
Prenatally Diagnosed Infantile Myofibroma of Sartorius Muscle—A Differential for Soft Tissue Masses in Early Infancy

 and
and
Abstract
:1. Introduction
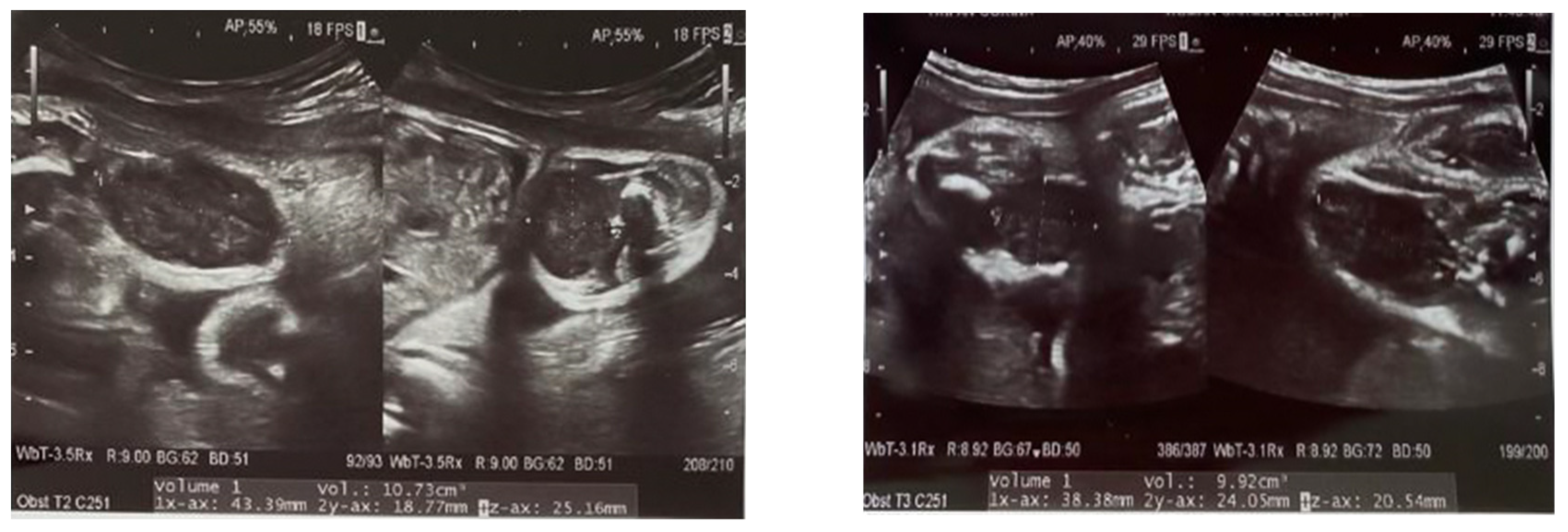
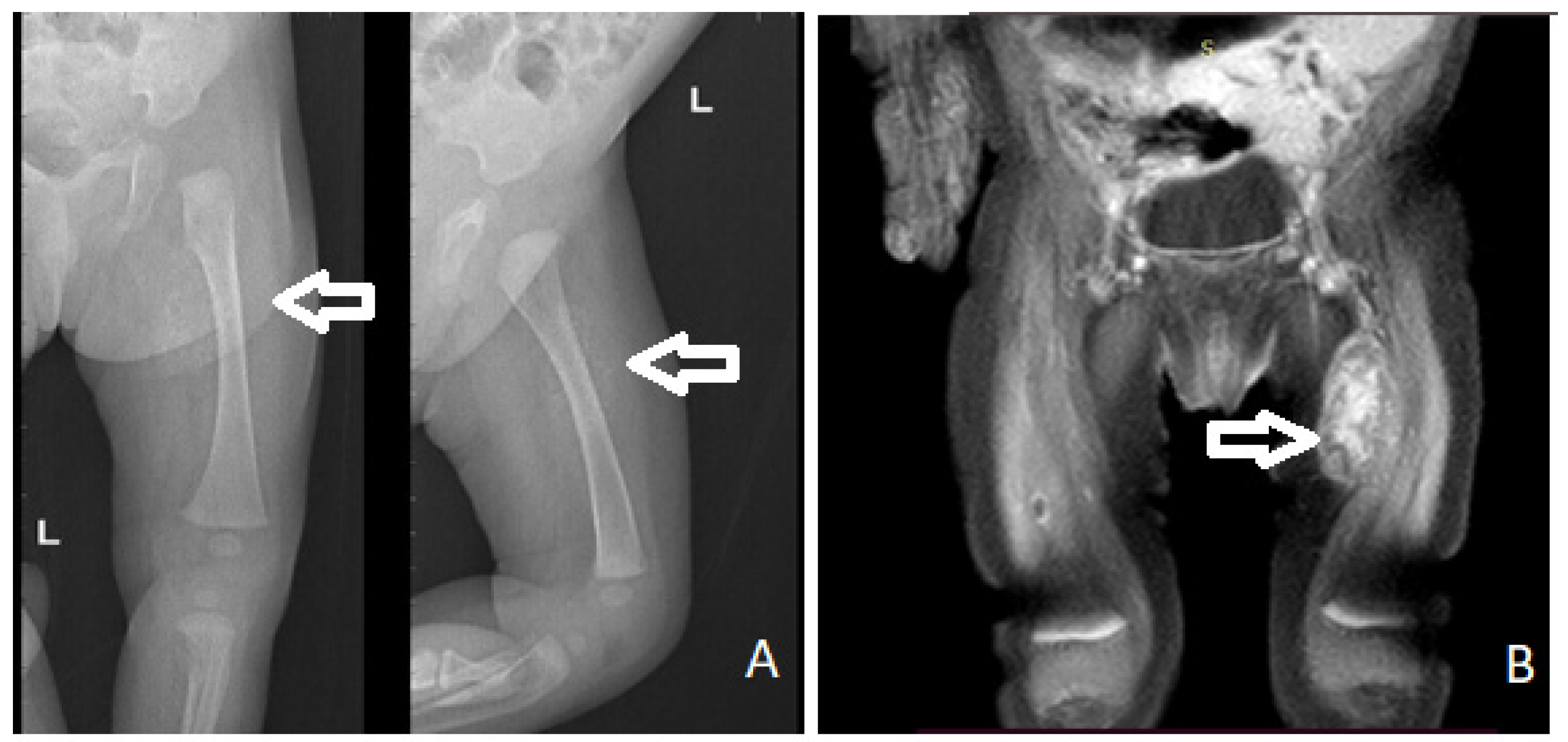
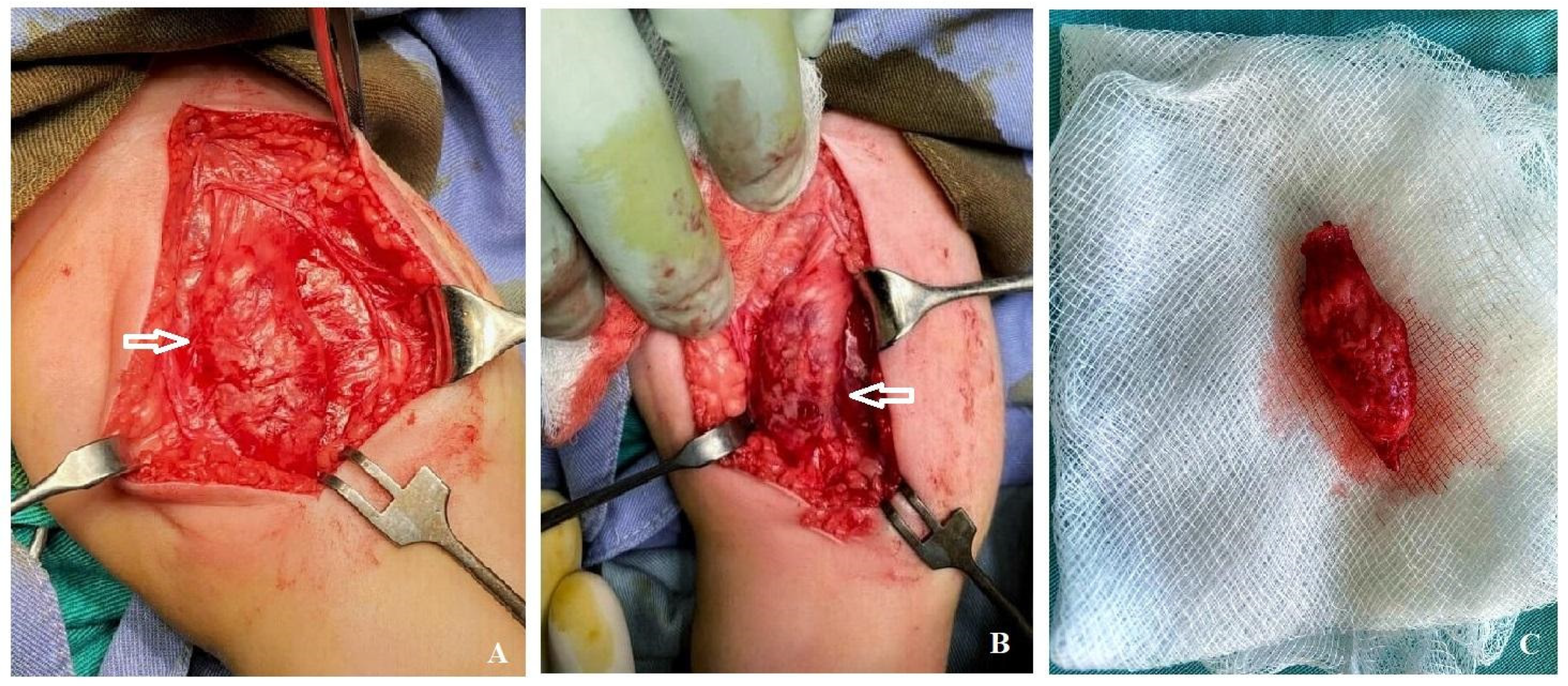
2. Case Report
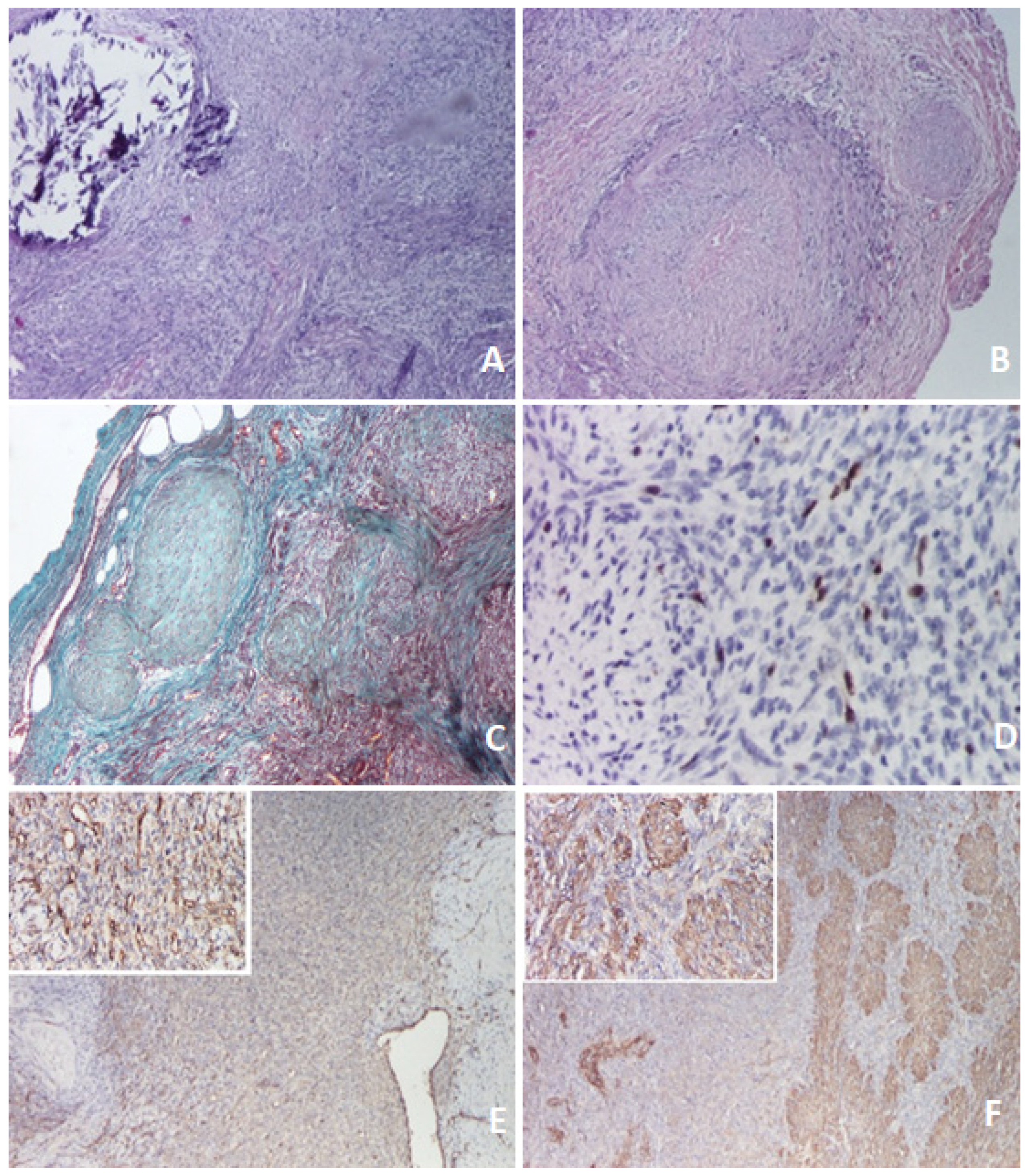
3. Discussion–Literature Review
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PRef | GA (Weeks) | Fetal Gender (M/F) | Location & Size (cm) | Prenatal Investigation (Type & Findings) | Management | Outcome Follow Up |
|---|---|---|---|---|---|---|
| Nishioka, 1999 [32] | 37 | M | Chest wall (5.0 × 5.5 × 2.5) | US: tumor recognized on the chest wall. | Resection and skin graft at 7 days of life. | No recurrence at 11 months. |
| Kubota, 1999 [14] | 36 | F | Left upper arm (8 × 7 × 5) | US: solid and spherical, slightly inhomogeneous, moderately echogenic mass, well demarcated with no evidence of bone invasion. | Resection of tumor at 2 months of age. | No evidence of tumor or functional disorder 3 years after surgery. |
| Meizner, 2000 [15] | 30 | F | Paraspinal (4.2 × 7.5 × 3.5) | US (30 w): solid mass on the left side of the spine extending from T7 to L5 of the vertebral column; no Doppler blood flow. MRI (31 w): well-defined mass, no direct connection between the mass and spinal cord elements. | Pregnanacy termination (32 w) via fetocide injection. | On autopsy, a large lump on the left side of the spine, with no connection to the spinal canal. Visceral involvement was also noted in the liver and retroperitoneum. |
| Wataganara, 2007 [33] | 35 | F | Anterior fetal neck, extending onto the anterior chest wall (10.8 × 9.2) | US (35 w): homogeneous solid mass. MRI: in midsagittal plane, a huge solid mass on the anterior portion of the neck and chest was observed, with low signal intensity mass. Nasopharynx, oropharynx, and trachea intact and filled with amniotic fluid position. | Spontaneously ruptured membranes at 35 weeks. EXIT procedure Resection at 11 days of age. | At 7 months of age, no signs of recurrence. |
