
Different Rates of the SLC26A4-Related Hearing Loss in Two Indigenous Peoples of Southern Siberia (Russia)

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.1.1. Patients
2.1.2. Control Samples
2.1.3. Ethics Statement
2.2. Molecular Analysis
2.2.1. Mutation Analysis of the SLC26A4 Gene
2.2.2. Screening of Pathogenic SLC26A4 Variants in Control Samples
2.3. Bioinformatics Tools
2.3.1. Bioinformatics Prediction Tools
2.3.2. 3D Modeling of Pendrin Molecule Structure
2.4. Statistical Methods
3. Results
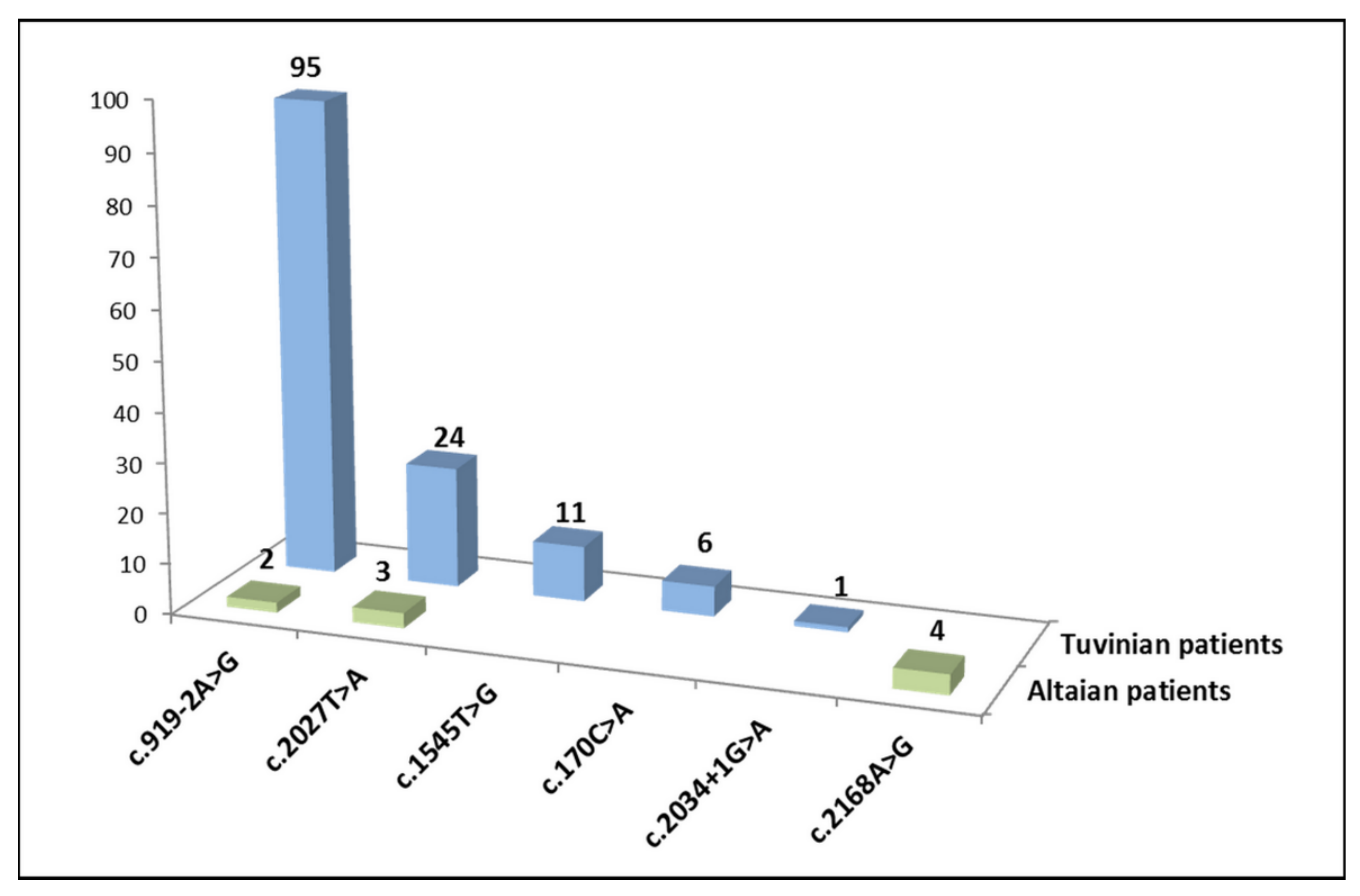
3.1. SLC26A4 Genotypes of Patients

{kind=link}
{kind=link}
{kind=link}
| SLC26A4 Genotypes | Tuvinian Patents (n = 220) | Altaian Patents (n = 93) | ||
|---|---|---|---|---|
| Homozygotes | ||||
| 1 | c.[919-2A>G];[919-2A>G] p.[splice acceptor variant];[splice acceptor variant] | intronic region between exons 7 and 8 | 30 | - |
| 2 | c.[2027T>A];[2027T>A] p.[Leu676Gln];[Leu676Gln] | exon 17 | 4 | - |
| 3 | c.[2168A>G];[2168A>G] p.[His723Arg];[His723Arg] | exon 19 | - | 2 |
