
Preclinical Evaluation of Artesunate as an Antineoplastic Agent in Ovarian Cancer Treatment

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Artesunate
2.2. Cell Viability Assays
2.3. 3D Organoids
2.3.1. Tumor Organoid (TO) Development
2.3.2. Chemosensitivity Screens
2.3.3. Gene Expression Analysis
2.4. DNA Damage Assay
2.5. Flow Cytometric Analysis of Cell Cycle
2.6. Drug Administration Sequence Assay
3. Results
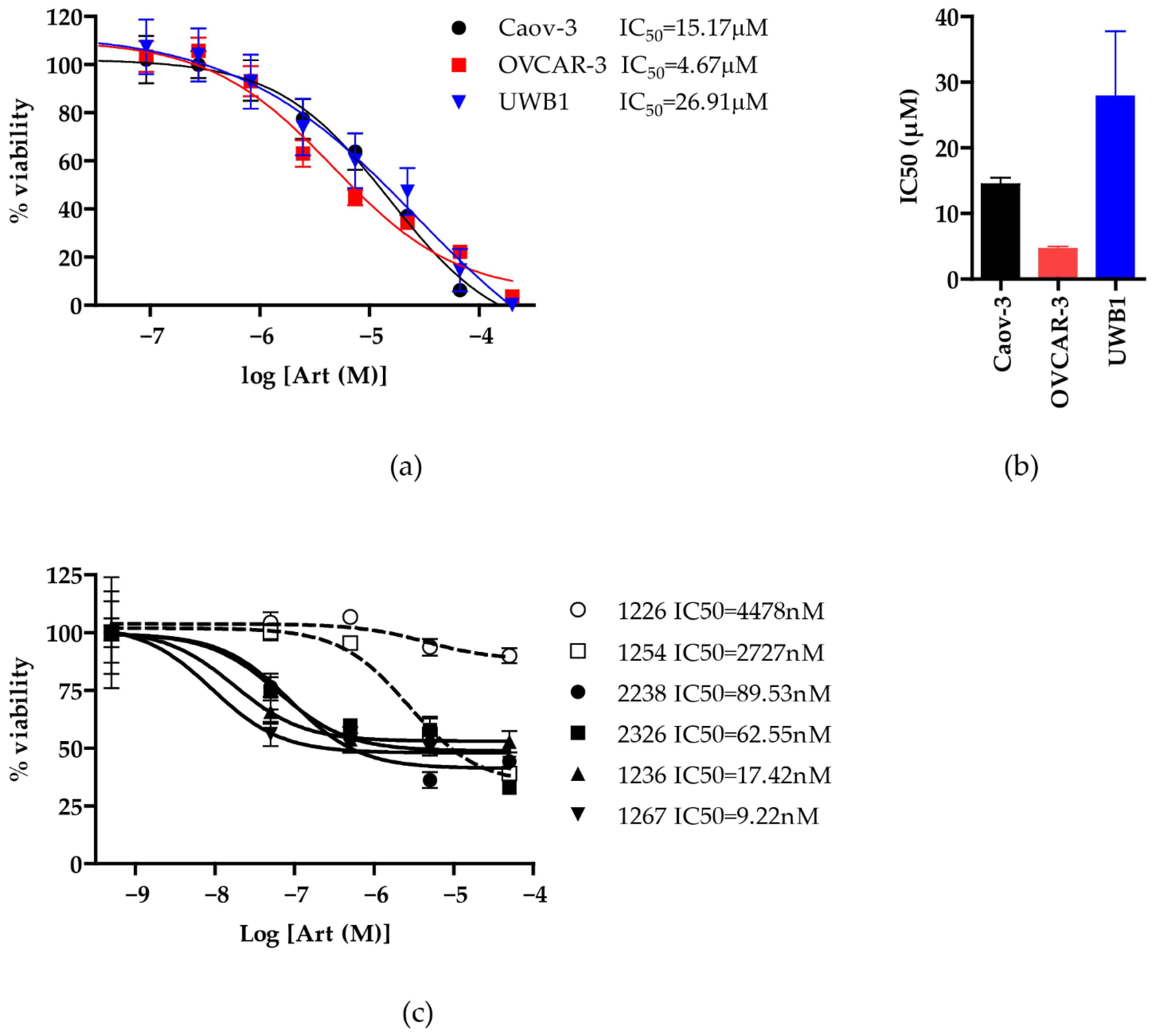
3.1. Artesunate Has Antineoplastic Activity in Ovarian Cancer
