation for DNA Panel
2.4. Sequencing Analysis Using the Ion S5XL
2.5. Sanger Sequencing
2.6. Bioinformatic Analysis
2.7. Statistical Analysis
3. Results
3.1. Clinicopathological Features of Studied Patients
3.2. Quality Control Metrics of Raw Sequencing Data
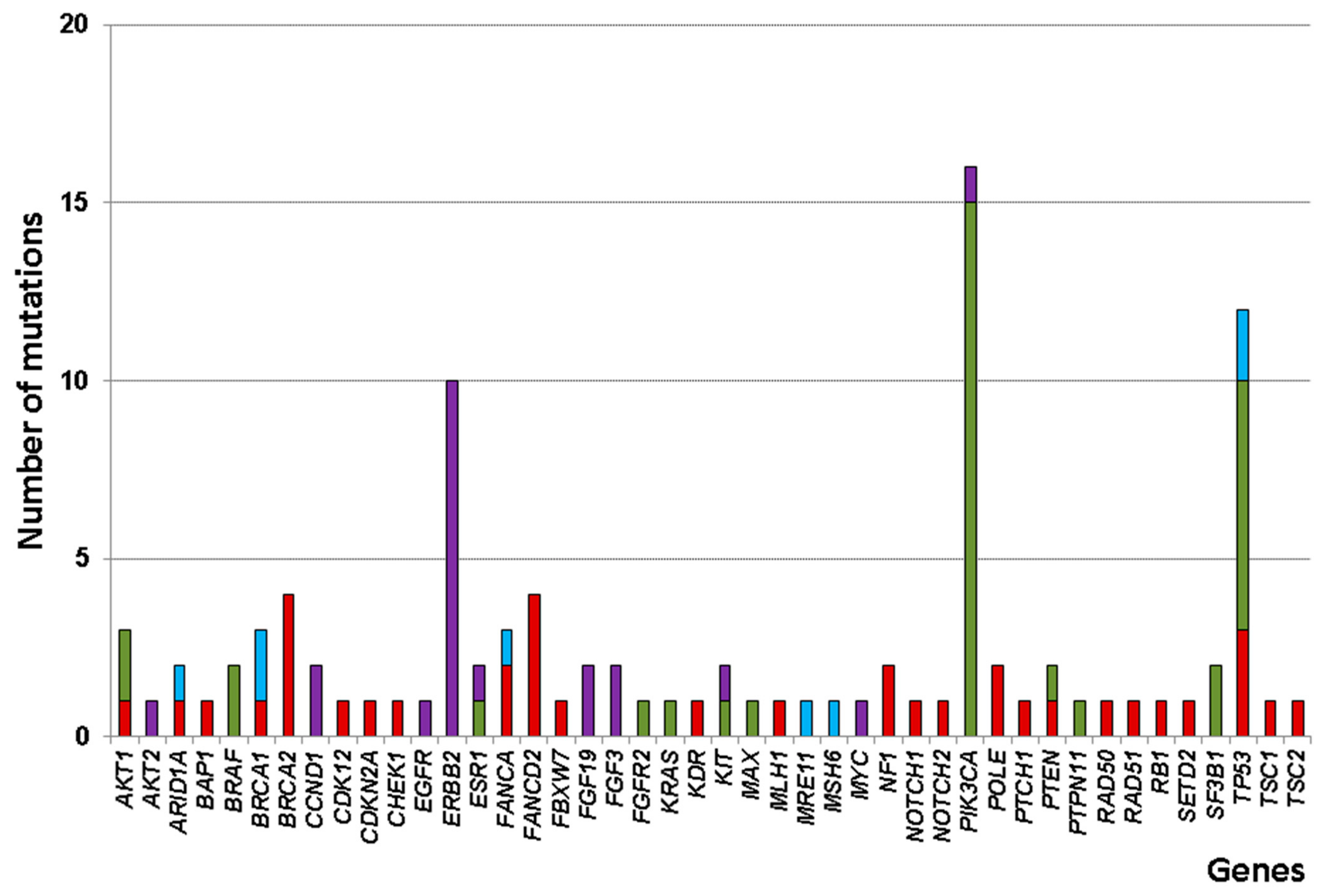
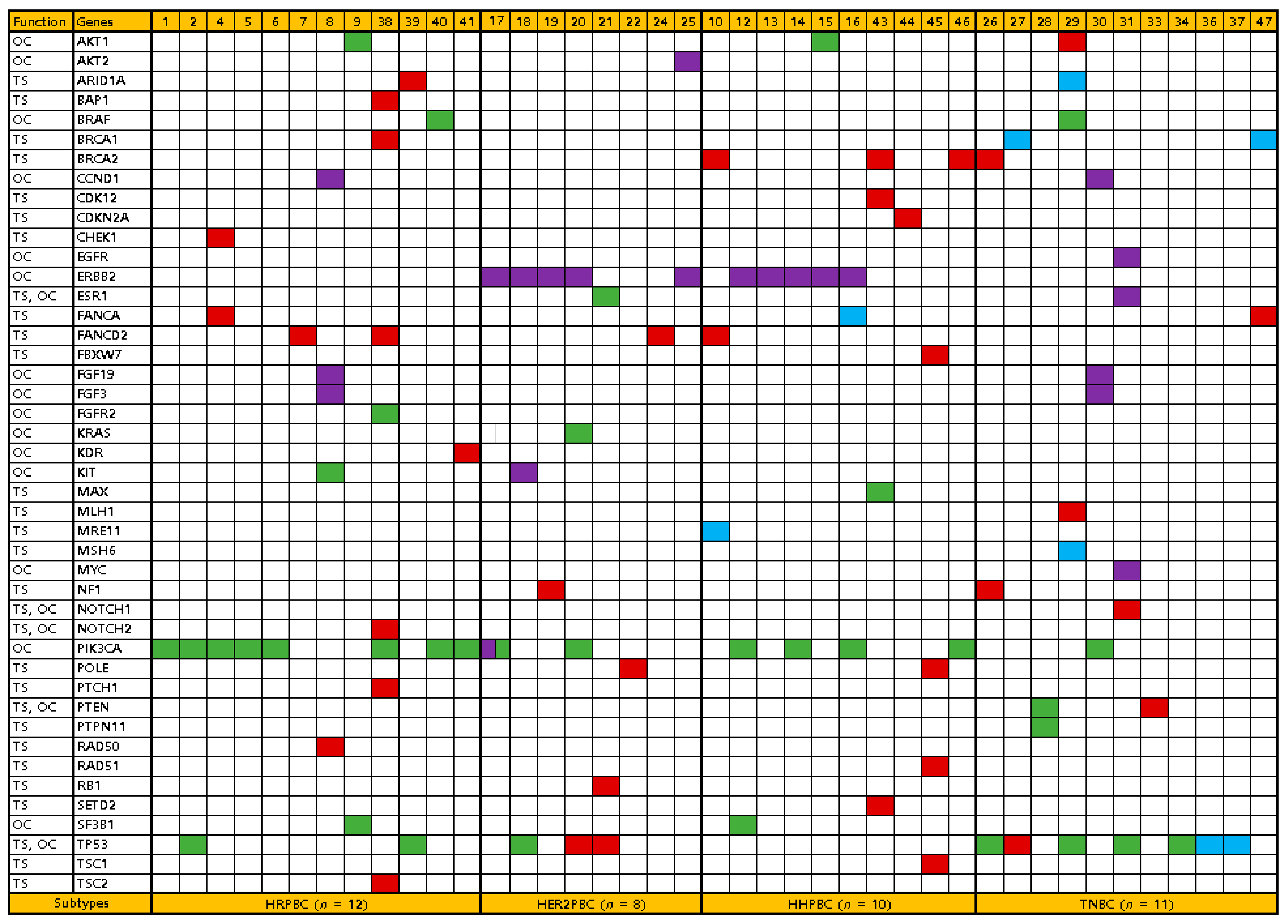
3.3. Somatic Alteration Profiles
3.4. Comparison of Somatic Alteration Profiles between Breast Cancer Subtypes
3.5. Ki-67 Expression and PIK3CA/ TP53 Mutation Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.Y.; Kim, Y.S.; Kim, Z.; Kim, H.Y.; Kim, H.J.; Park, S.; Bae, S.Y.; Yoon, K.H.; Lee, S.B.; Lee, S.K.; et al. Breast Cancer Statistics in Korea in 2017: Data from a Breast Cancer Registry. J. Breast Cancer 2020, 23, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Loibl, S.; Treue, D.; Budczies, J.; Weber, K.; Stenzinger, A.; Schmitt, W.D.; Weichert, W.; Jank, P.; Furlanetto, J.; Klauschen, F.; et al. Mutational Diversity and Therapy Response in Breast Cancer: A Sequencing Analysis in the Neoadjuvant GeparSepto Trial. Clin. Cancer Res. 2019, 25, 3986–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Golubnitschaja, O.; Filep, N.; Yeghiazaryan, K.; Blom, H.J.; Hofmann-Apitius, M.; Kuhn, W. Multi-omic approach decodes paradoxes of the triple-negative breast cancer: Lessons for predictive, preventive and personalised medicine. Amino Acids 2018, 50, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Reis-Filho, J.S. Basal-like breast cancer and the BRCA1 phenotype. Oncogene 2006, 25, 5846–5853. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Geenen, J.J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H.M. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin. Pharmacokinet. 2018, 57, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, C.I. Racial disparities in breast cancer diagnosis and treatment by hormone receptor and HER2 status. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1666–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niyomnaitham, S.; Parinyanitikul, N.; Roothumnong, E.; Jinda, W.; Samarnthai, N.; Atikankul, T.; Suktitipat, B.; Thongnoppakhun, W.; Limwongse, C.; Pithukpakorn, M. Tumor mutational profile of triple negative breast cancer patients in Thailand revealed distinctive genetic alteration in chromatin remodeling gene. PeerJ 2019, 7, e6501. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, L.; Huang, B.; Wang, Y.; Ji, L.; Wu, J.; Di, G.; Liu, G.; Yu, K.; Shao, Z.; et al. The prognostic and predictive potential of Ki-67 in triple-negative breast cancer. Sci. Rep. 2020, 10, 225. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Reddy, N.G.; Barkoh, B.A.; Handal, B.; Kanagal-Shamanna, R.; Greaves, W.O.; Medeiros, L.J.; Aldape, K.D.; et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J. Mol. Diagn. 2013, 15, 607–622. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Aldape, K.; Lu, X.; Manekia, J.; Abraham, R.; Reddy, N.G.; Barkoh, B.A.; Veliyathu, J.; et al. Clinical massively parallel next-generation sequencing analysis of 409 cancer-related genes for mutations and copy number variations in solid tumours. Br. J. Cancer 2014, 111, 2014–2023. [Google Scholar] [CrossRef] [Green Version]
- Siddharth, S.; Sharma, D. Racial Disparity and Triple-Negative Breast Cancer in African-American Women: A Multifaceted Affair between Obesity, Biology, and Socioeconomic Determinants. Cancers 2018, 10, 514. [Google Scholar] [CrossRef] [Green Version]
- Ademuyiwa, F.O.; Tao, Y.; Luo, J.; Weilbaecher, K.; Ma, C.X. Differences in the mutational landscape of triple-negative breast cancer in African Americans and Caucasians. Breast Cancer Res. Treat. 2017, 161, 491–499. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yao, S.; Zhao, H.; Hu, Q.; Kwan, M.L.; Roh, J.M.; Ambrosone, C.B.; Kushi, L.H.; Liu, S.; Zhu, Q. Early-onset triple-negative breast cancer in multiracial/ethnic populations: Distinct trends of prevalence of truncation mutations. Cancer Med. 2019, 8, 1845–1853. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Lévy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef] [PubMed]