| Muraoka, 2008 [20] | 32 | F | Spleen (5.7 × 3.9) | US (32 w): abdominal tumor, 5.7 × 3.9 cm; (34 w): grown to 6.0 × 5.5 cm. MRI (36 w): left upper abdominal tumor. | Splenectomy on day 20 of life. | No recurrence at 3 years of age. |
| Arabin, 2009 [18] | 13 | F | Superficial head | Monoamniotic twins. 13 w: echodense tumor on the side of one twin’s head. 18 w: vascularization of the tumor. 30 w: intestinal dilatation (same twin). MRI: excluded connections to the ventricles or brain. | Superficial head tumor resected and laparotomy for abdominal mass. | Affected twin died from sepsis 12 days after birth; Unaffected twin normal at follow up. |
| Yeniel, 2013 [34] | 32 | F | Left lung (4.6 × 2.7) | US: solid mass detected in the parenchyma of the left lung; Color Doppler: avascular. | Thoracotomy and complete mass resection on day 2 of life because of respiratory distress syndrome. | No recurrence or symptoms 1 year after surgery. |
| Zhang, 2014 [17] | 38 | M | Right side paraspinal, a pseudoulcerate plaque (2.4 × 2.2 × 1.2) | US: mass on the fetal right back (T3-8), hypoechoic with sporadic hyperecho. Color Doppler flow: intermittent blood flow inside and around the mass. MRI (next day after US): normal result, with no mass. | Resection of the mass 3 months after birth. | No recurrence at 2 years after surgery. |
| Coleman, 2016 [35] | 29 | F | Posterior medial aspect of the left thigh of her fetus (3.1 × 3 × 3.8 at 29 w; 5.6 × 4.8 × 5.3 at 33 w) | US (29 w): First visualization. US (33 w): Mass increased in size, hypoechoic compared to the adjacent subcutaneous fat, but isoechoic compared to muscle. The underlying bone was intact. MRI (same day with the last echo) defined