Low-Grade Gliomas in Patients with Noonan Syndrome: Case-Based Review of the Literature


 , , , , ,
, , , , ,  , , , , ,
, , , , ,
Abstract
:1. Introduction
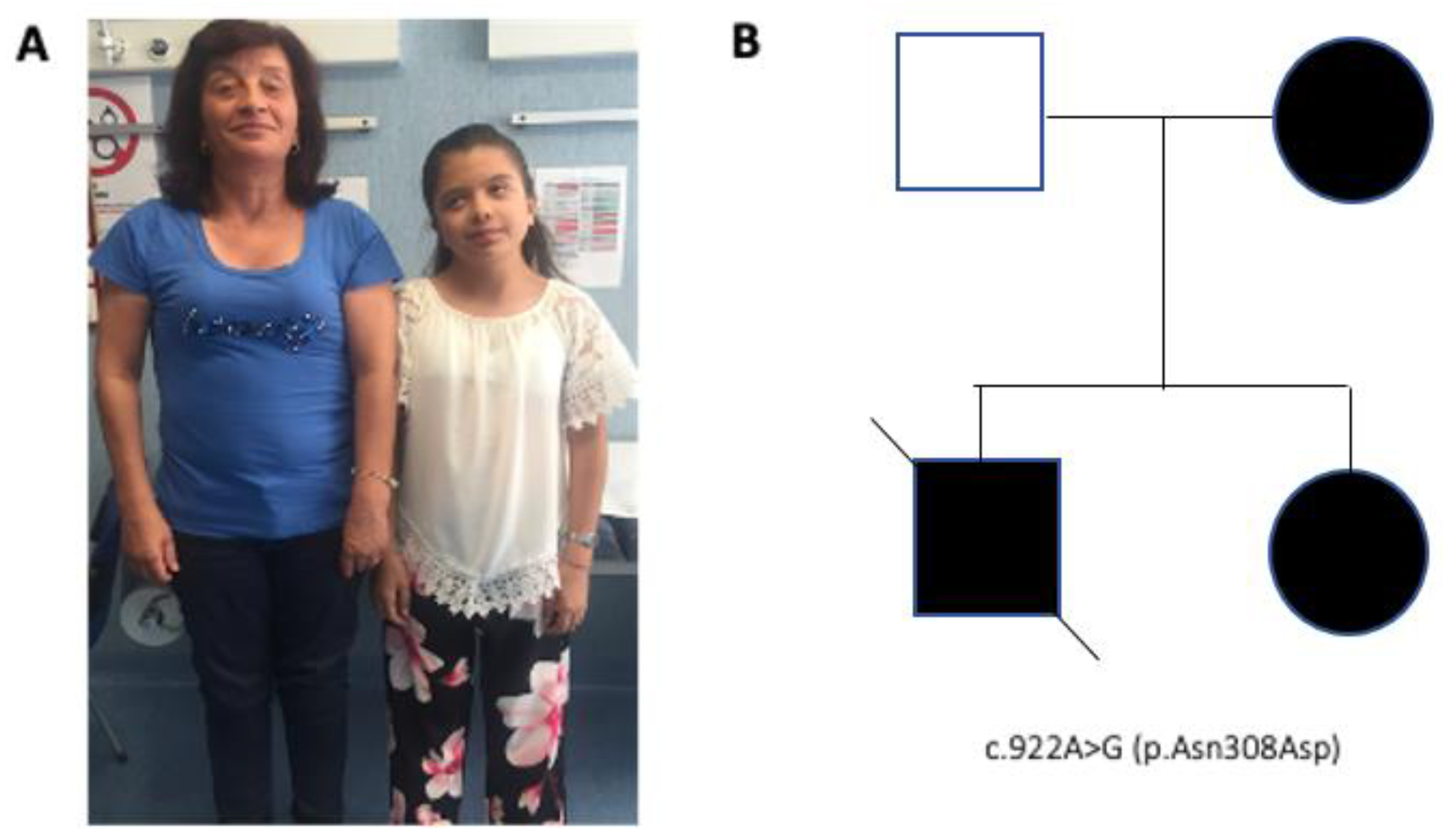
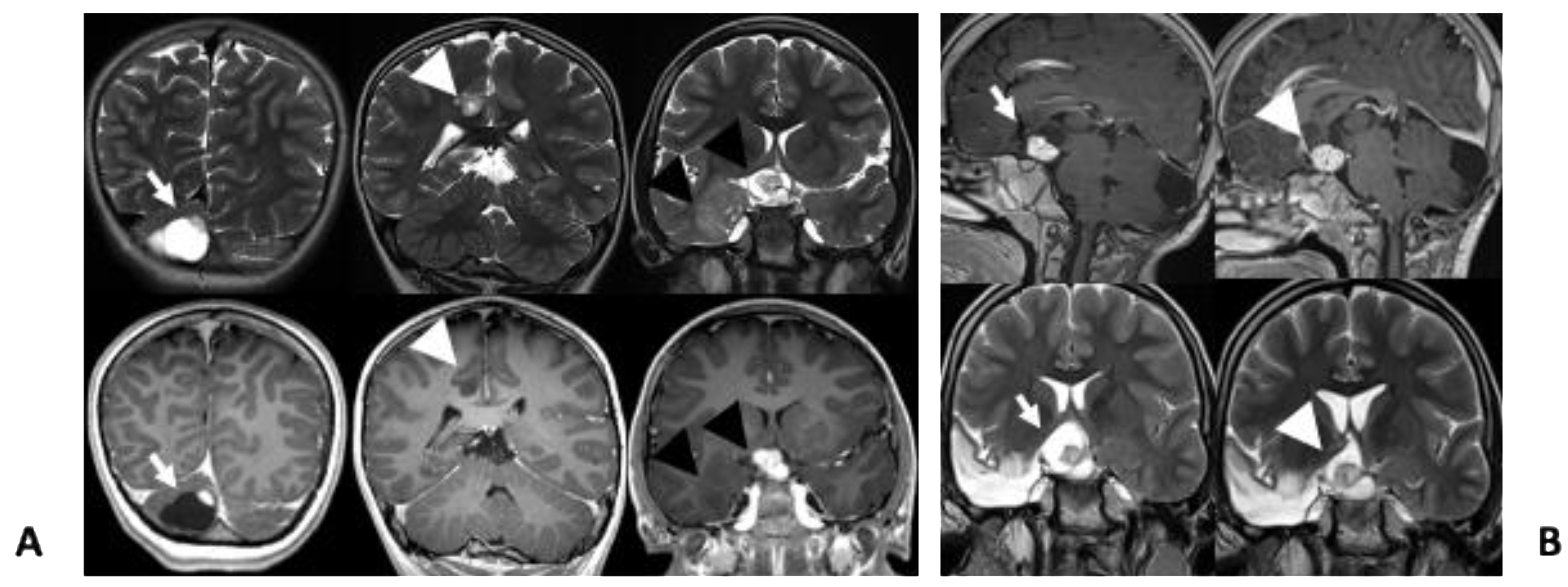
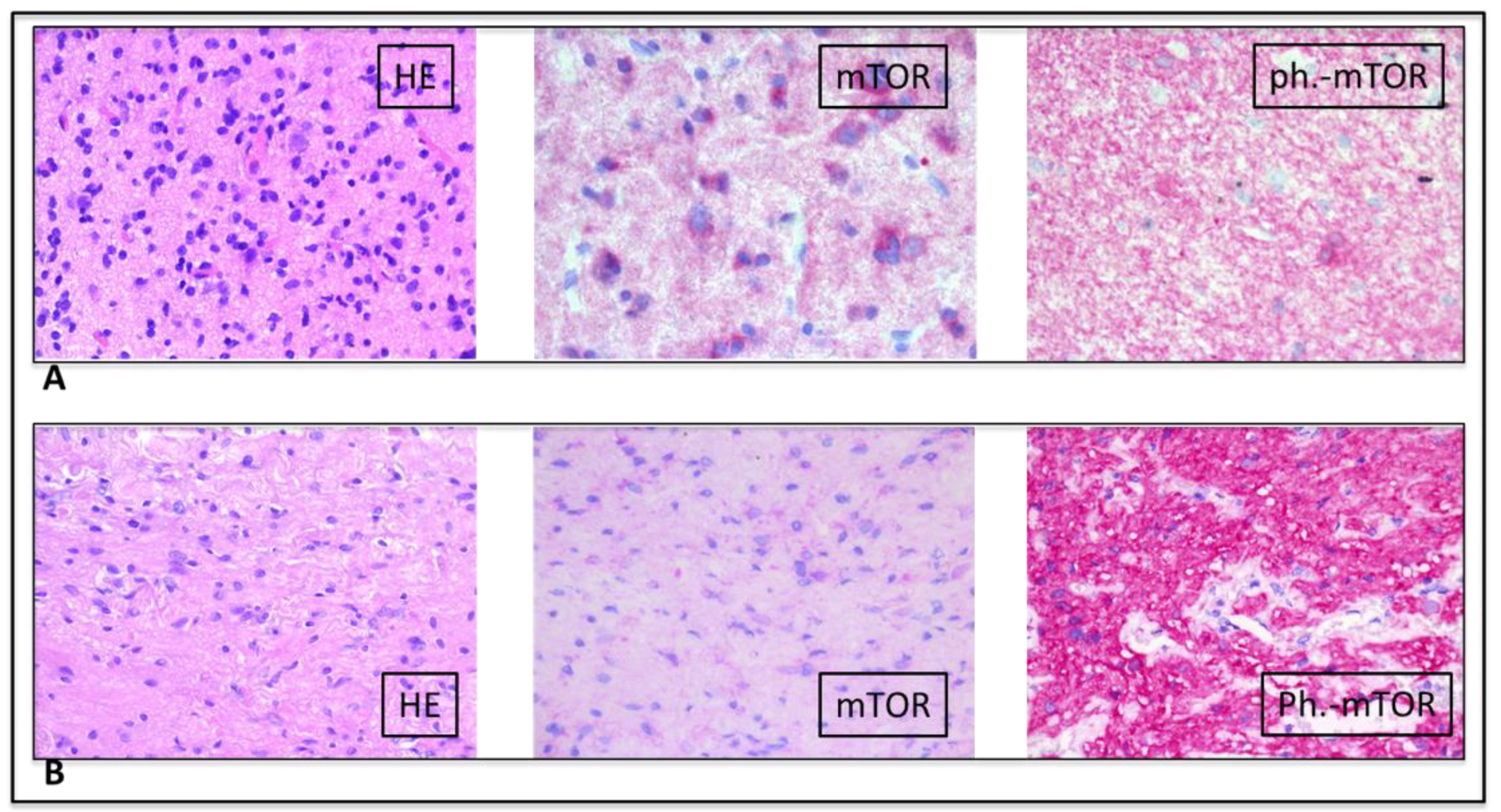
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, S.; de Luca, A.; Stellacci, E.; Rossi, C.; Checquolo, S.; Lepri, F.; Caputo, V.; Silvano, M.; Buscherini, F.; Consoli, F.; et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 2010, 87, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athota, J.P.; Bhat, M.; Nampoothiri, S.; Gowrishankar, K.; Narayanachar, S.G.; Puttamallesh, V.; Farooque, M.O.; Shetty, S. Molecular and clinical studies in 107 Noonan syndrome affected individuals with PTPN11 mutations. BMC Med. Genet. 2020, 21, 50. [Google Scholar] [CrossRef] [PubMed]
- Franz, D.N.; Belousova, E.; Sparagana, S.P.; Bebin, E.M.; Frost, M.; Kuperman, R.; Witt, O.; Kohrman, M.; Flamini, J.R.; Wu, J.Y.; et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): A multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013, 381, 125–132. [Google Scholar] [CrossRef]
- Smpokou, P.; Zand, D.J.; Rosenbaum, K.N.; Summar, M.L. Malignancy in Noonan syndrome and related disorders. Clin. Genet. 2015, 88, 516–522. [Google Scholar] [CrossRef]
- Jongmans, M.C.J.; van der Burgt, I.; Hoogerbrugge, P.M.; Noordam, K.; Yntema, H.G.; Nillesen, W.M.; Kuiper, R.P.; Ligtenberg, M.J.L.; van Kessel, A.G.; van Krieken, J.H.; et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur. J. Hum. Genet. 2011, 19, 870–874. [Google Scholar] [CrossRef] [Green Version]
- Kratz, C.P.; Rapisuwon, S.; Reed, H.; Hasle, H.; Rosenberg, P.S. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am. J. Med. Genet. C Semin Med. Genet. 2011, 157C, 83–89. [Google Scholar] [CrossRef]
- Kratz, C.P.; Franke, L.; Peters, H.; Kohlschmidt, N.; Kazmierczak, B.; Finckh, U.; Bier, A.; Eichhorn, B.; Blank, C.; Kraus, C.; et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br. J. Cancer 2015, 112, 1392–1397. [Google Scholar] [CrossRef] [Green Version]