| 4 | c.[170C>A];[170C>A] p.[Ser57Ter];[Ser57Ter] | exon 3 | 1 | - |
| Total | 35 | 2 | ||
| Compound heterozygotes | ||||
| 5 | c.[919-2A>G];[2027T>A] p.[splice acceptor variant];[Leu676Gln] | intronic region between exons 7 and 8/exon 17 | 14 | 2 |
| 6 | c.[919-2A>G];[1545T>G] * p.[splice acceptor variant];[Phe515Leu] * | intronic region between exons 7 and 8/exon 14 | 8 | - |
| 7 | c.[170C>A];[919-2A>G] p.[Ser57Ter];[splice acceptor variant] | exon 3/intronic region between exons 7 and 8 | 3 | - |
| 8 | c.[919-2A>G];[2034+1G>A] p.[splice acceptor variant];[splice donor variant] | intronic region between exons 7 and 8/intronic region between exons 17 and 18 | 1 | - |
| 9 | c.[1545T>G] *;[2027T>A] p.[Phe515Leu] *;[Leu676Gln] | exons 14/17 | 1 | - |
| Total | 27 | 2 | ||
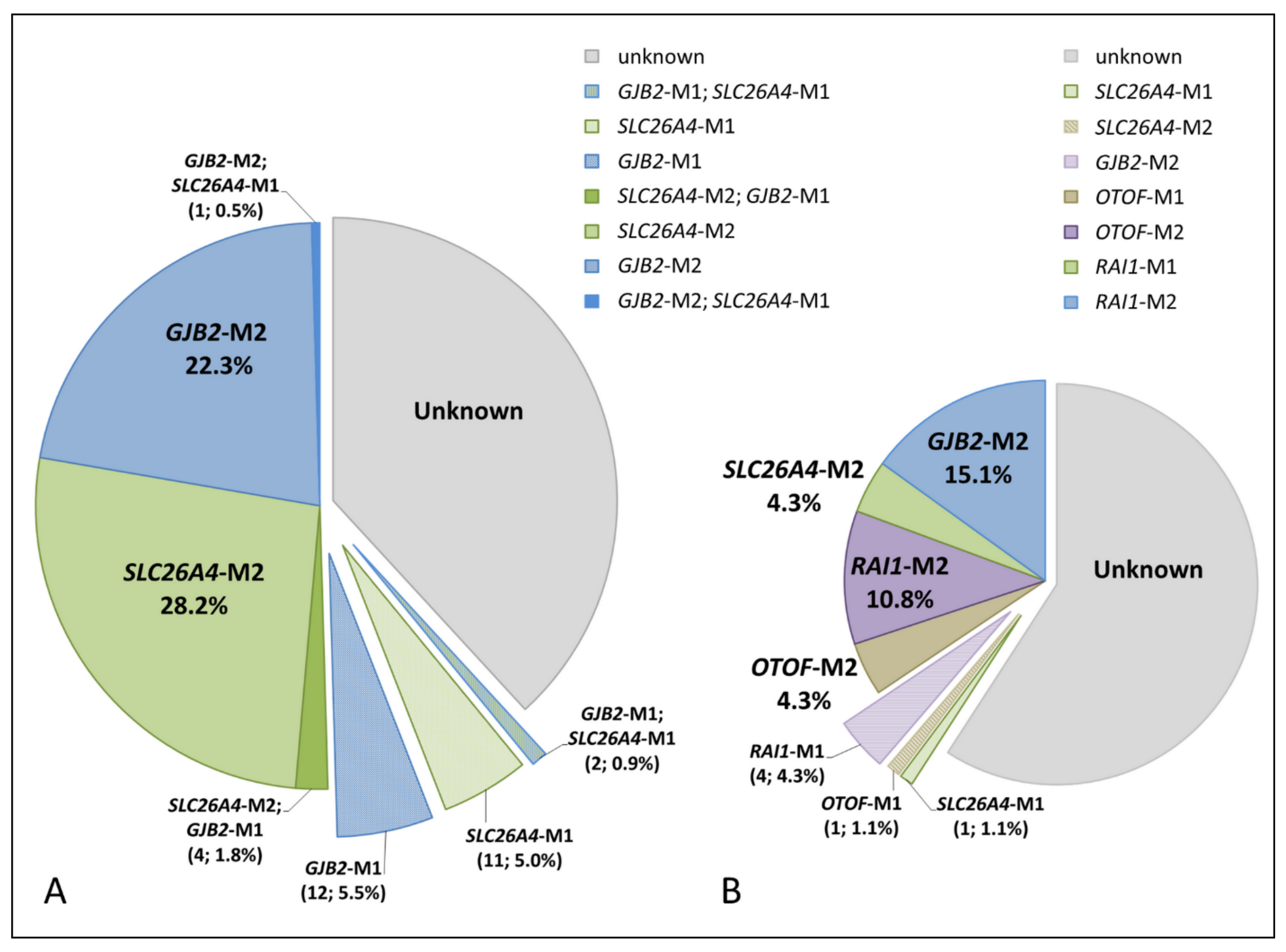
| Biallelic SLC26A4 mutations in total | 62 (28.2%) | 4 (4.3%) | ||
| Single heterozygotes | ||||