the mass as relatively well-circumscribed, superficial location that replaced the medial subcutaneous tissues of the thigh. | Surgical excision on hospital day 5 without complication, though it was found to involve the investing fascia of the gracilis and sartorius muscles. No direct involvement of the major neurovascular structures. | No functional or strength deficits in the lower extremity at the age of 9 months. |
| Vemavarapu, 2017 [36] | 32 | F | Left arm (9 × 5) | US: well circumscribed mass, arising from the left arm, just beside the humerus, but the osseous structure normal; Color Doppler: moderate blood flow in the solid part of the mass. MRI: improvised the detection and evaluation. | Resection of tumor. | Normal upper function and no recurrence 1 year after surgery. |
| Pekar-Zlotin, 2018 [37] | 34 | - | Multiple tumors | US: multiorgan involvement, with masses predominantly in the lower extremities, heart, abdominal cavity, and neck. Normal Doppler studies in the major fetal arterial and venous circulations, as well as normal amniotic fluid volume and unremarkable appearance of the placenta. | Inoperable tumors. | The infant died of cardiac failure 30 days after birth. |
| Rekawek, 2019 [16] | 36 | M | Lower thoracic spine (3.5 × 2.3) | US: avascular, heterogeneous mass of soft tissues, without cord involvement. MRI: thick-walled partly cystic lesion in the right paraspinal muscles of the lower thorax. | Biopsy on day 7 of life. | MRI at one year of age: multiple soft masses decreased in size compared to prior imaging, and three additional lesions: crus of the left diaphragam, left posterior chest wall, and left shoulder. |
