Quadruplicate Synchronous Adenocarcinoma of the Colon with Distant Metastases—Long-Term Molecular Follow-Up by KRAS and TP53 Mutational Profiling

Abstract
:1. Introduction
2. Materials and Methods
3. Results
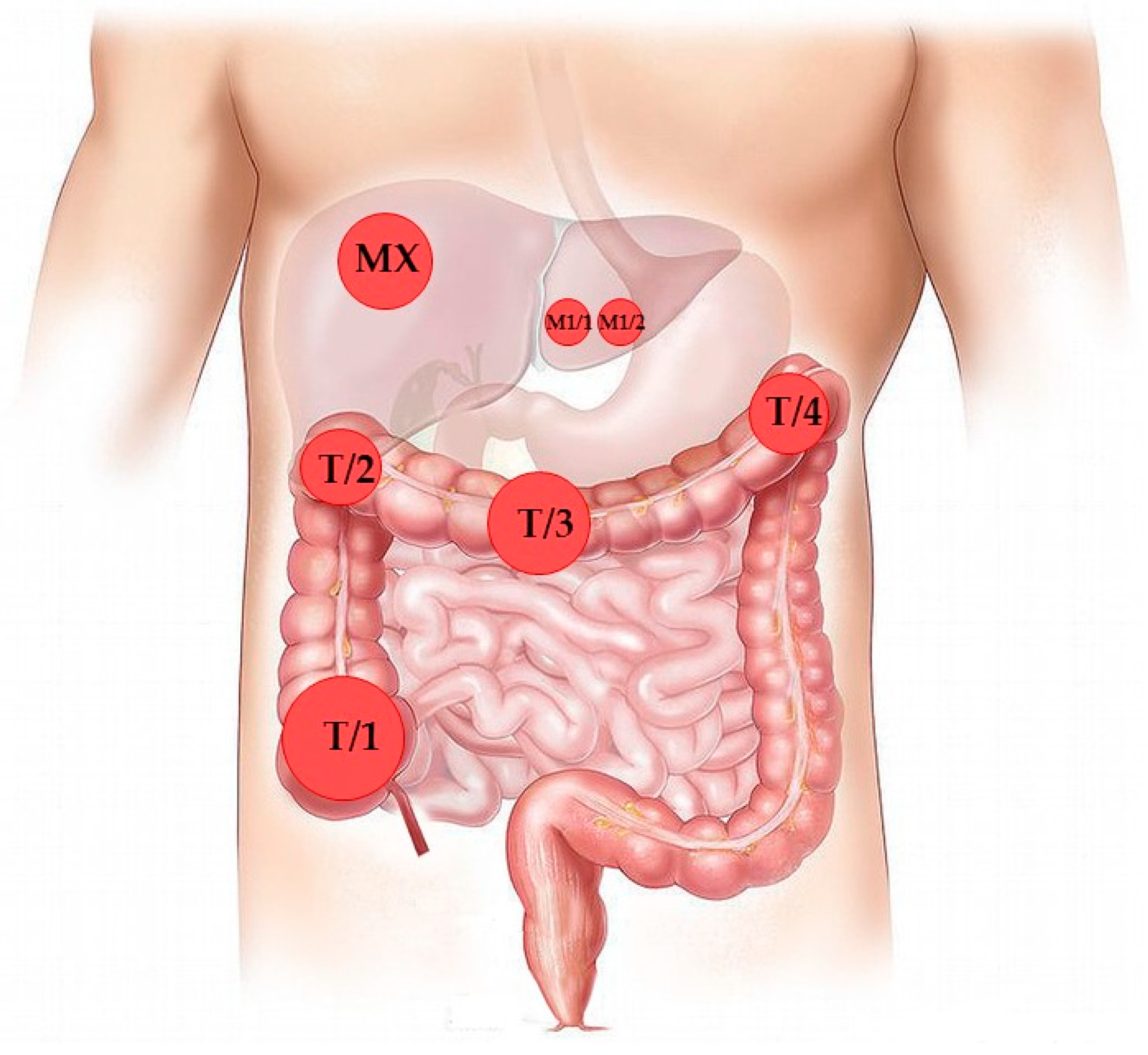
3.1. Case Presentation and Clinical Follow-Up
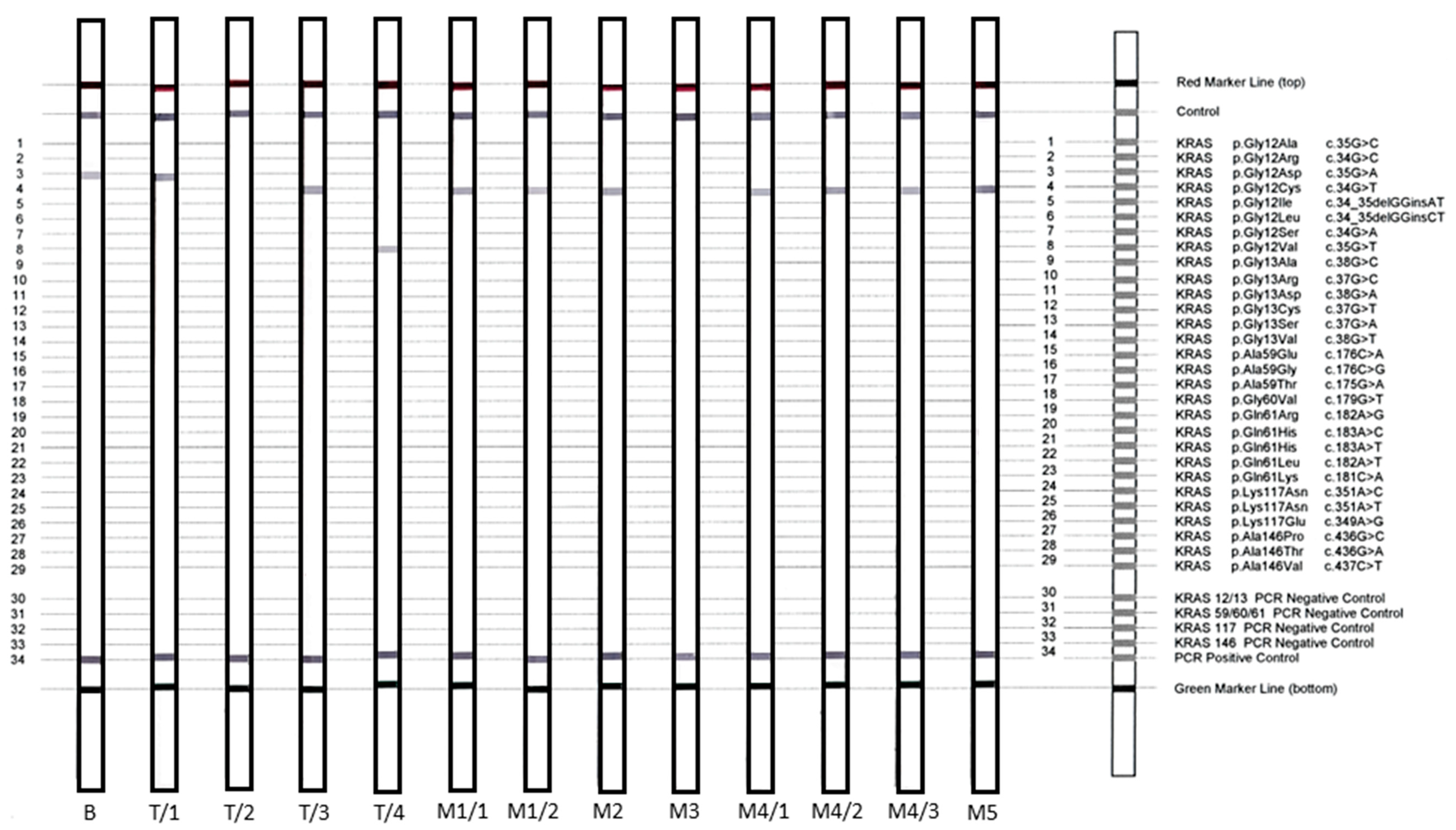
3.2. NGS-Based Mutation Profiling
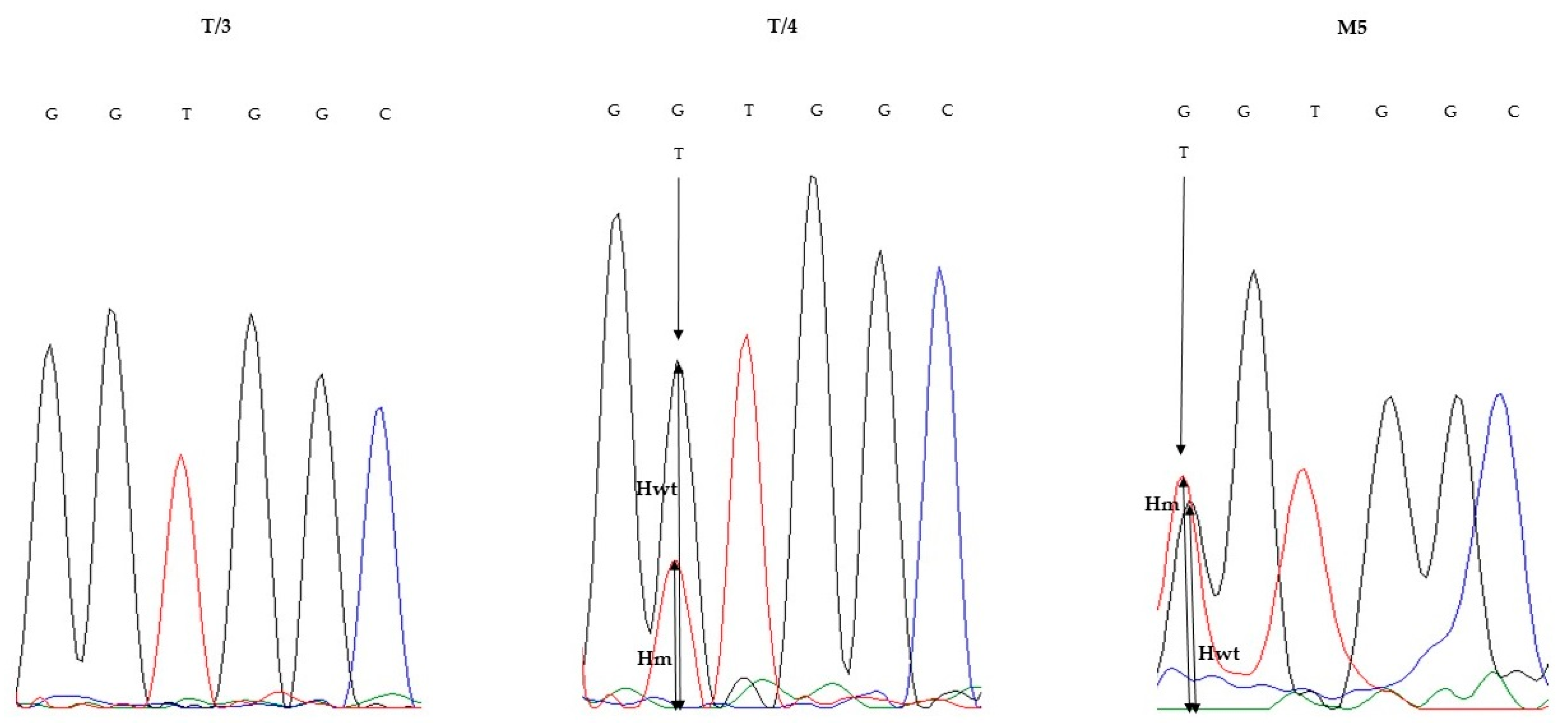
3.3. KRAS Validation Studies
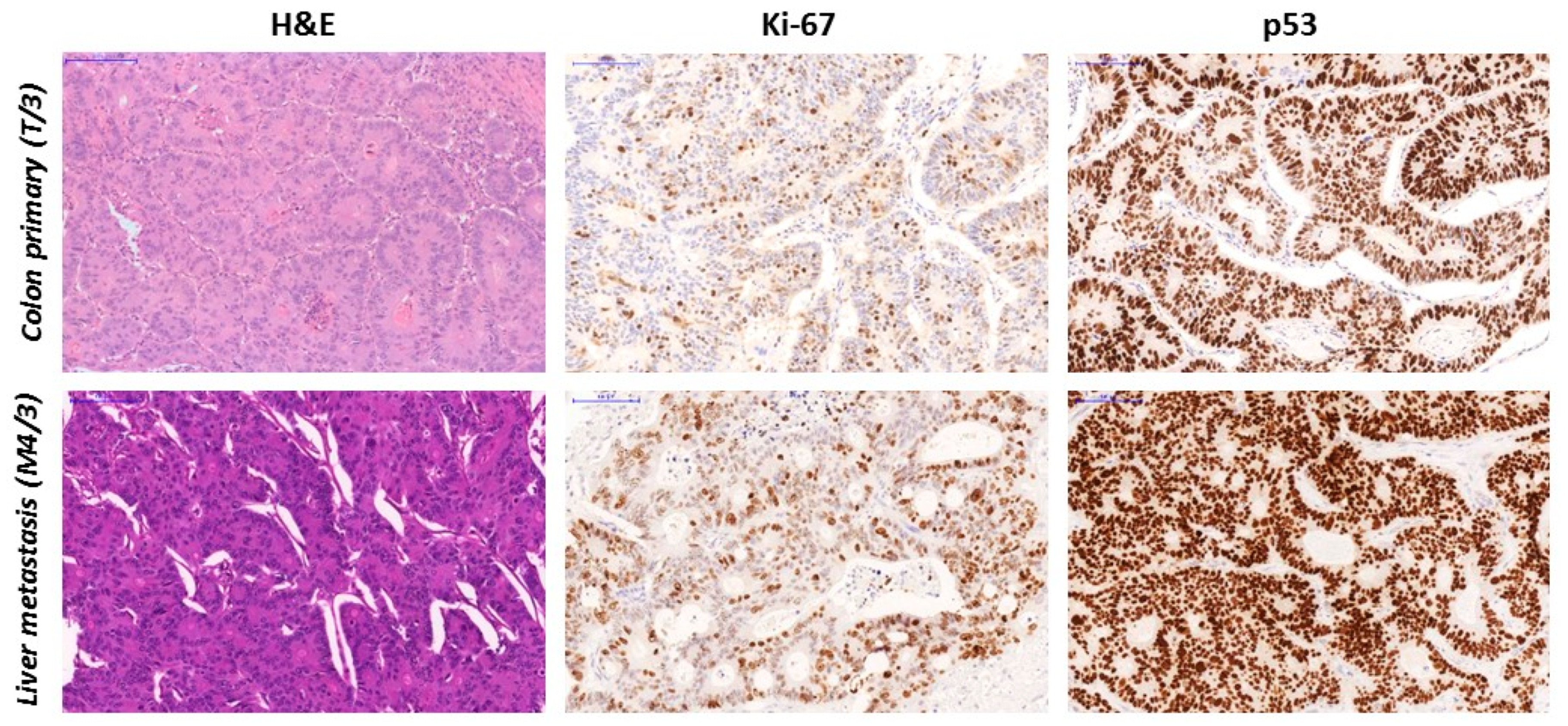
3.4. TP53 Gene Sequencing and p53 sTatus
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Lam, A.K.-Y.; Chan, S.S.-Y.; Leung, M. Synchronous colorectal cancer: Clinical, pathological and molecular implications. World J. Gastroenterol. 2014, 20, 6815–6820. [Google Scholar] [CrossRef] [PubMed]
- Pajares, J.A.; Perea, J. Multiple primary colorectal cancer: Individual or familial predisposition? World J. Gastrointest. Oncol. 2015, 7, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Duong, H.-Q. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy (review). Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, T.; McMechan, D.; Talukder, A.; Herline, A. Colorectal cancer screening and surveillance in individuals at increased risk. Am. Fam. Physician 2018, 97, 111–116. [Google Scholar]