| 10 | c.[919-2A>G];[?] p.[splice acceptor variant];[?] | intronic region between exons 7 and 8 | 9 | - |
| 11 | c.[1545T>G] *;[?] p.[Phe515Leu] *;[?] | exon 14 | 2 | - |
| 12 | c.[170C>A];[?] p.[Ser57Ter];[?] | exon 3 | 1 | - |
| 13 | c.[2027T>A];[?] p.[Leu676Gln];[?] | exon 17 | 1 | 1 |
| Total | 13 (5.9%) | 1 (1.1%) | ||
| SLC26A4 Variants | Location | Molecular Consequence | dbSNP ID | ClinVar (2021) | ||
|---|---|---|---|---|---|---|
| Nucleotide | Amino Acid | |||||
| 1 | c.170C>A | p.Ser57Ter | exon 3 | nonsense variant | rs111033200 | pathogenic |
| 2 | c.919-2A>G | splice acceptor variant | intronic region between exons 7 and 8 | splice acceptor | rs111033313 | pathogenic |
| 3 | c.1545T>G * | p.Phe515Leu | exon 14 | missense variant | not presented | not presented |
| 4 | c.2027T>A | p.Leu676Gln | exon 17 | missense variant | rs111033318 | pathogenic/likely pathogenic |
| 5 | c.2034+1G>A | splice donor variant | intronic region between exons 17 and 18 | splice donor | rs759683649 | likely pathogenic |