- Loh, M.L.; Vattikuti, S.; Schubbert, S.; Reynolds, M.G.; Carlson, E.; Lieuw, K.H.; Cheng, J.W.; Lee, C.M.; Stokoe, D.; Bonifas, J.M.; et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 2004, 103, 2325–2331. [Google Scholar] [CrossRef]
- Tartaglia, M.; Martinelli, S.; Iavarone, I.; Cazzaniga, G.; Spinelli, M.; Giarin, E.; Petrangeli, V.; Carta, C.; Masetti, R.; Arico, M.; et al. Somatic PTPN11 mutations in childhood acute myeloid leukaemia. Br. J. Haematol. 2005, 129, 333–339. [Google Scholar] [CrossRef]
- Miyamoto, D.; Miyamoto, M.; Takahashi, A.; Yomogita, Y.; Higashi, H.; Kondo, S.; Hatakeyama, M. Isolation of a distinct class of gain-of-function SHP-2 mutants with oncogenic RAS-like transforming activity from solid tumors. Oncogene 2008, 27, 3508–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentiresalj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.L.; Bardelli, A.; Sagarbaker, D.J.; et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004, 64, 8816–8820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar] [PubMed]
- Tartaglia, M.; Martinelli, S.; Stella, L.; Bocchinfuso, G.; Flex, E.; Cordeddu, V.; Zampino, G.; van der Burgt, I.; Palleschi, A.; Petrucci, T.C.; et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am. J. Hum. Genet. 2006, 78, 279–290. [Google Scholar] [CrossRef]
- McWilliams, G.; SantaCruz, K.; Hart, B.; Clericuzio, C. Occurrence of DNET and other brain tumors in Noonan syndrome warrants caution with growth hormone therapy. Am. J. Med. Genet. 2015, 170 Pt A, 195–201. [Google Scholar] [CrossRef]
- Siegfried, A.; Cances, C.; Denuelle, M.; Loukh, N.; Tauber, M.; Cave, H.; Delisle, M. Noonan syndrome, PTPN11 mutations, and brain tumors. A clinical report and review of the literature. Am. J. Med. Genet. 2017, 173, 1061–1065. [Google Scholar] [CrossRef]
- Bangalore Krishna, K.; Pagan, P.; Escobar, O.; Popovic, J. Occurrence of Cranial Neoplasms in Pediatric Patients with Noonan Syndrome Receiving Growth Hormone: Is Screening with Brain MRI prior to Initiation of Growth Hormone Indicated? Horm. Res. Paediatr. 2017, 88, 423–426. [Google Scholar] [CrossRef]