| Evens, 2021 [38] | 33 | - | Visceral organ involvement Lung masses | US: dilated bowel loop. MRI: suggested the dilated loop of bowel was colon, given the internal high T1 signal compatible with meconium. Additionally, multiple, solid-appearing lung masses were noted, which had not been previously visualized on ultrasound. After thorough review of the MRI images of the entire fetus, no primary lesion was found. No masses were visualized in the extremities. | The baby had not passed meconium after 1 day of life, therefor an exploratory laparotomy: nodules on the bowel serosa, resulting in a bowel obstruction. Owing the visceral organ involvement, chemotherapy was considered; however, it was ultimately decided to observe the patient and treat only if there was radiologic progression and/or clinical deterioration. |
References
- Scheper, M.A.; DiFabio, V.E.; Sauk, J.J.; Nikitakis, N.G. Myofibromatosis: A case report with a unique clinical presentation. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2005, 99, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Pelluard-Nehmé, F.; Coatleven, F.; Carles, D.; Alberti, E.M.; Briex, M.; Dallay, D. Multicentric infantile myofibromatosis: Two perinatal cases. Eur. J. Pediatr. 2007, 166, 997–1001. [Google Scholar] [CrossRef]
- Venkatesh, V.; Kumar, B.P.; Kumar, K.A.; Mohan, A.P. Myofibroma—A rare entity with unique clinical presentation. J. Maxillofac. Oral Surg. 2015, 14 (Suppl. 1), 64–68. [Google Scholar] [CrossRef] [Green Version]
- NORD Rare Disease Database. Available online: https://rarediseases.org/rare-diseases/infantile-myofibromatosis/ (accessed on 1 November 2021).
- Hausbrandt, P.A.; Leithner, A.; Beham, A.; Bodo, K.; Raith, J.; Windhager, R. A rare case of infantile myofibromatosis and review of literature. J. Pediatr. Orthop. B 2010, 19, 122–126. [Google Scholar] [CrossRef]
- Stout, A.P. Juvenile fibromatoses. Cancer 1954, 7, 953–978. [Google Scholar] [CrossRef]
- Kauffman, S.L.; Stout, A.P. Congenital mesenchymal tumours. Cancer 1965, 18, 460–476. [Google Scholar] [CrossRef]