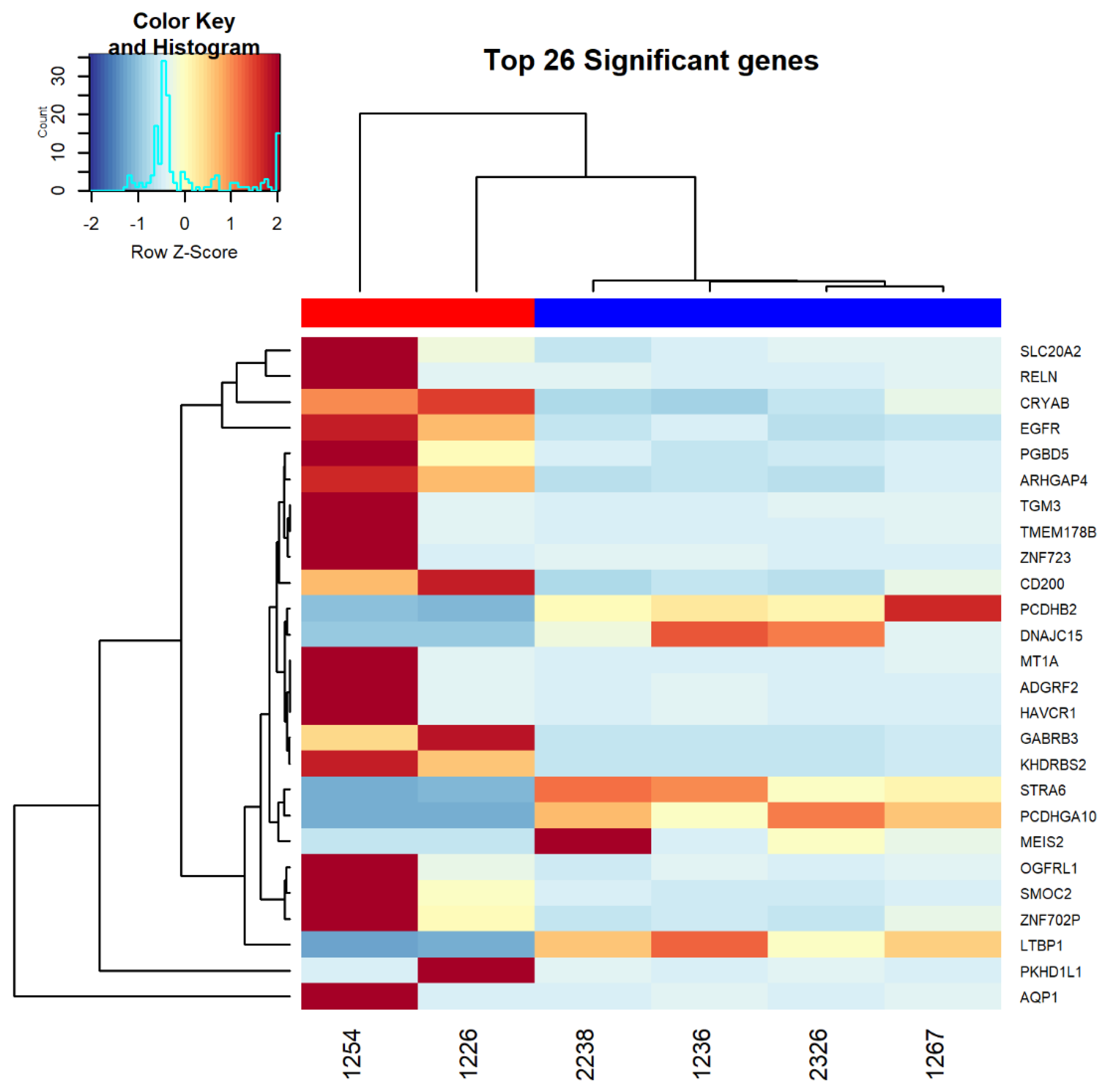
3.2. RNAseq Analysis Comparing Resistant and Sensitive Organoids
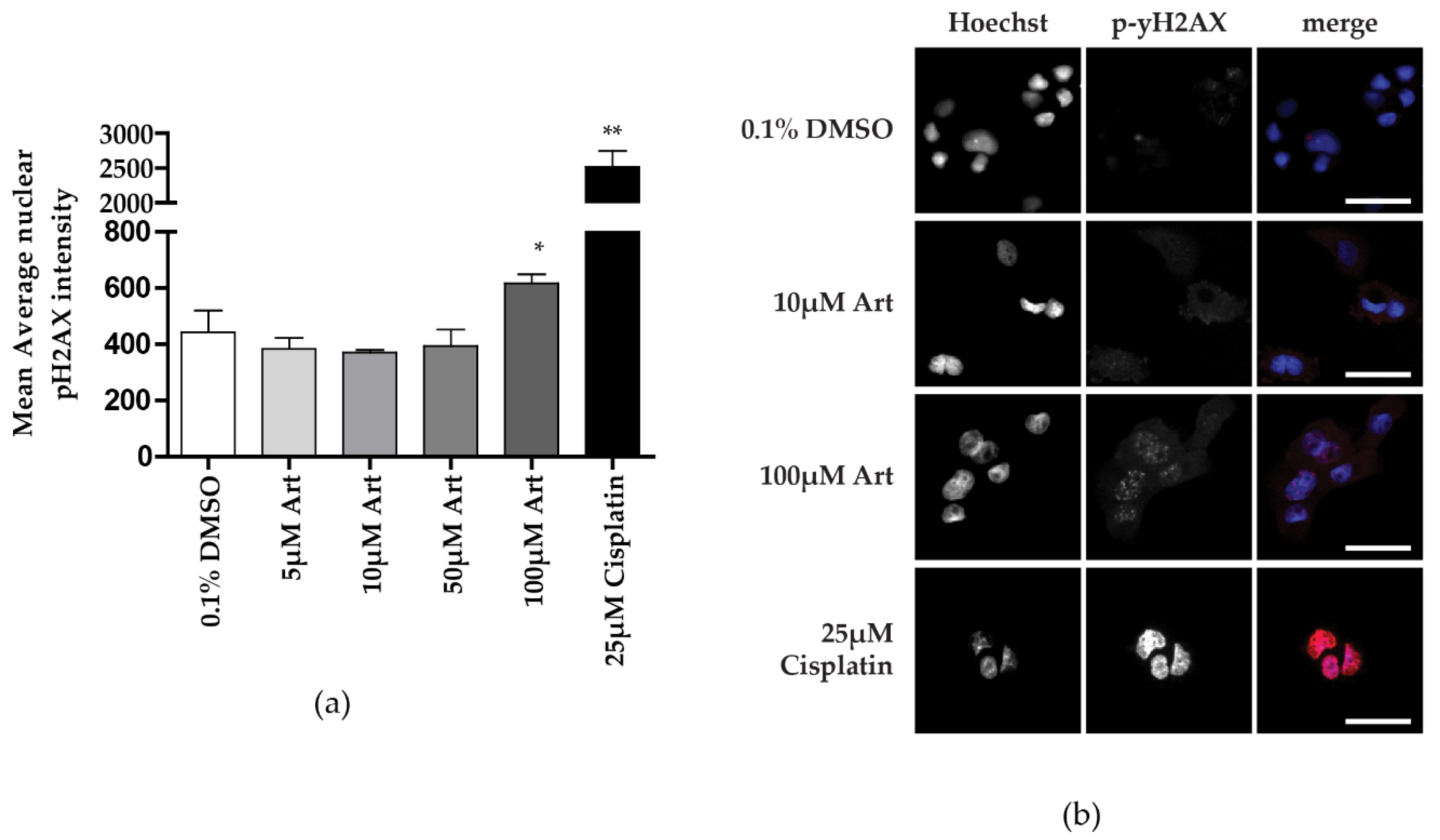
3.3. Clinically Relevant Concentrations of Artesunate Induce G1 Arrest, but Not DNA Damage
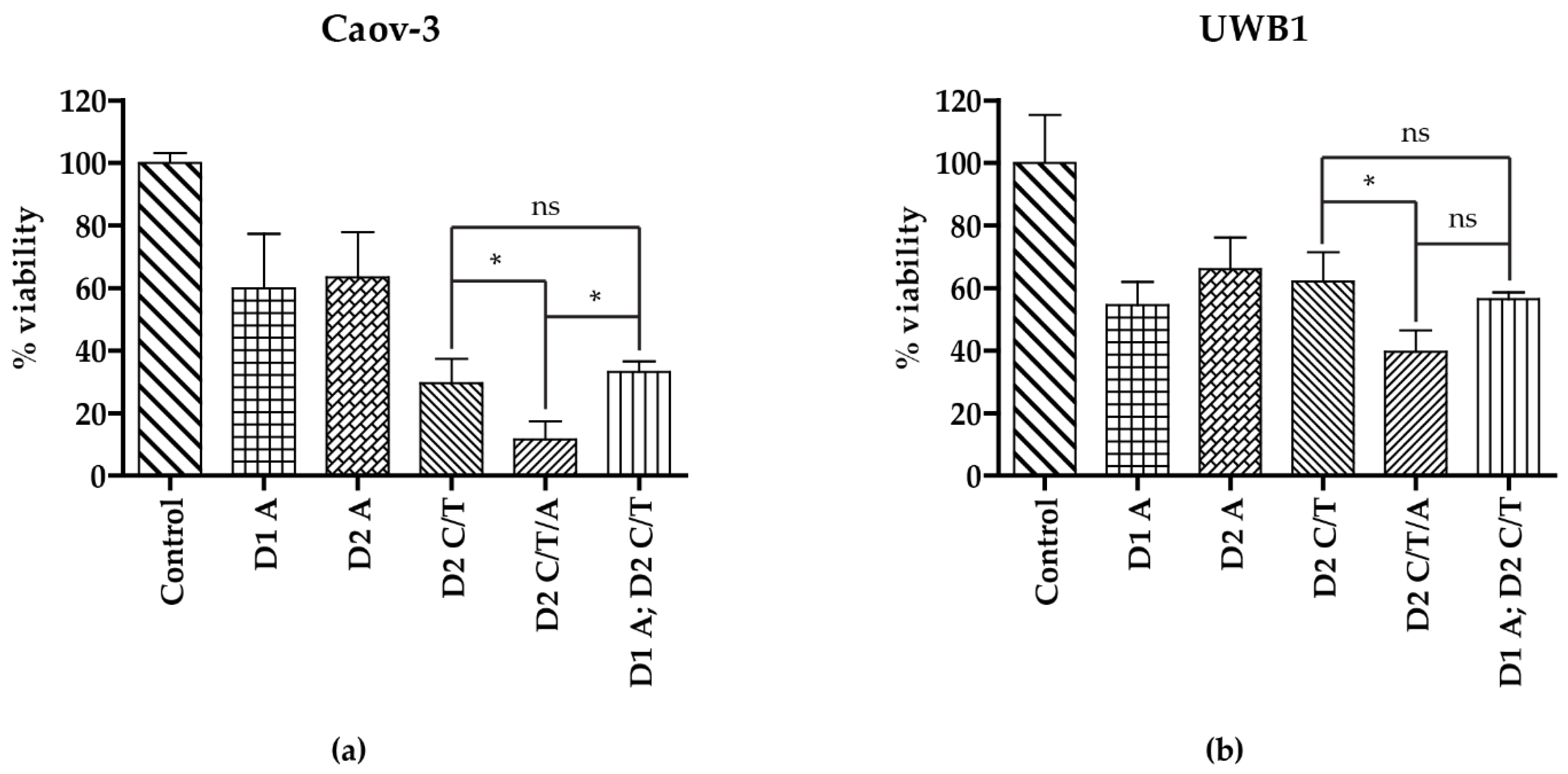
3.4. Addition of Artesunate to Carboplatin and Paclitaxel Improves Antineoplastic Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Cancer Statistics Center. 2021. Available online: http://cancerstatisticscenter.cancer.org (accessed on 23 November 2020).