- Elayadi, M.; Ansari, M.; Kuhnol, C.D.; Bendel, A.; Sturm, D.; Pietsch, T.; Kramm, C.M.; von Bueren, A.O. Occurrence of high-grade glioma in Noonan syndrome: Report of two cases. Pediatr. Blood Cancer 2019, 66, e27625. [Google Scholar] [CrossRef]
- Rickert, C.H.; Paulus, W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv. Syst. 2001, 17, 503–511. [Google Scholar] [CrossRef]
- Bessis, D.; Miquel, J.; Bourrat, E.; Chiaverini, C.; Moricepicard, F.; Abadie, C.; Manna, F.; Baumann, C.; Best, M.; Blanchet, P.; et al. Dermatological manifestations in Noonan syndrome: A prospective multicentric study of 129 patients positive for mutation. Br. J. Dermatol. 2019, 180, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Sarkozy, A.; de Zorzi, A.; Pacileo, G.; Limongelli, G.; Mingarelli, R.; Calabro, R.; Marino, B.; Dallapiccola, B. Leopard syndrome: Clinical diagnosis in the first year of life. Am. J. Med. Genet. A 2006, 140, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Lepri, F.; Baban, A.; Dentici, M.L.; Versacci, P.; Capolino, R.; Ferese, R.; de Luca, A.; Tartaglia, M.; Marino, B.; et al. RASopathies: Clinical Diagnosis in the First Year of Life. Mol. Syndromol. 2011, 1, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villani, A.; Greer, M.C.; Kalish, J.M.; Nakagawara, A.; Nathanson, K.L.; Pajtler, K.W.; Pfister, S.M.; Walsh, M.F.; Wasserman, J.D.; Zelley, K.; et al. Recommendations for Cancer Surveillance in Individuals with RASopathies and Other Rare Genetic Conditions with Increased Cancer Risk. Clin. Cancer Res. 2017, 23, e83–e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selter, M.; Dresel, R.; Althaus, J.; Bartels, M.B.; Dittrich, S.; Geb, S.; Hoche, F.; Qirshi, M.; Vlaho, S.; Zielen, S.; et al. Dysembryoplastic neuroepithelial tumor (DNET) in a patient with Noonan syndrome. Neuropediatrics 2010, 41, P1356. [Google Scholar] [CrossRef]
- Bendel, A.; Hansen, M.; Dugan, S.; Mendelsohn, N. Dysembyoplastic Neuroepithelial Tumor in Two Relatives with Noonan Syndrome And A PTPN 11 Mutation. Neuro. Oncol. 2012, 14, 156. [Google Scholar] [CrossRef] [Green Version]