- Hall, J.F. Management of malignant adenomas. Clin. Colon Rectal Surg. 2015, 28, 215–219. [Google Scholar] [CrossRef] [Green Version]
- D’Haene, N.; Fontanges, Q.; De Nève, N.; Blanchard, O.; Melendez, B.; Delos, M.; Dehou, M.-F.; Maris, C.; Nagy, N.; Rousseau, E.; et al. Clinical application of targeted next-generation sequencing for colorectal cancer patients: A multicentric Belgian experience. Oncotarget 2018, 9, 20761–20768. [Google Scholar] [CrossRef]
- Margonis, G.A.; Kim, Y.; Spolverato, G.; Ejaz, A.; Gupta, R.; Cosgrove, D.; Anders, R.; Karagkounis, G.; Choti, M.A.; Pawlik, T.M. Association between specific mutations in KRAS codon 12 and colorectal liver metastasis. JAMA Surg. 2015, 150, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis comparing the efficacy of anti-EGFR monoclonal antibody therapy between KRAS G13D and other KRAS mutant metastatic colorectal cancer tumours. Eur. J. Cancer Oxf. Engl. 1990 2016, 55, 122–130. [Google Scholar] [CrossRef]
- Abdelnaby, A.Y.; Moussa, G.I.; Abdou, S.M.; Abdul Baki, E.A.; Salem, M.L.; Sharnobi, G.E.; Gharib, F.; Farghal, E.E.; Watany, M.; Soliman, S.M.; et al. Genetic variations of selected genes using target deep sequencing in colorectal cancer patients. J. Cancer Sci. Ther. 2017, 9, 683. [Google Scholar] [CrossRef] [Green Version]