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and Cisplatin Compared with Paclitaxel and Cisplatin in Patients with Stage III and Stage IV Ovarian Cancer. N. Engl. J. Med. 1996, 334, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R. Phase III Trial of Carboplatin and Paclitaxel Compared With Cisplatin and Paclitaxel in Patients With Optimally Resected Stage III Ovarian Cancer: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef]
- Bookman, M.A. GOG0182-ICON5: 5-arm phase III randomized trial of paclitaxel (P) and carboplatin (C) vs combinations with gemcitabine (G), PEG-lipososomal doxorubicin (D), or topotecan (T) in patients (pts) with advanced-stage epithelial ovarian (EOC) or primary peritoneal (PPC) carcinoma. J. Clin. Oncol. 2006, 24, 5002. [Google Scholar] [CrossRef]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of Bevacizumab in the Primary Treatment of Ovarian Cancer. New Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Barakat, R.R.; Markman, M.; Randall, M. Principles and Practice of Gynecologic Oncology, 5th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009. [Google Scholar]
- Golenser, J.; Waknine, J.H.; Krugliak, M.; Hunt, N.H.; Grau, G.E. Current perspec-tives on the mechanism of action of artemisinins. Int. J. Parasitol. 2006, 36, 1427–1441. [Google Scholar] [CrossRef]
- Crespo-Ortiz, M.P.; Wei, M.Q. Antitumor activity of artemisinin and its deriva-tives: From a well-known antimalarial agent to a potential anticancer drug. J. Biomed. Biotechnol. 2012. [Google Scholar] [CrossRef]
- Byakika-Kibwika, P.; Lamorde, M.; Mayito, J.; Nabukeera, L.; Mayanja-Kizza, H.; Katabira, E.; Hanpithakpong, W.; Obua, C.; Pakker, N.; Lindegårdh, N.; et al. Pharmacokinetics and pharmacodynamics of intravenous artesunate during severe malaria treatment in Ugandan adults. Malar. J. 2012, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef]
- Efferth, T.; Sauerbrey, A.; Olbrich, A.; Gebhart, E.; Rauch, P.; Weber, H.O.; Hengstler, J.G.; Halatsch, M.-E.; Volm, M.; Tew, K.D.; et al. Molecular Modes of Action of Artesunate in Tumor Cell Lines. Mol. Pharmacol. 2003, 64, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-H.; Zhou, H.-J.; Wang, W.-Q.; Wu, G.-D. Antimalarial dihydroartemisinin also inhibits angiogenesis. Cancer Chemother. Pharmacol. 2004, 53, 423–432. [Google Scholar] [CrossRef]
- Gao, P.; Wang, L.-L.; Liu, J.; Dong, F.; Song, W.; Liao, L.; Wang, B.; Zhang, W.; Zhou, X.; Xie, Q.; et al. Dihydroartemisinin inhibits endothelial cell tube formation by suppression of the STAT3 signaling pathway. Life Sci. 2020, 242, 117221. [Google Scholar] [CrossRef]
- Greenshields, A.L.; Shepherd, T.G.; Hoskin, D.W. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 2017, 56, 75–93. [Google Scholar] [CrossRef]