- Delisle, M.; Siegfried, A.; Tauber, M.; Cave, H.; Loukh, N.; Boetto, S.; Bertozzi, A.; Urocoste, E.; Cances, C. Dysembryoplastic neuroepithelial tumor (DNET) and Noonan syndrome. A case report. In Proceedings of the Brain Pathology. Abstracts of the XVIII International Congress of Neuropathology, Rio de Janeiro, Brazil, 14–18 September 2014. [Google Scholar]
- Sherman, C.B.; Ali-Nazir, A.; Gonzales-Gomez, I.; Finlay, J.L.; Dhall, G. Primary mixed glioneuronal tumor of the central nervous system in a patient with noonan syndrome: A case report and review of the literature. J. Pediatr. Hematol. Oncol. 2009, 31, 61–64. [Google Scholar] [CrossRef]
- Schuettpelz, L.G.; Mcdonald, S.; Whitesell, K.; Desruisseau, D.M.; Grange, D.K.; Gurnett, C.A.; Wilson, D.B. Pilocytic astrocytoma in a child with Noonan syndrome. Pediatr. Blood Cancer 2009, 53, 1147–1149. [Google Scholar] [CrossRef]
- Fryssira, H.; Leventopoulos, G.; Psoni, S.; Kitsioutzeli, S.; Stavrianeas, N.G.; Kanavakis, E. Tumor development in three patients with Noonan syndrome. Eur. J. Pediatr. 2008, 167, 1025–1031. [Google Scholar] [CrossRef]
- de Jong, M.; Schieving, J.; Goraj, B. Remarkable intra-cerebral lesions on MRI in a patient with Noonan syndrome. Eur. J. Radiol. Extra. 2011, 78, e17–e19. [Google Scholar] [CrossRef]
- Karafin, M.; Jallo, G.I.; Ayars, M.; Eberhart, C.G.; Rodriguez, F.J. Rosette forming glioneuronal tumor in association with Noonan syndrome: Pathobiological implications. Clin. Neuropathol. 2011, 30, 297–300. [Google Scholar] [PubMed]
- Takagi, M.; Miyashita, Y.; Koga, M.; Ebara, S.; Arita, N.; Kasayama, S. Estrogen deficiency is a potential cause for osteopenia in adult male patients with Noonan’s syndrome. Calcif. Tissue Int. 2000, 66, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Sanford, R.A.; Bowman, R.; Tomita, T.; De Leon, G.; Palka, P. A 16-year-old male with Noonan’s syndrome develops progressive scoliosis and deteriorating gait. Pediatr. Neurosurg. 1999, 30, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Fort, J.A.; Yachnis, A.T.; Williams, C.A. Optic nerve pilomyxoid astrocytoma in a patient with Noonan syndrome. Pediatr. Blood Cancer 2015, 62, 1084–1086. [Google Scholar] [CrossRef]