| 6 | c.2168A>G | p.His723Arg | exon 19 | missense variant | rs121908362 | pathogenic/likely pathogenic |
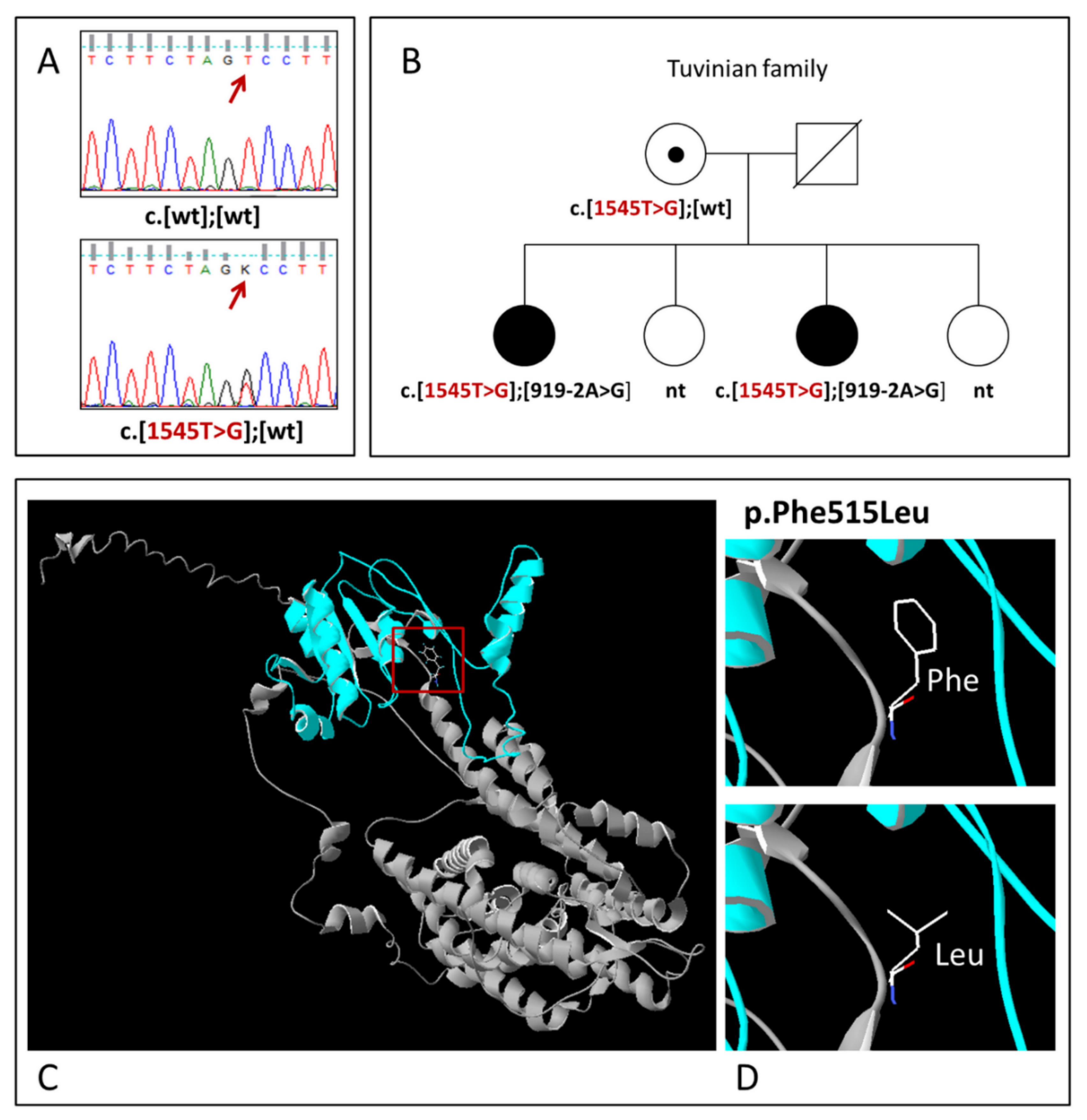
3.2. Novel SLC26A4 Variant c.1545T>G (p.Phe515Leu)

3.3. Carrier Frequency of Pathogenic SLC26A4 Variants in Tuvinian and Altaian Control Samples
3.4. Computed Tomography (CT) of the Temporal Bones in Tuvinian Patients
4. Discussion
4.1. The SLC26A4-Related HL in Tuvinian and Altaian Patients
4.2. Pathogenic SLC26A4 Variants in Tuvinians and Altaians
4.3. Comparative Analysis of Genetic Causes of HL in Tuvinian and Altaian Patients
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/en/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 1 April 2021).
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef]
- Van Camp, G.; Smith, R.J.H. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org (accessed on 4 October 2021).
- MITOMAP: A Human Mitochondrial Genome Database. 2019. Available online: http://www.mitomap.org (accessed on 1 November 2021).