- Vermaat, J.S.; Nijman, I.; Koudijs, M.J.; Gerritse, F.L.; Scherer, S.J.; Mokry, M.; Roessingh, W.M.; Lansu, N.; de Bruijn, E.; van Hillegersberg, R.; et al. Primary colorectal cancers and their subsequent hepatic metastases are genetically different: Implications for selection of patients for targeted treatment. Clin. Cancer Res. 2012, 18, 688–699. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Haq, F.; Kim, D.; Jun, C.; Jo, H.-J.; Ahn, S.-M.; Lee, W.-S. Comparative genomic analysis of primary and synchronous metastatic colorectal cancers. PLoS ONE 2014, 9, e90459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogita, A.; Yoshioka, Y.; Sakai, K.; Togashi, Y.; Sogabe, S.; Nakai, T.; Okuno, K.; Nishio, K. Inter- and intra-tumor profiling of multi-regional colon cancer and metastasis. Biochem. Biophys. Res. Commun. 2015, 458, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Blank, A.; Roberts, D.E.; Dawson, H.; Zlobec, I.; Lugli, A. Tumor heterogeneity in primary colorectal cancer and corresponding metastases. Does the apple fall far from the tree? Front. Med. 2018, 5, 234. [Google Scholar] [CrossRef] [Green Version]
- Dócs, O.; Fazakas, F.; Horváth, N.L.; Tóth, L.; András, C.; Horváth, Z.; Méhes, G. Changes of KRAS exon 2 codon 12/13 mutation status in recurrent colorectal cancer. Pathol. Oncol. Res. 2015, 21, 399–404. [Google Scholar] [CrossRef]