- Martinelli, S.; Carta, C.; Flex, E.; Binni, F.; Cordisco, E.L.; Moretti, S.; Puxeddu, E.; Tonacchera, M.; Pinchera, A.; Mcdowell, H.P.; et al. Activating PTPN11 mutations play a minor role in pediatric and adult solid tumors. Cancer Genet. Cytogenet. 2006, 166, 124–129. [Google Scholar] [CrossRef]
- Gnekow, A.; Kandels, D.; van Tilburg, C.M.; Azizi, A.; Opocher, E.; Stokland, T.; Driever, P.H.; Meeteren, A.Y.N.S.; Thomale, U.W.; Schuhmann, M.U.; et al. SIOP-E-BTG and GPOH Guidelines for Diagnosis and Treatment of Children and Adolescents with Low Grade Glioma. Klin. Padiatr. 2019, 231, 107–135. [Google Scholar]
- Goodden, J.; Pizer, B.; Pettorini, B.; Williams, D.; Blair, J.; Didi, M.; Thorp, N.; Mallucci, C. The role of surgery in optic pathway/hypothalamic gliomas in children. J. Neurosurg. Pediatr. 2014, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yu, W.; Zhang, J.; Chan, R.J.; Loh, M.L.; Zhang, Z.; Bunting, K.D.; Qu, C. Inhibition of the Gab2/PI3K/mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia 2017, 31, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Catanzaro, G.; Besharat, Z.M.; Miele, E.; Chiacchiarini, M.; Po, A.; Carai, A.; Marras, C.E.; Antonelli, M.; Badiali, M.; Raso, A.; et al. The miR-139-5p regulates proliferation of supratentorial paediatric low-grade gliomas by targeting the PI3K/AKT/mTORC1 signalling. Neuropath. Appl. Neuro. 2018, 44, 687–706. [Google Scholar] [CrossRef] [Green Version]
- Krueger, D.A.; Care, M.M.; Holland, K.D.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.; Byars, A.W.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef]
- Tartaglia, M.; Gelb, B.D.; Zenker, M. Noonan syndrome and clinically related disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 161–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| No. | Reference | Age | Gender | Noonan Syndrome Diagnosis | Brain Tumor Diagnosis | Location |
|---|---|---|---|---|---|---|
| 1 | McWilliams et al. [16] | 8 | M | PTPN11 p.Glu139Asp | DNET | Temporal lobe and cerebellum |
