
Development and Validation of a Targeted ‘Liquid’ NGS Panel for Treatment Customization in Patients with Metastatic Colorectal Cancer

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Enrollment and Data Collection
2.2. Sampling of Blood and Plasma Preparation
2.3. cfDNA Isolation and Quantitation
2.4. Library Preparation and Targeted NGS
2.5. NGS Data Analysis
2.6. BEAMing Digital PCR
2.7. Statistics
3. Results
3.1. Study Population
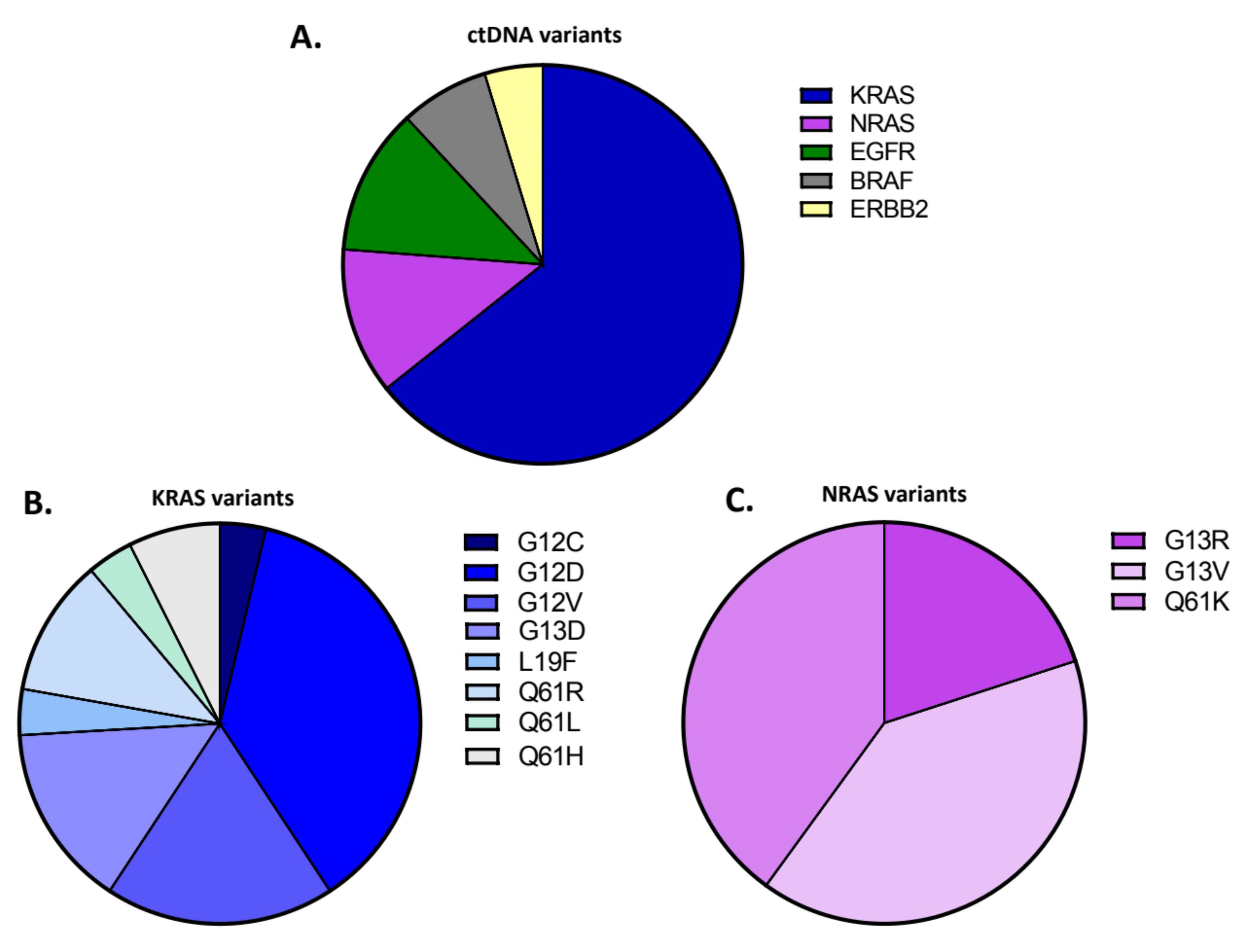
3.2. Development, Application and Evaluation of the NGS Assay
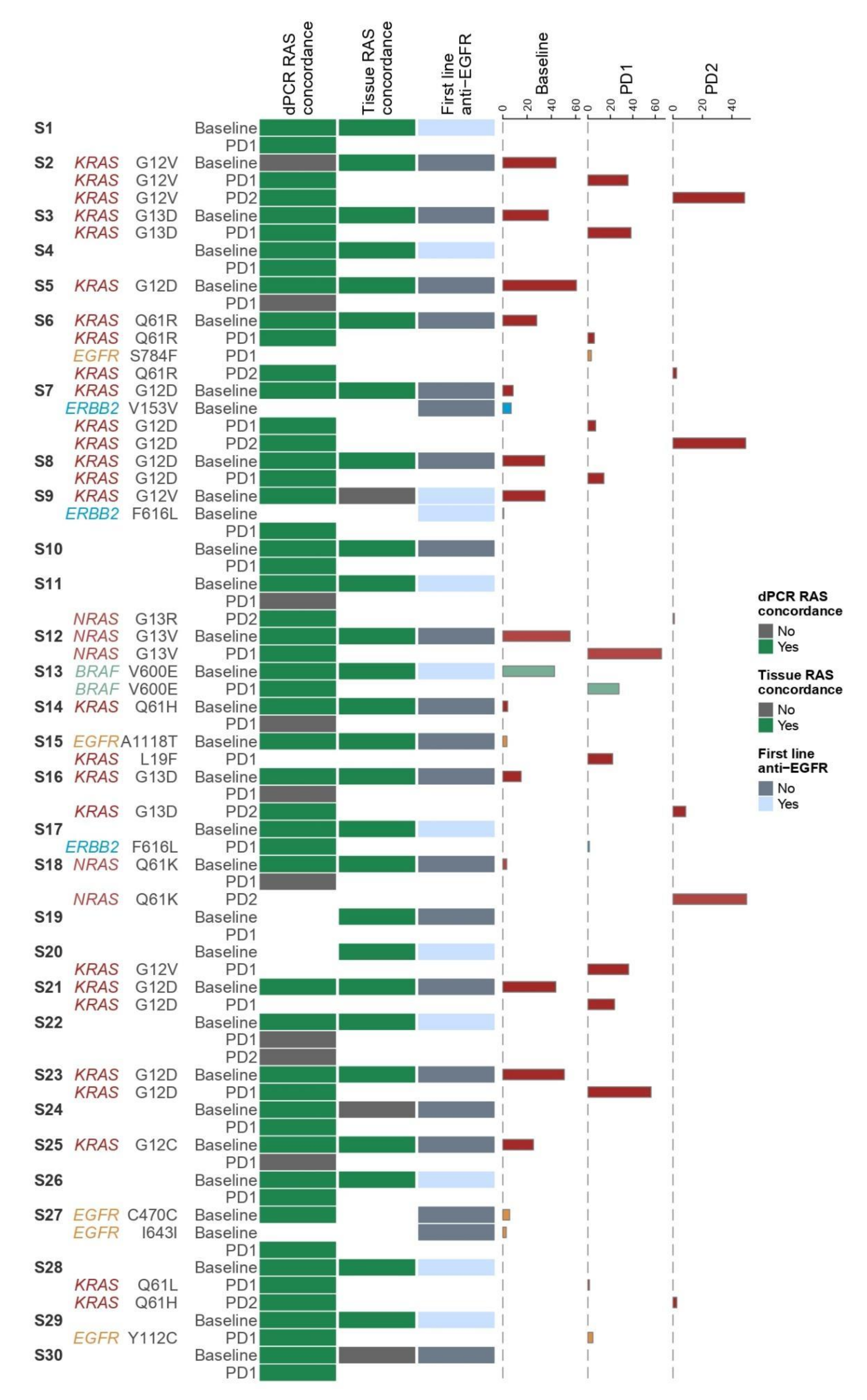
3.3. Validation of the NGS Plasma Assay by BEAMing dPCR
3.4. Concordance Analysis of RAS Testing in Tissue and Plasma
3.5. Association of Clinical Parameters with ctDNA
3.6. Cost and Turn-around Time of NGS Assay versus dPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Erratum: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2020, 70, 313. [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Foncillas, J.; Alba, E.; Aranda, E.; Díaz-Rubio, E.; López-López, R.; Tabernero, J.; Vivancos, A. Incorporating BEAMing technology as a liquid biopsy into clinical practice for the management of colorectal cancer patients: An expert taskforce review. Ann. Oncol. 2017, 28, 2943–2949. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; van Krieken, J.H.; Aderka, D.; Aguilar, E.A.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Final results from PRIME: Randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann. Oncol. 2014, 25, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Li, J.; Wang, L.; Xu, J.; Cheng, Y.; Bai, Y.; Li, W.; Xu, N.; Lin, L.-Z.; Wu, Q.; et al. Efficacy and Tolerability of First Line Cetuximab Plus Leucovorin, Fluorouracil, and Oxaliplatin (FOLFOX-4) versus FOLFOX-4 in Patients with RAS Wild-Type Metastatic Colorectal Cancer: The Open-Label, Randomized, Phase III TAILOR Trial. J. Clin. Oncol. 2018, 36, 3031–3039. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chien, C.-R.C.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Dias, M.M.; Wiese, M.; Kichenadasse, G.; McKinnon, R.; Karapetis, C.; Sorich, M. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br. J. Cancer 2015, 112, 1888–1894. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Wang, C.; Fakih, M. Targeting KRAS in Colorectal Cancer. Curr. Oncol. Rep. 2021, 23, 28. [Google Scholar] [CrossRef]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, A.; Cañadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef] [Green Version]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; A Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef]