- Konstat-Korzenny, E.; Ascencio-Aragón, J.A.; Niezen-Lugo, S.; Vázquez-López, R. Artemisinin and Its Synthetic Derivatives as a Possible Therapy for Cancer. Med Sci. 2018, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Slezakova, S.; Ruda-Kucerova, J. Anticancer Activity of Artemisinin and its Derivatives. Anticancer. Res. 2017, 37, 5995–6003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilaoui, M.; Mouse, H.A.; Jaafari, A.; Zyad, A. Differential Effect of Artemisinin Against Cancer Cell Lines. Nat. Prod. Bioprospecting 2014, 4, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Deeken, J.F.; Wang, H.; Hartley, M.; Cheema, A.K.; Smaglo, B.; Hwang, J.J.; He, A.R.; Weiner, L.M.; Marshall, J.L.; Giaccone, G.; et al. A phase I study of intravenous artesunate in patients with advanced solid tumor ma-lignancies. Cancer Chemother. Pharmacol. 2008, 81, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Ge, C.-M.; Meng, Q.-H.; Cao, J.-P.; Tong, J.; Fan, S.-J. Dihydroartemisinin is an inhibitor of ovarian cancer cell growth. Acta Pharmacol. Sin. 2007, 28, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Hou, D.; Liu, Q.; Wu, T.; Guo, H.; Zhang, X.; Zou, Y.; Liu, Z.; Liu, J.; Wei, J.; et al. Artesunate sensitizes ovarian cancer cells to cisplatin by downregulat-ing RAD51. Cancer Biol. Ther. 2015, 16, 1548–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.J.; Mangler, M.; Sehouli, J.; et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organ-oids. Nat. Commun. 2015, 6, 8989. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, W.; Bolker, B.; Lodewijk, B.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Mar-tin, M.; Arni, M.; Steffen, N.M. Gplots: Various R Programming Tools for Plotting Data. R Package Version. 2. 2005. Available online: https://cran.r-project.org/web/packages/gplots/index.html (accessed on 23 November 2020).
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, X.L.; Zhang, G.H.; Gao, Y.F. Artesunate promotes Th1 differen-tiation from CD4+ T cells to enhance cell apoptosis in ovarian cancer via miR-142. Braz. J. Med. Biol. Res. 2019, 52, e7992. [Google Scholar] [CrossRef]
- Berdelle, N.; Nikolova, T.; Quiros, S.; Efferth, T.; Kaina, B. Artesunate induces oxi-dative DNA damage, sustained DNA double-strand breaks, and the ATM/ATR damage re-sponse in cancer cells. Mol. Cancer Ther. 2011, 10, 2224–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, J.; Tomar, S.; Pulliam, N.; Xiong, S.; Perkins, S.M.; Karpf, A.R.; Mitra, S.; Nephew, K.P.; Mitra, A.K. Functional characterization of a panel of high-grade serous ovarian cancer cell lines as representative experimental models of the disease. Oncotarget 2016, 7, 32810–32820. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gao, S.; Zhu, J.; Zheng, Y.; Zhang, H.; Sun, H. Dihydroartemisinin induces apoptosis and inhibits proliferation, migration, and invasion in epithelial ovarian cancer via inhibition of the hedgehog signaling pathway. Cancer Med. 2018, 7, 5704–5715. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, H.; Guo, Y.; Wang, K.; Wang, X.; Guo, Z. Towards rational design of RAD51-targeting prodrugs: Platinum(IV)-artesunate conjugates with enhanced cytotoxi-city against BRCA-proficient ovarian and breast cancer cells. Chem. Commun (Camb) 2018, 54, 11717–11720. [Google Scholar] [CrossRef]