| 2 | Jongmans et al. [6] | 10 | Ukn | PTPN11 c.179G > C; p.Gly60Ala | DNET | Temporal lobe |
| 3 | Selter et al. [25] Pellegrin et al. [17] | 13 | M | PTPN11 exon 3 ** | DNET | Left parietal lobe |
| 4 | Bendel et al. [26] | 17 | M | PTPN11 c.174C > G; p.Asn58Lys | DNET | Occipital cortex |
| 5 | Bendel et al. [26] | 37 | M | Maternal uncle of Case 4 | DNET | Unknown |
| 6 | Delisle et al. [27] | 12 | M | PTPN11 mutation ** | DNET | Temporal lobe and thalamus |
| 7 | Krishna et al. [18] | 9 | M | PTPN11 p.Asp61Gly | DNET | Temporal lobe and cerebellum |
| 8 | Pellegrin et al. [17] | 13 | M | N/A | DNET | Right parieto-occipital cortex |
| 9 | Pellegrin et al. [17] | 13 | M | PTPN11 exon 3 mutation ** | DNET (MRI) | Left parietal lobe |
| 10 | Kratz et al. [8] | 6 | F | PTPN11 p.Asn308Asp | DNET | Unknown |
| 11 | Siegfried et al. [17] | 16 | M | PTPN11 c.922A > G; p.Asn308Asp | DNET | Left temporal and frontal lobe, right thalamus |
| 12 | Rankin et al. [26] | 10 | M | PTPN11 c.1403C > T; p.Thr468Met | Medulloblastoma | Cerebellum |
| 13 | Jongmans el al [6] | 18 | F | PTPN11 c.417G > C; p.Glu139Asp | Oligodendroglioma | Hypothalamus |
| 14 | Sherman et al. [28] | 6 | M | PTPN11 c.172A > G; p.Asn58Asp | Low-grade mixed glioneuronal tumor | Suprasellar cisterns, sella turcica and hypothalamus and diffuse leptomeningeal disease |
| 15 | Schuettpelz et al. [29] | 8 | M | PTPN11 c.1471C > T and c.1472C > T; p.Pro491Phe | Pilocytic astrocytoma | Sellar/suprasellar with extension to prepontine region to the level of the pontomedullary junction |
| 16 | Fryssira et al. [30] | 11 | F | Clinical diagnosis | Pilocytic astrocytoma | Sellar/suprasellar |
| 17 | De Jongo et al. [31] | 21 | M | PTPN11 mutation ** | Multiple indeterminate lesions on MRI | Multiple: supratentorial, infratentorial, cortical and subortical, thalamus |
| 18 | Karafin et al. [32] | 18 | M | Clinical diagnosis | Rosette forming glioneuronal tumor | Fourth ventricle |
| 19, 20, 21 | Rush et al. * [17] | Ukn | M | PTPN11 mutation ** | Low grade astrocytoma | Suprasellar and thalamic region |
| 22 | Kratz et al. [19] | 7 | M | PTPN11 p.Gly60Ala | Pilocytic astrocytoma | Right optic nerve |