- Morelli, M.P.; Overman, M.J.; Dasari, A.; Kazmi, S.M.A.; Mazard, T.; Vilar, E.; Morris, V.K.; Lee, M.S.; Herron, D.; Eng, C.; et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann. Oncol. 2015, 26, 731–736. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Emburgh, B.O.; Arena, S.; Siravegna, G.; Lazzari, L.; Crisafulli, G.; Corti, G.; Mussolin, B.; Baldi, F.; Buscarino, M.; Bartolini, A.; et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 2016, 7, 13665. [Google Scholar] [CrossRef] [PubMed]
- Pectasides, E.; Bass, A.J. ERBB2 emerges as a new target for colorectal cancer. Cancer Discov. 2015, 5, 799–801. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Jauhri, M.; Bhatnagar, A.; Gupta, S.; Shokeen, Y.; Minhas, S.; Aggarwal, S. Targeted molecular profiling of rare genetic alterations in colorectal cancer using next-generation sequencing. Med. Oncol. 2016, 33, 106. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Sartore-Bianchi, A.; Marsoni, S.; Hurwitz, H.; McCall, S.; Penault-Llorca, F.; Srock, S.; Bardelli, A.; Trusolino, L. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann. Oncol. 2018, 29, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Loree, J.M.; Bailey, A.M.; Johnson, A.M.; Yu, Y.; Wu, W.; Bristow, C.A.; Davis, J.S.; Shaw, K.R.; Broaddus, R.; Banks, K.C.; et al. Molecular Landscape of ERBB2/ERBB3 Mutated Colorectal Cancer. J. Natl. Cancer Inst. 2018, 110, 1409–1417. [Google Scholar] [CrossRef] [Green Version]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet. Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wildtype, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet. Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Strickler, J.H.; Zemla, T.; Ou, F.-S.; Cercek, A.; Wu, C.; Sanchez, F.; Hubbard, J.; Jaszewski, B.; Bandel, L.; Schweitzer, B.; et al. Trastuzumab and tucatinib for the treatment of HER2 amplified metastatic colorectal cancer (mCRC): Initial results from the MOUNTAINEER trial. Ann. Oncol. 2019, 30, v200. [Google Scholar] [CrossRef]