- Chung, E.B.; Enzinger, F.M. Infantile myofibromatosis. Cancer 1981, 48, 1807–1818. [Google Scholar] [CrossRef]
- Smith, K.J.; Skelton, H.G.; Barrett, T.L.; Lupton, G.P.; Graham, J.H. Cutaneous myofibroma. Mod. Pathol. 1989, 2, 603–609. [Google Scholar]
- Daimaru, Y.; Hashimoto, H.; Enjoji, M. Myofibromatosis in adults (adult counterpart of infantile myofibromatosis). Am. J. Surg. Pathol. 1989, 13, 859–865. [Google Scholar] [CrossRef]
- Wu, W.; Chen, J.; Cao, X.; Yang, M.; Zhu, J.; Zhao, G. Solitary infantile myofibromatosis in the bones of the upper extremities: Two rare cases and a review of the literature. Oncol. Lett. 2013, 6, 1406–1408. [Google Scholar] [CrossRef] [Green Version]
- Martignetti, J.A.; Tian, L.; Li, D.; Ramirez, M.C.; Camacho-Vanegas, O.; Camacho, S.C.; Guo, Y.; Zand, D.J.; Bernstein, A.M.; Masur, S.K.; et al. Mutations in PDGFRB Cause Autosomal-Dominant Infantile Myofibromatosis. Am. J. Hum. Genet. 2013, 92, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Stenman, G.; Nadal, N.; Persson, S.; Gunterberg, B.; Angervall, L. del(6)(q12q15) as the sole cytogenetic anomaly in a case of solitary infantile myofibromatosis. Oncol. Rep. 1999, 6, 1101–1104. [Google Scholar] [CrossRef]
- Kubota, A.; Imano, M.; Yonekura, T.; Hirooka, S.; Nose, K.; Oyanagi, H.; Nakayama, M. Infantile myofibromatosis of the triceps detected by prenatal sonography. J. Clin. Ultrasound 1999, 27, 147–150. [Google Scholar] [CrossRef]
- Meizner, I.; Shalev, J.; Mashiach, R.; Vardimon, D.; Ben-Raphael, Z. Prenatal ultrasound diagnosis of infantile myofibromatosis —A case report. Ultrasound Obstet. Gynecol. 2000, 16, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Rekawek, P.; Coleman, B.G.; Kamath, A.; Stone, J.L. Prenatal sonography of multicentric infantile myofibromatosis: Case report and review of the literature. J. Clin. Ultrasound 2019, 47, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cheng, D.; Wu, M.; Ge, L.; Ma, X. Diagnosis of infantile myofibromatosis with pseudo-ulcerated plaque using prenatal ultrasound: A case report. Exp. Ther. Med. 2014, 8, 1769–1771. [Google Scholar] [CrossRef]