- Uscanga-Perales, G.I.; Santuario-Facio, S.K.; Sanchez-Dominguez, C.N.; Cardona-Huerta, S.; Muñoz-Maldonado, G.E.; Ruiz-Flores, P.; Barcenas-Walls, J.R.; Osuna-Rosales, L.E.; Rojas-Martinez, A.; Gonzalez-Guerrero, J.F.; et al. Genetic alterations of triple negative breast cancer (TNBC) in women from Northeastern Mexico. Oncol. Lett. 2019, 17, 3581–3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.T.; Lenkiewicz, E.; Malasi, S.; Basu, A.; Yearley, J.H.; Annamalai, L.; McCullough, A.E.; Kosiorek, H.E.; Narang, P.; Wilson Sayres, M.A.; et al. The association of genomic lesions and PD-1/PD-L1 expression in resected triple-negative breast cancers. Breast Cancer Res. 2018, 20, 71. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Jin, J.; Ji, W.; Guan, X. Therapeutic landscape in mutational triple negative breast cancer. Mol. Cancer 2018, 17, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayor, S. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Von Wahlde, M.K.; Timms, K.M.; Chagpar, A.; Wali, V.B.; Jiang, T.; Bossuyt, V.; Saglam, O.; Reid, J.; Gutin, A.; Neff, C.; et al. Intratumor Heterogeneity of Homologous Recombination Deficiency in Primary Breast Cancer. Clin. Cancer Res. 2017, 23, 1193–1199. [Google Scholar] [CrossRef] [Green Version]
- Atchley, D.P.; Albarracin, C.T.; Lopez, A.; Valero, V.; Amos, C.I.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Arun, B.K. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J. Clin. Oncol. 2008, 26, 4282–4288. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, M.; Mountzios, G.; Papadimitriou, C.A. The role of PARP inhibition in triple-negative breast cancer: Unraveling the wide spectrum of synthetic lethality. Cancer Treat. Rev. 2018, 67, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Su, J.C.; Mar, A.C.; Wu, S.H.; Tai, W.T.; Chu, P.Y.; Wu, C.Y.; Tseng, L.M.; Lee, T.C.; Chen, K.F.; Liu, C.Y.; et