- Kavuri, S.M.; Jain, N.; Galimi, F.; Cottino, F.; Leto, S.M.; Migliardi, G.; Searleman, A.; Shen, W.; Monsey, J.D.; Trusolino, L.; et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015, 5, 832–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Li, G.; Sun, X.; Ni, S.; Tan, C.; Xu, M.; Huang, D.; Ren, F.; Li, D.; Wei, P.; et al. MET amplification, expression, and exon 14 mutations in colorectal adenocarcinoma. Hum. Pathol. 2018, 77, 108–115. [Google Scholar] [CrossRef]
- Pilotto, S.; Gkountakos, A.; Carbognin, L.; Scarpa, A.; Tortora, G.; Bria, E. MET exon 14 juxtamembrane splicing mutations: Clinical and therapeutical perspectives for cancer therapy. Ann. Transl. Med. 2017, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chang, S.; Li, G.; Sun, Y. Application of liquid biopsy in precision medicine: Opportunities and challenges. Front. Med. 2017, 11, 522–527. [Google Scholar] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, A.; Morris, V.K.; Allegra, C.J.; Atreya, C.; Benson, A.B., 3rd; Boland, P.; Chung, K.; Copur, M.S.; Corcoran, R.B.; Deming, D.A.; et al. ctDNA applications and integration in colorectal cancer: An NCI Colon and Rectal-Anal Task Forces whitepaper. Nat. Rev. Clin. Oncol. 2020, 17, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [Green Version]
- Volckmar, A.L.; Sültmann, H.; Riediger, A.; Fioretos, T.; Schirmacher, T.; Endris, V.; Stenzinger, A.; Dietz, S. A field guide for cancer diagnostics using cell-free DNA: From principles to practice and clinical applications. Genes Chromosomes Cancer 2018, 57, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Du, M.; Wang, L. Bioinformatics Analysis for Circulating Cell-Free DNA in Cancer. Cancers 2019, 11, 805. [Google Scholar] [CrossRef] [Green Version]
- Kivioja, T.; Vähärautio, A.; Karlsson, K.; Bonke, M.; Enge, M.; Linnarsson, S.; Taipale, J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 2011, 9, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Houtgast, E.J.; Sima, V.-M.; Bertels, K.; Al-Ars, Z. Hardware acceleration of BWA-MEM genomic short read mapping for longer read lengths. Comput. Biol. Chem. 2018, 75, 54–64. [Google Scholar] [CrossRef]