- Del Castillo, F.J.; del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Shaukat, S.; Liu, X.Z.; Hahn, S.H.; Naz, S.; Ghosh, M.; Kim, H.N.; Moon, S.K.; Abe, S.; Tukamoto, K.; et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: Global implications for the epidemiology of deafness. J. Med. Genet. 2003, 40, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, S.; Blons, H.; Jonard, L.; Feldmann, D.; Chauvin, P.; Loundon, N.; Sergent-Allaoui, A.; Houang, M.; Joannard, A.; Schmerber, S.; et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur. J. Hum. Genet. 2006, 14, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.J.; Zhao, Y.L.; Rao, S.Q.; Guo, Y.F.; Yuan, H.; Zong, L.; Guan, J.; Xu, B.C.; Wang, D.Y.; Han, M.K.; et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin. Genet. 2007, 72, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, M.; Nishio, S.Y.; Usami, S. Deafness Gene Study Consortium. Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: A large cohort study. J. Hum. Genet. 2014, 59, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, K.; Nishio, S.Y.; Hattori, M.; Usami, S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. S1), 61S–76S. [Google Scholar] [CrossRef]
- Erdenechuluun, J.; Lin, Y.-H.; Ganbat, K.; Bataakhuu, D.; Makhbal, Z.; Tsai, C.-Y.; Lin, Y.-H.; Chan, Y.-H.; Hsu, C.-J.; Hsu, W.-C.; et al. Unique spectra of deafness-associated mutations in Mongolians provide insights into the genetic relationships among Eurasian populations. PLoS ONE 2018, 13, e0209797. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Tsai, C.Y.; Lin, Y.H.; Chen, P.Y.; Lin, P.H.; Cheng, Y.F.; Wu, C.M.; Lin, Y.H.; Lee, C.Y.; Erdenechuluun, J.; et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes 2019, 10, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, L.A.; Glaser, B.; Beck, J.C.; Idol, J.R.; Buchs, A.; Heyman, M.; Adawi, F.; Hazani, E.; Nassir, E.; Baxevanis, A.D.; et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat. Genet. 1997, 17, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Everett, L.A.; Morsli, H.; Wu, D.K.; Green, E.D. Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc. Natl. Acad. Sci. USA 1999, 96, 9727–9732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mount, D.B.; Romero, M.F. The SLC26 gene family of multifunctional anion exchangers. Pflug. Arch. 2004, 447, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Griffith, A.J. Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss. Hum. Genet. 2021. [Google Scholar] [CrossRef]
- Valvassori, G.E.; Clemis, J.D. The large vestibular aqueduct syndrome. Laryngoscope 1978, 88, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Boston, M.; Halsted, M.; Meinzen-Derr, J.; Bean, J.; Vijayasekaran, S.; Arjmand, E.; Choo, D.; Benton, C.; Greinwald, J. The large vestibular aqueduct: A new definition based on audiologic and computed tomography correlation. Otolaryngol. Head Neck Surg. 2007, 136, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Vijayasekaran, S.; Halsted, M.J.; Boston, M.; Meinzen-Derr, J.; Bardo, D.M.; Greinwald, J.; Benton, C. When is the vestibular aqueduct enlarged? A statistical analysis of the normative distribution of vestibular aqueduct size. AJNR Am. J. Neuroradiol. 2007, 28, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Dror, A.A.; Brownstein, Z.; Avraham, K.B. Integration of human and mouse genetics reveals pendrin function in hearing and deafness. Cell Physiol. Biochem. 2011, 28, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rasp, G.; Sarikas, A.; Dossena, S. Genetic Determinants of Non-Syndromic Enlarged Vestibular Aqueduct: A Review. Audiol. Res. 2021, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Hutchin, T.; Coy, N.N.; Conlon, H.; Telford, E.; Bromelow, K.; Blaydon, D.; Taylor, G.; Coghill, E.; Brown, S.; Trembath, R.; et al. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK—Implications for genetic testing. Clin. Genet. 2005, 68, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Pourová, R.; Janousek, P.; Jurovcík, M.; Dvoráková, M.; Malíková, M.; Rasková, D.; Bendová, O.; Leonardi, E.; Murgia, A.; Kabelka, Z.; et al. Spectrum and frequency of SLC26A4 mutations among Czech patients with early hearing loss with and without Enlarged Vestibular Aqueduct (EVA). Ann. Hum. Genet. 