- Maru, Y.; Tanaka, N.; Itami, M.; Hippo, Y. Efficient use of patient-derived organ-oids as a preclinical model for gynecologic tumors. Gynecol. Oncol. 2019, 154, 189–198. [Google Scholar] [CrossRef]
- Gotsbacher, M.P.; Cho, S.M.; Kim, N.H.; Liu, F.; Kwon, H.J.; Karuso, P. Reverse Chemical Proteomics Identifies an Unanticipated Human Target of the Antimalarial Ar-tesunate. ACS Chem. Biol. 2019, 14, 636–643. [Google Scholar] [CrossRef]
- von Hagens, C.; Walter-Sack, I.; Goeckenjan, M.; Storch-Hagenlocher, B.; Sertel, S.; Elsasser, M.; Remppis, B.A.; Munzinger, J.; Edler, L.; Efferth, T.; et al. Long-term add-on therapy (compassionate use) with oral artesunate in patients with metastatic breast cancer after participating in a phase I study (ARTIC M33/2). Phytomedicine 2019, 54, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ni, W.; Deng, Z.; Liu, M.; She, L.; Xie, Q. Targeting nasopharyngeal carcino-ma by artesunate through inhibiting Akt/mTOR and inducing oxidative stress. Fundam. Clin. Pharmacol. 2017, 31, 301–310. [Google Scholar] [CrossRef] [PubMed]
- König, M.; Von Hagens, C.; Hoth, S.; Baumann, I.; Walter-Sack, I.; Edler, L.; Sertel, S. Investigation of ototoxicity of artesunate as add-on therapy in patients with metastatic or locally advanced breast cancer: New audiological results from a prospective, open, uncontrolled, monocentric phase I study. Cancer Chemother. Pharmacol. 2016, 77, 413–427. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Category | Pathway ID | Pathway Description | p-Value | Differentially Expressed Genes |
|---|---|---|---|---|
| ROS/oxidative phosphorylation | GO:0000302 | response to reactive oxygen species | ↑ 0.0008 | ↑ AQP1, ↑ CRYAB, ↑ EGFR |
| GO:0006979 | response to oxidative stress | ↑ 0.0056 | ↑ AQP1, ↑ CRYAB, ↑ EGFR | |
| GO:0034614 | cellular response to reactive oxygen species | ↑ 0.0077 | ↑ AQP1, ↑ EGFR | |
| GO:2000377 | regulation of reactive oxygen species metabolic process | ↑ 0.0098 | ↑ CRYAB, ↑ EGFR | |
| GO:0072593 | reactive oxygen species metabolic process | ↑ 0.0212 | ↑ CRYAB, ↑ EGFR | |
| GO:0034599 | cellular response to oxidative stress | ↑ 0.0241 | ↑ AQP1, ↑ EGFR | |
| GO:2000378 | negative regulation of reactive oxygen species metabolic process | ↑ 0.045 | ↑ CRYAB | |
| GO:0090324 | negative regulation of oxidative phosphorylation | ↓ 0.0018 | ↓ DNAJC15 | |
| GO:0002082 | regulation of oxidative phosphorylation | ↓ 0.0074 | ↓ DNAJC15 | |
| GO:0006119 | oxidative phosphorylation | ↓ 0.0352 | ↓ DNAJC15 | |
| mitochondrial functon/cellular respiration | GO:1902957 | negative regulation of mitochondrial electron transport, NADH to ubiquinone | ↓ 0.0005 | ↓ DNAJC15 |
| GO:1905447 | negative regulation of mitochondrial ATP synthesis coupled electron transport | ↓ 0.0005 | ↓ DNAJC15 | |
| GO:1901856 | negative regulation of cellular respiration | ↓ 0.0010 | ↓ DNAJC15 | |
| GO:1902956 | regulation of mitochondrial electron transport, NADH to ubiquinone | ↓ 0.0013 | ↓ DNAJC15 | |
| GO:1905446 | regulation of mitochondrial ATP synthesis coupled electron transport | ↓ 0.0018 | ↓ DNAJC15 | |