- Xu, C.; Gu, X.; Padmanabhan, R.; Wu, Z.; Peng, Q.; DiCarlo, J.; Wang, Y. smCounter2: An accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics 2019, 35, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Diehl, F.; Dressman, D.; Vogelstein, B.; Kinzler, K.W. BEAMing up for detection and quantification of rare sequence variants. Nat. Methods 2006, 3, 95–97. [Google Scholar] [CrossRef]
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loree, J.M.; Wang, Y.; Syed, M.A.; Sorokin, A.V.; Coker, O.; Xiu, J.; Weinberg, B.A.; Vanderwalde, A.M.; Tesfaye, A.; Raymond, V.M.; et al. Clinical and Functional Characterization of Atypical KRAS/NRAS Mutations in Metastatic Colorectal Cancer. Clin. Cancer Res. 2021, 27, 4587–4598. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Yang, Y.; Huang, H.; Kuang, X.; Yang, X. HER2-amplified metastatic lung adenocarcinoma responds to fourth-line pyrotinib therapy: A case report. Mol. Clin. Oncol. 2021, 15, 213. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wildtype BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef]
- Rad, R.; Cadiñanos, J.; Rad, L.; Varela, I.; Strong, A.; Kriegl, L.; Constantino-Casas, F.; Eser, S.; Hieber, M.; Seidler, B.; et al. A genetic progression model of Braf (V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell 2013, 24, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Elez, E.; Chianese, C.; Sanz-García, E.; Martinelli, E.; Noguerido, A.; Mancuso, F.; Caratù, G.; Matito, J.; Grasselli, J.; Cardone, C.; et al. Impact of circulating tumor DNA mutant allele fraction on prognosis in RAS-mutant metastatic colorectal cancer. Mol. Oncol. 2019, 13, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- IjzermJan, M.; Boer, J.; Azad, A.; Degeling, K.; Geoghegan, J.; Hewitt, C.; Hollande, F.; Lee, B.; To, Y.; Tothill, R.; et al. Towards Routine Implementation of Liquid Biopsies in Cancer Management: It Is Always Too Early, until Suddenly It Is Too Late. Diagnostics 2021, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front. Oncol. 2019, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- Galvano, A.; Taverna, S.; Badalamenti, G.; Incorvaia, L.; Castiglia, M.; Barraco, N.; Passiglia, F.; Fulfaro, F.; Beretta, G.; Duro, G.; et al. Detection of RAS mutations in circulating tumor DNA: A new weapon in an old war against colorectal cancer. A systematic review of literature and meta-analysis. Adv. Med. Oncol. 2019, 11, 1758835919874653. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, H.; Kotani, D.; Bando, H.; Kato, T.; Oki, E.; Shinozaki, E.; Sunakawa, Y.; Yamazaki, K.; Yuki, S.; Nakamura, Y.; et al. REMARRY and PURSUIT trials: Liquid biopsy-guided rechallenge with anti-epidermal growth factor receptor (EGFR) therapy with panitumumab plus irinotecan for patients with plasma RAS wild-type metastatic colorectal cancer. BMC Cancer 2021, 21, 674. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Lenz, H.-J.; Köhne, C.-H.; Heinemann, V.; Tejpar, S.; Melezínek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Vidal, J.; Bellosillo, B.; Santos Vivas, C.; García-Alfonso, P.; Carrato, A.; Cano, M.T.; García-Carbonero, R.; Élez, E.; Losa, F.; Massutí, B.; et al. Ultra-selection of metastatic colorectal cancer patients using next generation sequencing to improve clinical efficacy of anti-EGFR therapy. Ann. Oncol. 2019, 30, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, J. Concordance of blood-and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2017, 28, 1294–1301. [Google Scholar] [CrossRef]
- Santos, C.; Azuara, D.; Garcia-Carbonero, R.; Alfonso, P.G.; Carrato, A.; Elez, M.E.; Gomez, A.; Losa, F.; Montagut, C.; Massuti, B.; et al. Optimization of RAS/BRAF Mutational Analysis Confirms Improvement in Patient Selection for Clinical Benefit to Anti-EGFR Treatment in Metastatic Colorectal Cancer. Mol. Cancer Ther. 2017, 16, 1999–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.; Kim, S.; Kang, J.-K.; Kim, H.-P.; Jang, H.; Han, H.; Kim, H.; Kim, M.J.; Lee, K.-H.; Ryoo, S.-B.; et al. Circulating tumor DNA sequencing in colorectal cancer patients treated with first-line chemotherapy with anti-EGFR. Sci. Rep. 2021, 11, 16333. [Google Scholar] [CrossRef]
- Awad, M.M.; Shengwu, L.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, I.; Ačkar, L.; Mossahebi Mohammadi, P.; Pantel, K. Liquid biopsies: Potential and challenges. Int. J. Cancer 2021, 148, 528–545. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | n (%) |
|---|---|
| Gender | |
| Male | 20 (67%) |
| Female | 10 (33%) |
| Age in years | |
| Mean | 68 |
| Range | 43–83 |
| Primary tumor location | |
| Right colon | 16 (53%) |
| Left colon/Rectum | 14 (47%) |
| First line chemotherapy | |
| Oxaliplatin-based doublet | 21 (70%) |
| Irinotecan-based doublet | 4 (13%) |
| Other 1 | 5 (17%) |