al. Disrupting VEGF-A paracrine and autocrine loops by targeting SHP-1 suppresses triple negative breast cancer metastasis. Sci. Rep. 2016, 6, 28888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.Y.; Chen, K.F.; Chao, T.I.; Chu, P.Y.; Huang, C.T.; Huang, T.T.; Yang, H.P.; Wang, W.L.; Lee, C.H.; Lau, K.Y.; et al. Sequential combination of docetaxel with a SHP-1 agonist enhanced suppression of p-STAT3 signaling and apoptosis in triple negative breast cancer cells. J. Mol. Med. 2017, 95, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Su, J.C.; Huang, T.T.; Chu, P.Y.; Huang, C.T.; Wang, W.L.; Lee, C.H.; Lau, K.Y.; Tsai, W.C.; Yang, H.P.; et al. Sorafenib analogue SC-60 induces apoptosis through the SHP-1/STAT3 pathway and enhances docetaxel cytotoxicity in triple-negative breast cancer cells. Mol. Oncol. 2017, 11, 266–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Features | Total (n = 48) | HRPBC (n = 14) | HER2PBC (n = 9) | HHPBC (n = 11) | TNBC (n = 14) | p Value * |
|---|---|---|---|---|---|---|
| Age (Mean ± SD), year | 62.4 ± 11.2 | 64.4 ± 11.0 | 63.6 ± 10.7 | 63.1 ± 12.1 | 59.1 ± 11.4 | 0.638 |
| <50 | 7 | 1 | 1 | 1 | 4 | 0.365 |
| ≥50 | 41 | 13 | 8 | 10 | 10 | |
| Familial history | 4 | 1 | 0 | 0 | 3 | 0.173 |
| Postmenopause | 41 | 13 | 8 | 10 | 10 | 0.365 |
| BMI (Mean ± SD), kg/m2 | 24.7 ± 3.6 | 24.9 ± 3.0 | 23.7 ± 4.5 | 24.8 ± 4.0 | 25.0 ± 3.6 | 0.846 |
| Primary tumor size | 0.402 | |||||
| 1 | 24 | 9 | 3 | 4 | 8 | |
| 2 | 22 | 4 | 6 | 6 | 6 | |
| 3 | 1 | 1 | 0 | 0 | 0 | |
| 4 | 1 | 0 | 0 | 1 | 0 | |
| Lymph node metastasis | 0.601 | |||||
| 0 | 33 | 9 | 6 | 7 | 11 | |
| 1 | 8 | 1 | 1 | 3 | 3 | |
| 2 | 4 | 2 | 1 | 1 | 0 | |
| 3 | 3 | 2 | 1 | 0 | 0 | |
| Pathologic stage | 0.214 | |||||
| I | 20 | 7 | 2 | 3 | 8 | |
| II | 20 | 3 | 5 | 6 | 6 | |
| III | 8 | 4 | 2 | 2 | 0 | |
| Ki67, % | 0.004 | |||||
| <30 | 23 | 13 | 2 | 6 | 2 | |
| ≥30 | 25 | 1 | 7 | 5 | 12 | |
| Type of breast surgery | 0.590 | |||||
| Breast conservation | 29 | 10 | 4 | 6 | 9 | |