- Arabin, B.; Hack, K.; Nooij, L.; Nikkels, P. Dis-crepant findings in a monoamniotic twin pregnancy affected by infantile myofibromatosis. Ultrasound Obstet. Gynecol. 2009, 33, 488–490. [Google Scholar] [CrossRef]
- Alamo, L.; Beck-Popovic, M.; Gudinchet, F.; Meuli, R. Congenital tumors: Imaging when life just begins. Insights Imaging 2011, 2, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, I.; Ohno, Y.; Kamitamari, A.; Okada, M.; Moriuchi, H.; Kanematsu, T. Congenital occurrence of solitary infantile myofibromatosis of the spleen. J. Pediatr. Surg. 2008, 43, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Thunnissen, B.T.; Bax, N.M.; Rövekamp, M.H.; Beek, F.J.; van Gorp, J. Infantile myofibromatosis: An unusual presentation and a review of the literature. Eur. J. Pediatr. Surg. 1993, 3, 179–181. [Google Scholar] [CrossRef]
- Wiswell, T.E.; Davis, J.; Cunnigham, B.E.; Solenberger, R.; Thomas, P.J. Infantile myofibromatosis: The most common fibrous tumor of infancy. J. Pediatr. Surg. 1988, 23, 315–318. [Google Scholar] [CrossRef]
- Koujok, K.; Ruiz, R.E.; Hernandez, R.J. Myofibromatosis: Imaging characteristics. Pediatr. Radiol. 2005, 35, 374–380. [Google Scholar] [CrossRef] [Green Version]
- Fraitag, S.; Boccara, O. What to Look Out for in a Newborn with Multiple Papulonodular Skin Lesions at Birth. Dermatopathology 2021, 8, 390–417. [Google Scholar] [CrossRef]
- Allen, P.W. The fibromatosis: A clinicopathologic classification based on 140 cases. Review articles, part 2. Am. J. Surg. Pathol. 1977, 1, 305–321. [Google Scholar] [CrossRef]
- Fukasawa, Y.; Ishikura, H.; Takada, A.; Yokoyama, S.; Imamura, M.; Yoshiki, T.; Sato, H. Massive apoptosis in infantile myofibromatosis: A putative mechanism of tumor regression. J. Pathol. 1994, 144, 480–485. [Google Scholar]