| First line targeted agent | |
| Anti-angiogenic | 15 (50%) |
| Anti-EGFR | 11 (37%) |
| None | 4 (13%) |
| Total | 30 (100%) |
| Gene | Exon(s) | Codon Position | Number of Hotspots |
|---|---|---|---|
| KRAS | 2 | 12, 13 | 2 |
| 3 | 59, 61 | 2 | |
| 4 | 117, 146 | 2 | |
| NRAS | 2 | 12, 13 | 2 |
| 3 | 59, 61 | 2 | |
| 4 | 117, 146 | 2 | |
| MET | 14 | 1003 | 1 |
| BRAF | 15 | 600 | 1 |
| ERBB2 | 8 | 309, 310 | 2 |
| 19 | 740, 755, 767, 769 | 4 | |
| 20 | 773, 776, 777 | 3 | |
| 21 | 824, 842, 874 | 3 | |
| EGFR | All exons (1–28) including 12 | 451, 464, 465, 467, 491, 492 | 6 |
| Plasma ctDNA RAS Status by dPCR | |||||||
|---|---|---|---|---|---|---|---|
| RAS | Mutant | WT | Total | PPA | NPA | OPA | |
| Plasma ctDNA RAS status by NGS | Mutant | 26 | 1 | 27 | 100 × 26/35 = 74.3% | 100 × 25/26 = 96.2% | 100 × 51/61 = 83.6% |
| WT | 9 | 25 | 34 | ||||
| Total | 35 | 26 | 61 | ||||
| Tissue RAS Status | |||||||
|---|---|---|---|---|---|---|---|
| RAS | Mutant | WT | Total | PPA | NPA | OPA | |
| Plasma ctDNA RAS status by NGS | Mutant | 13 | 1 | 14 | 100 × 13/16 = 81.2% | 100 × 13/14 = 92.8% | 100 × 26/30 = 86.7% |
| WT | 3 | 13 | 16 | ||||
| Total | 16 | 14 | 30 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kastrisiou, M.; Zarkavelis, G.; Kougioumtzi, A.; Sakaloglou, P.; Kostoulas, C.; Georgiou, I.; Batistatou, A.; Pentheroudakis, G.; Magklara, A. Development and Validation of a Targeted ‘Liquid’ NGS Panel for Treatment Customization in Patients with Metastatic Colorectal Cancer. Diagnostics 2021, 11, 2375. https://doi.org/10.3390/diagnostics11122375
Kastrisiou M, Zarkavelis G, Kougioumtzi A, Sakaloglou P, Kostoulas C, Georgiou I, Batistatou A, Pentheroudakis G, Magklara A. Development and Validation of a Targeted ‘Liquid’ NGS Panel for Treatment Customization in Patients with Metastatic Colorectal Cancer. Diagnostics. 2021; 11(12):2375. https://doi.org/10.3390/diagnostics11122375
Chicago/Turabian StyleKastrisiou, Myrto, George Zarkavelis, Anastasia Kougioumtzi, Prodromos Sakaloglou, Charilaos Kostoulas, Ioannis Georgiou, Anna Batistatou, George Pentheroudakis, and Angeliki Magklara. 2021. "Development and Validation of a Targeted ‘Liquid’ NGS Panel for Treatment Customization in Patients with Metastatic Colorectal Cancer" Diagnostics 11, no. 12: 2375. https://doi.org/10.3390/diagnostics11122375