| Total mastectomy | 19 | 4 | 5 | 5 | 5 | |
| Type of axillary surgery | 0.757 | |||||
| Sentinel node biopsy | 34 | 9 | 7 | 7 | 11 | |
| Axillary dissection | 14 | 5 | 2 | 4 | 3 | |
| Adjuvant chemotherapy | 40 | 8 | 9 | 9 | 14 | 0.009 |
| Adjuvant radiotherapy | 36 | 13 | 5 | 7 | 11 | 0.168 |
| Hormone therapy | 25 | 14 | 0 | 11 | 0 | <0.001 |
| HER2 target therapy | 17 | 0 | 8 | 9 | 0 | <0.001 |
| Features | Total (n = 48) | HRPBC (n = 14) | HER2PBC (n = 9) | HHPBC (n = 11) | TNBC (n = 14) | p Value * |
|---|---|---|---|---|---|---|
| Alteration number | 0.942 | |||||
| 0 | 7 | 2 | 1 | 1 | 3 | |
| 1 | 12 | 4 | 2 | 2 | 4 | |
| ≥2 | 29 | 8 | 6 | 8 | 7 | |
| Alteration type | 0.983 | |||||
| Missense | 35 | 15 | 5 | 7 | 8 | |
| Nonsense | 37 | 12 | 6 | 11 | 8 | |
| Frameshift | 8 | 0 | 0 | 2 | 6 | |
| Amplification | 22 | 3 | 8 | 5 | 6 | |
| Frequent alteration | ||||||
| ERBB2 | 10 | 0 | 5 | 5 | 0 | 0.085 |
| PIK3CA | 15 | 8 | 2 | 4 | 1 | 0.037 |
| TP53 | 12 | 2 | 3 | 0 | 7 | 0.024 |
| Gene role dominance | 0.010 | |||||
| Oncogene | 17 | 7 | 4 | 5 | 1 | |
| Tumor suppressor | 20 | 4 | 3 | 4 | 9 | |
| Codominant | 4 | 1 | 1 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, W.Y.; Lee, J.; Kim, B.K.; Kim, J.O.; Park, J. Distinct Somatic Alteration Features Identified by Gene Panel Sequencing in Korean Triple-Negative Breast Cancer with High Ki67 Expression. Diagnostics 2021, 11, 416. https://doi.org/10.3390/diagnostics11030416
Sun WY, Lee J, Kim BK, Kim JO, Park J. Distinct Somatic Alteration Features Identified by Gene Panel Sequencing in Korean Triple-Negative Breast Cancer with High Ki67 Expression. Diagnostics. 2021; 11(3):416. https://doi.org/10.3390/diagnostics11030416
Chicago/Turabian StyleSun, Woo Young, Jina Lee, Bong Kyun Kim, Jong Ok Kim, and Joonhong Park. 2021. "Distinct Somatic Alteration Features Identified by Gene Panel Sequencing in Korean Triple-Negative Breast Cancer with High Ki67 Expression" Diagnostics 11, no. 3: 416. https://doi.org/10.3390/diagnostics11030416