| 23 | Takagi at al. [33] | 20 | M | Clinical diagnosis | Glioma | Unknown |
| 24 | Standford et al. [34] | 16 | M | Clinical diagnosis | Pilocytic astrocytoma | Intramedullary spinal cord lesion involving the cervical medullary junction and descending to the C2-C3 disc space level |
| 25 | Nair et al. [35] | 14 | M | PTPN11 c.417G > C in exon 4 | Pilomyxoid astrocytoma | Right optic nerve |
| 26 | Bendel and Pond. [19] | 14 | F | PTPN11 c.922A > G; p.Asn308Asp | High grade glioma | Left brainstem/cerebellar peduncle |
| 27 | Martinelli et al. [36] | 24 | Ukn | PTPN11 c.64A > G; pThr22Ala | Oligodendroglioma grade II | Unknown |
| 28 | El Ayadi et al. [19] | 14 | F | PTPN11 c.922A > G; p.Asn308Asp | Anaplastic astocytoma | Left brainstem/cerebellar peduncle |
| 29 | El Ayadi et al. [19] | 9 | M | PTPN11 c.5C > T; p.Thr2lle | Anaplastic astocytoma | Third ventricle, cerebellum and fornix |
| 30 | Our case | 9 | F | PTPN11 c.922A > G; p.Asn308Asp | Pilocytic astrocitoma and glioneuronal tumor | Cerebellum Right temporal lobe |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lodi, M.; Boccuto, L.; Carai, A.; Cacchione, A.; Miele, E.; Colafati, G.S.; Diomedi Camassei, F.; De Palma, L.; De Benedictis, A.; Ferretti, E.; et al. Low-Grade Gliomas in Patients with Noonan Syndrome: Case-Based Review of the Literature. Diagnostics 2020, 10, 582. https://doi.org/10.3390/diagnostics10080582
Lodi M, Boccuto L, Carai A, Cacchione A, Miele E, Colafati GS, Diomedi Camassei F, De Palma L, De Benedictis A, Ferretti E, et al. Low-Grade Gliomas in Patients with Noonan Syndrome: Case-Based Review of the Literature. Diagnostics. 2020; 10(8):582. https://doi.org/10.3390/diagnostics10080582
Chicago/Turabian StyleLodi, Mariachiara, Luigi Boccuto, Andrea Carai, Antonella Cacchione, Evelina Miele, Giovanna Stefania Colafati, Francesca Diomedi Camassei, Luca De Palma, Alessandro De Benedictis, Elisabetta Ferretti, and et al. 2020. "Low-Grade Gliomas in Patients with Noonan Syndrome: Case-Based Review of the Literature" Diagnostics 10, no. 8: 582. https://doi.org/10.3390/diagnostics10080582