2010, 74, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Nonose, R.W.; Lezirovitz, K.; de Mello Auricchio, M.T.B.; Batissoco, A.C.; Yamamoto, G.L.; Mingroni-Netto, R.C. Mutation analysis of SLC26A4 (Pendrin) gene in a Brazilian sample of hearing-impaired subjects. BMC Med. Genet. 2018, 19, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koohiyan, M. A systematic review of SLC26A4 mutations causing hearing loss in the Iranian population. Int. J. Pediatr. Otorhinolaryngol. 2019, 125, 1–5. [Google Scholar] [CrossRef]
- Anwar, S.; Riazuddin, S.; Ahmed, Z.M.; Tasneem, S.; Ateeq-ul-Jaleel; Khan, S.Y.; Griffith, A.J.; Friedman, T.B.; Riazuddin, S. SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred’s syndrome in Pakistanis. J. Hum. Genet. 2009, 54, 266–270. [Google Scholar] [CrossRef]
- Chandru, J.; Jeffrey, J.M.; Pavithra, A.; Vanniya, S.P.; Devi, G.N.; Mahalingam, S.; Karthikeyen, N.P.; Srisailapathy, C.R.S. Genetic analysis of SLC26A4 gene (pendrin) related deafness among a cohort of assortative mating families from southern India. Eur. Arch. Otorhinolaryngol. 2020, 277, 3021–3035. [Google Scholar] [CrossRef]
- Han, J.J.; Nguyen, P.D.; Oh, D.Y.; Han, J.H.; Kim, A.R.; Kim, M.Y.; Park, H.R.; Tran, L.H.; Dung, N.H.; Koo, J.W.; et al. Elucidation of the unique mutation spectrum of severe hearing loss in a Vietnamese pediatric population. Sci. Rep. 2019, 9, 1604. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.F.; Liu, X.W.; Guan, J.; Han, M.K.; Wang, D.Y.; Zhao, Y.L.; Rao, S.Q.; Wang, Q.J. GJB2, SLC26A4 and mitochondrial DNA A1555G mutations in prelingual deafness in Northern Chinese subjects. Acta Otolaryngol. 2008, 128, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Huang, Z.; Tao, Z.; Li, X.; Li, L.; Li, Y.; Wu, H.; Yang, T. Molecular etiology of hearing impairment associated with nonsyndromic enlarged vestibular aqueduct in East China. Am. J. Med. Genet. A 2013, 161A, 2226–2233. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.B.; Tang, S.H.; Li, H.Z.; Xu, C.Y.; Chen, C.; Xu, Y.Z.; Ding, L.R.; Xu, X.Q. Mutation analysis of common deafness-causing genes among 506 patients with nonsyndromic hearing loss from Wenzhou city, China. Int. J. Pediatr. Otorhinolaryngol. 2019, 122, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Han, Y.; Zhang, F.; Bai, X.; Wang, H. Mutation spectrum and hotspots of the common deafness genes in 314 patients with nonsyndromic hearing loss in Heze area, China. Acta Otolaryngol. 2019, 139, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Chai, Y.; Chen, P.; He, L.; Wang, X.; Wu, H.; Yang, T. Mono-allelic mutations of SLC26A4 is over-presented in deaf patients with non-syndromic enlarged vestibular aqueduct. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1351–1353. [Google Scholar] [CrossRef]
- Rah, Y.C.; Kim, A.R.; Koo, J.W.; Lee, J.H.; Oh, S.H.; Choi, B.Y. Audiologic presentation of enlargement of the vestibular aqueduct according to the SLC26A4 genotypes. Laryngoscope 2015, 125, E216–E222. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, H.; Liu, D.; Zhang, J.; Yang, Z.; Zhang, S.; Liu, H.; Li, R.; Tian, Y.; Zeng, B.; et al. Increased diagnosis of enlarged vestibular aqueduct by multiplex PCR enrichment and next-generation sequencing of the SLC26A4 gene. Mol. Genet. Genom. Med. 2021, 9, e1734. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Guo, Y.; Wang, C.; Wang, Y.; Liu, X. A systematic review and meta-analysis of common mutations of SLC26A4 gene in Asian populations. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.J.; Yao, J.; Wei, Q.J.; Xing, G.Q.; Cao, X. Diagnostic Value of SLC26A4 Mutation Status in Hereditary Hearing Loss With EVA: A PRISMA-Compliant Meta-Analysis. Medicine 2015, 94, e2248. [Google Scholar] [CrossRef]
- Mongush, M.V. Tuvans of Mongolia and China. Int. J. Cent. Asian Stud. 1996, 1, 225–243. [Google Scholar]