- Mazières, J.; Brugger, W.; Cappuzzo, F.; Middel, P.; Frosch, A.; Bara, I.; Klingelschmitt, G.; Klughammer, B. Evaluation of EGFR protein expression by immunohistochemistry using H-score and the magnification rule: Re-analysis of the SATURN study. Lung Cancer Amst. Neth. 2013, 82, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Soh, J.; Okumura, N.; Lockwood, W.W.; Yamamoto, H.; Shigematsu, H.; Zhang, W.; Chari, R.; Shames, D.S.; Tang, X.; MacAulay, C.; et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS ONE 2009, 4, e7464. [Google Scholar] [CrossRef]
- Tariq, K.; Ghias, K. Colorectal cancer carcinogenesis: A review of mechanisms. Cancer Biol. Med. 2016, 13, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, Y.; Diaz-Meco, M.T.; Moscat, J. Serrated colorectal cancer: The road less travelled? Trends Cancer 2019, 5, 742–754. [Google Scholar] [CrossRef]
- Heinen, C.D. Mismatch repair defects and Lynch syndrome: The role of the basic scientist in the battle against cancer. DNA Repair 2016, 38, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Vecchio, F.D.; Mastroiaco, V.; Marco, A.D.; Compagnoni, C.; Capece, D.; Zazzeroni, F.; Capalbo, C.; Alesse, E.; Tessitore, A. Next-generation sequencing: Recent applications to the analysis of colorectal cancer. J. Transl. Med. 2017, 15, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jancik, S.; Drabek, J.; Berkovcova, J.; Xu, Y.Z.; Stankova, M.; Klein, J.; Kolek, V.; Skarda, J.; Tichy, T.; Grygarkova, I.; et al. A comparison of direct sequencing, pyrosequencing, high resolution melting analysis, TheraScreen DxS, and the K-ras StripAssay for detecting KRAS mutations in non small cell lung carcinomas. J. Exp. Clin. Cancer Res. CR 2012, 31, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time from Primary Surgery (months) | Type of Operation | Histology | Sample ID |
|---|---|---|---|
| October 2015 | Colon biopsy | Tubular adenoma with high-grade dysplasia | B |
| December 2015 | Subtotal colectomy and liver metastasectomy | Colon adenocarcinoma (four independent sites) | T1/1-4 |
| Liver metastasis | M1 (M1/1, M1/2) | ||
| 11 | Hepatic resection | Liver metastasis | M2 |
| 18 | Hepatic resection | Liver metastasis | M3 |
| 27 | Hepatic resection | Liver metastasis | M4 (M4/1, M4/2, M4/3) |
| 36 | Spinal resection and decompression | Adenocarcinoma metastasis | M5 |
| Samples | Tumor Cell Content (%) | NGS | NGS VAF (%) | StripAssay | Sanger Sequencing | Sanger VAF (%) |
|---|---|---|---|---|---|---|
| B | 50 | c.35G > A, p.Gly12Asp | 10.2 | c.35G > A, p.Gly12Asp | NMD b | 0 |
| T/1 | 30 | c.35G > A; p.Gly12Asp | 40.6 | c.35G > A; p.Gly12Asp | c.35G > A; p.Gly12Asp | 10 |
| T/2 | 50 | NMD a | 0 | NMD a | NMD a | 0 |