| GO:0005744 | TIM23 mitochondrial import inner membrane translocase complex | ↓ 0.0036 | ↓ DNAJC15 | |
| GO:0043457 | regulation of cellular respiration | ↓ 0.0076 | ↓ DNAJC15 | |
| GO:0006120 | mitochondrial electron transport, NADH to ubiquinone | ↓ 0.0135 | ↓ DNAJC15 | |
| GO:0042775 | mitochondrial ATP synthesis coupled electron transport | ↓ 0.0235 | ↓ DNAJC15 | |
| GO:0042773 | ATP synthesis coupled electron transport | ↓ 0.0238 | ↓ DNAJC15 | |
| GO:0006626 | protein targeting to mitochondrion | ↓ 0.0248 | ↓ DNAJC15 | |
| GO:1990542 | mitochondrial transmembrane transport | ↓ 0.0255 | ↓ DNAJC15 | |
| GO:0022904 | respiratory electron transport chain | ↓ 0.0285 | ↓ DNAJC15 | |
| GO:0098800 | inner mitochondrial membrane protein complex | ↓ 0.0332 | ↓ DNAJC15 | |
| GO:0072655 | establishment of protein localization to mitochondrion | ↓ 0.0345 | ↓ DNAJC15 | |
| GO:0045333 | cellular respiration | ↓ 0.0473 | ↓ DNAJC15 | |
| cell cycle progression | GO:1900087 | positive regulation of G1/S transition of mitotic cell cycle | ↑ 0.0302 | ↑ EGFR |
| GO:0045740 | positive regulation of DNA replication | ↑ 0.0302 | ↑ EGFR | |
| GO:1902808 | positive regulation of cell cycle G1/S phase transition | ↑ 0.0384 | ↑ EGFR | |
| GO:0045787 | positive regulation of cell cycle | ↑ 0.0391 | ↑ SMOC2, ↑ EGFR, ↓ MEIS2 | |
| response to metal ion | GO:0010038 | response to metal ion | ↑ 0.0034 | ↑ AQP1, ↑ MT1A, ↑ EGFR |
| GO:0071248 | cellular response to metal ion | ↑ 0.0005 | ↑ AQP1, ↑ MT1A, ↑ EGFR | |
| Cell death/apoptosis | GO:0060548 | negative regulation of cell death | ↑ 0.0011 | ↑ AQP1, ↑ GABRB3, ↑ CD200,↑ CRYAB, ↑ EGFR |
| GO:0043067 | regulation of programmed cell death | ↑ 0.0375 | ↑ AQP1, ↑ GABRB3, ↑ CRYAB,↑ EGFR | |
| ATP metabolic process | GO:1903579 | negative regulation of ATP metabolic process | ↓ 0.0064 | ↓ DNAJC15 |
| GO:1903578 | regulation of ATP metabolic process | ↓ 0.0300 | ↓ DNAJC15 | |
| response to antineoplastic agent | GO:0097327 | response to antineoplastic agent | ↑ 0.0029 | ↑ AQP1, ↑ EGFR |
| phosphatidylinositol-mediated signaling | GO:0048015 | phosphatidylinositol-mediated signaling | ↑ 0.0097 | ↑ RELN, ↑ EGFR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDowell, A., Jr.; Hill, K.S.; McCorkle, J.R.; Gorski, J.; Zhang, Y.; Salahudeen, A.A.; Ueland, F.; Kolesar, J.M. Preclinical Evaluation of Artesunate as an Antineoplastic Agent in Ovarian Cancer Treatment. Diagnostics 2021, 11, 395. https://doi.org/10.3390/diagnostics11030395
McDowell A Jr., Hill KS, McCorkle JR, Gorski J, Zhang Y, Salahudeen AA, Ueland F, Kolesar JM. Preclinical Evaluation of Artesunate as an Antineoplastic Agent in Ovarian Cancer Treatment. Diagnostics. 2021; 11(3):395. https://doi.org/10.3390/diagnostics11030395
Chicago/Turabian StyleMcDowell, Anthony, Jr., Kristen S. Hill, Joseph Robert McCorkle, Justin Gorski, Yilin Zhang, Ameen A. Salahudeen, Fred Ueland, and Jill M. Kolesar. 2021. "Preclinical Evaluation of Artesunate as an Antineoplastic Agent in Ovarian Cancer Treatment" Diagnostics 11, no. 3: 395. https://doi.org/10.3390/diagnostics11030395