- Chen, Z.; Zhang, Y.; Fan, A.; Zhang, Y.; Wu, Y.; Zhao, Q.; Zhou, Y.; Zhou, C.; Bawudong, M.; Mao, X.; et al. Brief communication: Y-chromosome haplogroup analysis indicates that Chinese Tuvans share distinctive affinity with Siberian Tuvans. Am. J. Phys. Anthropol. 2011, 144, 492–497. [Google Scholar] [CrossRef]
- Vainshtein, S.I.; Mannay-Ool, M.H. History of Tyva, 2nd ed.; Science: Novosibirsk, Russia, 2001. (In Russian) [Google Scholar]
- Mannai-ool, M.K. Tuvan People. The Origin and Formation of the Ethnos; Nauka Publ.: Novosibirsk, Russia, 2004; pp. 99–166. (In Russian) [Google Scholar]
- Potapov, L.P. Ethnical Structure and Origin of Altaians; Nauka: Leningrad, Russia, 1969. (In Russian) [Google Scholar]
- Posukh, O.; Pallares-Ruiz, N.; Tadinova, V.; Osipova, L.; Claustres, M.; Roux, A.-F. First molecular screening of deafness in the Altai Republic population. BMC Med. Genet. 2005, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzhemileva, L.U.; Posukh, O.L.; Tazetdinov, A.M.; Barashkov, N.A.; Zhuravskiĭ, S.G.; Ponidelko, S.N.; Markova, T.G.; Tadinova, V.N.; Fedorova, S.A.; Maksimova, N.R.; et al. Analysis of mitochondrial 12S rRNA and tRNA(Ser(UCN)) genes in patients with nonsyndromic sensorineural hearing loss from various regions of Russia. Genetika 2009, 45, 982–991. (In Russian) [Google Scholar] [CrossRef]
- Churbanov, A.Y.; Karafet, T.M.; Morozov, I.V.; Mikhalskaia, V.Y.; Zytsar, M.V.; Bondar, A.A.; Posukh, O.L. Whole Exome Sequencing Reveals Homozygous Mutations in RAI1, OTOF, and SLC26A4 Genes Associated with Nonsyndromic Hearing Loss in Altaian Families (South Siberia). PLoS ONE 2016, 11, e0153841. [Google Scholar] [CrossRef] [Green Version]
- Posukh, O.L.; Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Bondar, A.A.; Morozov, I.V.; Maximov, V.N.; Voevoda, M.I. Unique mutational spectrum of the GJB2 Gene and its pathogenic contribution to deafness in Tuvinians (Southern Siberia, Russia): A high prevalence of rare variant c.516G>C (p.Trp172Cys). Genes 2019, 10, 429. [Google Scholar] [CrossRef] [Green Version]
- Posukh, O.L.; (Institute of Cytology and Genetics, Novosibirsk, Russia). Personal communication, 2019.
- Danilchenko, V.Y.; (Institute of Cytology and Genetics, Novosibirsk, Russia). Personal communication, 2020.
- Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L. High Rates of Three Common GJB2 Mutations c.516G>C, c.-23+1G>A, c.235delC in Deaf Patients from Southern Siberia Are Due to the Founder Effect. Genes 2020, 11, 833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Dewan, K.; Wippold, F.J., 2nd; Lieu, J.E. Enlarged vestibular aqueduct in pediatric sensorineural hearing loss. Otolaryngol. Head Neck Surg. 2009, 140, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Coucke, P.J.; van Hauwe, P.; Everett, L.A.; Demirhan, O.; Kabakkaya, Y.; Dietrich, N.L.; Smith, R.J.; Coyle, E.; Reardon, W.; Trembath, R.; et al. Identification of two different mutations in the PDS gene in an inbred family with Pendred syndrome. J. Med. Genet. 1999, 36, 475–477. [Google Scholar]
- Dai, P.; Li, Q.; Huang, D.; Yuan, Y.; Kang, D.; Miller, D.T.; Shao, H.; Zhu, Q.; He, J.; Yu, F.; et al. SLC26A4 c.919-2A>G varies among Chinese ethnic groups as a cause of hearing loss. Genet. Med. 2008, 10, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hauwe, P.; Everett, L.A.; Coucke, P.; Scott, D.A.; Kraft, M.L.; Ris-Stalpers, C.; Bolder, C.; Otten, B.; de Vijlder, J.J.; Dietrich, N.L.; et al. Two frequent missense mutations in Pendred syndrome. Hum. Mol. Genet. 1998, 7, 1099–1104. [Google Scholar] [CrossRef] [Green Version]
- Usami, S.; Abe, S.; Weston, M.D.; Shinkawa, H.; van Camp, G.; Kimberling, W.J. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum. Genet. 1999, 104, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Lee, S.C.; Lee, H.K.; Park, H.J. Genetic screening of GJB2 and SLC26A4 in Korean cochlear implantees: Experience of Soree ear clinic. Clin. Exp. Otorhinolaryngol. 2012, 5 (Suppl. S1), S10–S13. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Yeh, T.H.; Chen, P.J.; Hsu, C.J. Prevalent SLC26A4 mutations in patients with enlarged vestibular aqueduct and/or Mondini dysplasia: A unique spectrum of mutations in Taiwan, including a frequent founder mutation. Laryngoscope 2005, 115, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Gillam, M.P.; Sidhaye, A.R.; Lee, E.J.; Rutishauser, J.; Stephan, C.W.; Kopp, P. Functional characterization of pendrin in a polarized cell system. Evidence for pendrin-mediated apical iodide efflux. J. Biol. Chem. 2004, 279, 13004–13010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.S.; Park, H.J.; Yoo, S.Y.; Namkung, W.; Jo, M.J.; Koo, S.K.; Park, H.Y.; Lee, W.S.; Kim, K.H.; Lee, M.G. Heterogeneity in the processing defect of SLC26A4 mutants. J. Med. Genet. 2008, 45, 411–419. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, J.; Shin, J.W.; Song, M.H.; Kim, S.H.; Lee, J.H.; Lee, K.A.; Shin, S.; Kim, U.K.; Bok, J.; et al. Correlation between genotype and phenotype in patients with bi-allelic SLC26A4 mutations. Clin. Genet. 2014, 86, 270–275. [Google Scholar] [CrossRef]
- Zhao, J.; Yuan, Y.; Huang, S.; Huang, B.; Cheng, J.; Kang, D.; Wang, G.; Han, D.; Dai, P. KCNJ10 may not be a contributor to nonsyndromic enlargement of vestibular aqueduct (NSEVA) in Chinese subjects. PLoS ONE 2014, 9, e108134. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.R.; Bashir, R.; Naz, S. SLC26A4 mutations in patients with moderate to severe hearing loss. Biochem. Genet. 2013, 51, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cengiz, F.B.; Yilmazer, R.; Olgun, L.; Sennaroglu, L.; Kirazli, T.; Alper, H.; Olgun, Y.; Incesulu, A.; Atik, T.; Huesca-Hernandez, F.; et al. Novel pathogenic variants underlie SLC26A4-related hearing loss in a multiethnic cohort. Int. J. Pediatr. Otorhinolaryngol. 2017, 101, 167–171. [Google Scholar] [CrossRef]
- Uzumcu, A.; Uyguner, O.; Ulubil-Emiroglu, M.; Hafiz, G.; Baserer, N.; Eris, H.; Basaran, S.; Wollnik, B. Compound heterozygosity for novel and known mutations in SLC26A4 cause large vestibular aqueduct. Balkan J. Med. Genet. 2006, 9, 105. [Google Scholar]
- Pryor, S.P.; Madeo, A.C.; Reynolds, J.C.; Sarlis, N.J.; Arnos, K.S.; Nance, W.E.; Yang, Y.; Zalewski, C.K.; Brewer, C.C.; Butman, J.A.; et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): Evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J. Med. Genet. 2005, 42, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pique, L.M.; Brennan, M.L.; Davidson, C.J.; Schaefer, F.; Greinwald, J., Jr.; Schrijver, I. Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. PeerJ 2014, 2, e384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, J.J.; de Bruijn, S.E.; Lanting, C.P.; Oostrik, J.; O’Gorman, L.; Mantere, T.; DOOFNL Consortium; Cremers, F.P.M.; Roosing, S.; Yntema, H.G.; et al. Exploring the missing heritability in subjects with hearing loss, enlarged vestibular aqueducts, and a single or no pathogenic SLC26A4 variant. Hum. Genet. 2021. [Google Scholar] [CrossRef]


| Pathogenic SLC26A4 Variants | Tuvinian Control Sample | Altaian Control Sample |
|---|---|---|
| c.919-2A>G | 5.1% (8/157) | nt |
| c.1545T>G | 2.0% (3/148) | nt |
| c.170C>A | 0% (0/100) | nt |
| c.2027T>A | 0% (0/157) | 0% (0/123) |
| c.2034+1G>A | 0% (0/157) | 0% (0/123) |
| c.2168A>G | nt | 0% (0/141) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danilchenko, V.Y.; Zytsar, M.V.; Maslova, E.A.; Bady-Khoo, M.S.; Barashkov, N.A.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L. Different Rates of the SLC26A4-Related Hearing Loss in Two Indigenous Peoples of Southern Siberia (Russia). Diagnostics 2021, 11, 2378. https://doi.org/10.3390/diagnostics11122378
Danilchenko VY, Zytsar MV, Maslova EA, Bady-Khoo MS, Barashkov NA, Morozov IV, Bondar AA, Posukh OL. Different Rates of the SLC26A4-Related Hearing Loss in Two Indigenous Peoples of Southern Siberia (Russia). Diagnostics. 2021; 11(12):2378. https://doi.org/10.3390/diagnostics11122378
Chicago/Turabian StyleDanilchenko, Valeriia Yu., Marina V. Zytsar, Ekaterina A. Maslova, Marita S. Bady-Khoo, Nikolay A. Barashkov, Igor V. Morozov, Alexander A. Bondar, and Olga L. Posukh. 2021. "Different Rates of the SLC26A4-Related Hearing Loss in Two Indigenous Peoples of Southern Siberia (Russia)" Diagnostics 11, no. 12: 2378. https://doi.org/10.3390/diagnostics11122378