- Schmidt, D. Fibrous tumors and tumor-like lesions of childhood: Diagnosis, differential diagnosis, and prognosis. Curr. Top. Pathol. 1995, 89, 175–191. [Google Scholar] [PubMed]
- Burton, J.L.; Lovell, C.R. Disorders of connective tissue. In Textbook of Dermatology, 6th ed.; Champion, R.H., Burton, J.L., Burns, D.A., Breathnach, S.M., Eds.; Blackwell Science: Malden, MA, USA, 1998; Volume 3, pp. 2048–2049. [Google Scholar]
- Kiel, K.D.; Suit, H.D. Radiation therapy in the treatment of aggressive fibromatosis (desmoid tumors). Cancer 1984, 54, 2051–2055. [Google Scholar] [CrossRef]
- Savasan, S.; Fulgenzi, L.A.; Rabah, R.; Mohamed, A.N.; Ravindranath, Y. Generalized infantile myofibromatosis in a patient with Turner’s syndrome: A trial of interferon-α. J. Pediatr. 1998, 133, 694–696. [Google Scholar] [CrossRef]
- Sramek, M.; Neradil, J.; Macigova, P.; Mudry, P.; Polaskova, K.; Slaby, O.; Noskova, H.; Sterba, J.; Veselska, R. Effects of Sunitinib and Other Kinase Inhibitors on Cells Harboring a PDGFRB Mutation Associated with Infantile Myofibromatosis. Int. J. Mol. Sci. 2018, 19, 2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, K.; Seguchi, T.; Yamamura, Y.; Tatsumura, M.; Sou, H.; Gondo, T.; Hoshii, Y.; Iwata, T. Infantile myofibromatosis identified by fetal ultrasound. Br. J. Dermatol. 1999, 140, 539–541. [Google Scholar] [CrossRef]
- Wataganara, T.; Ngerncham, S.; Kitsommart, R.; Fuangtharnthip, P. Fetal neck myofibroma. J. Med. Assoc. Thai. 2007, 90, 376–380. [Google Scholar] [PubMed]
- Yeniel, A.O.; Ergenoglu, A.M.; Zeybek, B.; Kazandi, M.; Akercan, F.; Ozcan, C.; Veral, A. Prenatal diagnosis of infantile myofibromatosis of the lung: A case report and review of the literature. J. Clin. Ultrasound 2013, 41, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Coleman, A.M.; Calvo-Garcia, M.A.; Zbojniewicz, A.M.; Alonso, M.; Lim, F.Y. Prenatal Diagnosis of Infantile Myofibroma with Postnatal Imaging Correlation. Fetal Diagn Ther. 2016, 40, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Vemavarapu, G.; Reddy, P.; Panigrahi, N.; Jayaram, H. Case Report of Prenatal Diagnosis of Fetal Left Arm Tumor (Infantile Myofibromatosis); The Fetal Medicine Foundation, 2017. Available online: https://fetalmedicine.org/abstracts/2017/var/pdf/abstracts/2017/2057.pdf (accessed on 4 November 2021).
- Pekar-Zlotin, M.; Levinsohn-Tavor, O.; Livneh, A.; Sher, O.; Melcer, Y.; Maymon, R. Gynecology and Oncology Fetal Myofibromatosis: A Challenge for Prenatal Diagnosis Mini Review of the English Literature. Obstet. Gynecol. Surv. 2019, 74, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Evens, A.; Gonzalez-Gomez, I.; Towbin, R.B.; Towbin, A.J.; Kucera, J.N. Infantile myofibromatosis: Prenatal and postnatal imaging features. Appl. Radiol. 2021, 50, 44–46. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popa, Ș.; Apostol, D.; Bîcă, O.; Benchia, D.; Sârbu, I.; Ciongradi, C.I. Prenatally Diagnosed Infantile Myofibroma of Sartorius Muscle—A Differential for Soft Tissue Masses in Early Infancy. Diagnostics 2021, 11, 2389. https://doi.org/10.3390/diagnostics11122389
Popa Ș, Apostol D, Bîcă O, Benchia D, Sârbu I, Ciongradi CI. Prenatally Diagnosed Infantile Myofibroma of Sartorius Muscle—A Differential for Soft Tissue Masses in Early Infancy. Diagnostics. 2021; 11(12):2389. https://doi.org/10.3390/diagnostics11122389
Chicago/Turabian StylePopa, Ștefan, Dan Apostol, Ovidiu Bîcă, Diana Benchia, Ioan Sârbu, and Carmen Iulia Ciongradi. 2021. "Prenatally Diagnosed Infantile Myofibroma of Sartorius Muscle—A Differential for Soft Tissue Masses in Early Infancy" Diagnostics 11, no. 12: 2389. https://doi.org/10.3390/diagnostics11122389