| T/3 | 30 | c.34G > T; p.Gly12Cys | 16.2 | c.34G > T; p.Gly12Cys | NMD | 0 |
| T/4 | 30 | c.35G > T; p.Gly12Val | 15.1 | c.35G > T; p.Gly12Val | c.35G > T; p.Gly12Val | 26.86 |
| M1/1 | 20 | NMD a | 0 | c.34G > T; p.Gly12Cys b | NMD a | 0 |
| M1/2 | 30 | c.34G > T; p.Gly12Cys | 19.1 | c.34G > T; p.Gly12Cys | c.34G > T; p.Gly12Cys | 45.89 |
| M2 | 20 | c.34G > T; p.Gly12Cys | 6.4 | c.34G > T; p.Gly12Cys | NMD | 0 |
| M3 | 30 | NMD a | 0 | NMD a | NMD a | 0 |
| M4/1 | 30 | c.34G > T; p.Gly12Cys | <2 | c.34G > T; p.Gly12Cys | NMD | 0 |
| M4/2 | c.34G > T; p.Gly12Cys | <2 | c.34G > T; p.Gly12Cys | c.34G > T; p.Gly12Cys | 9.77 | |
| 30 | c.35G > A; p.Gly12Asp | <2 | ||||
| c.35G > T; p.Gly12Val | <2 | |||||
| M4/3 | 30 | c.34G > T; p.Gly12Cys | 5.3 | c.34G > T; p.Gly12Cys | NMD | 0 |
| M5 | 50 | c.34G > T; p.Gly12Cys | 32.1 | c.34G > T; p.Gly12Cys | c.34G > T; p.Gly12Cys | 52 |
| Sample | NGS | NGS VAF (%) | Mitosis Index | p53 IHC (%) | p53 H-Score | p53 Interpretation | Clinical Significance (COSMIC) | Clinical Significance (ClinVar) |
|---|---|---|---|---|---|---|---|---|
| Biopsy | c.417G > A; p.Lys139Asn | 2.9 | 24 | 2 | 5 | WT | P | ND |
| c.472C > T; p.Arg158Cys | 3.8 | P | C | |||||
| c.1009C > T; p.Arg337Cys | 3.1 | P | P | |||||
| c.464C > T; p.Thr155Ile | 7.6 | N | ND | |||||
| T/1 | c.455C > T, p.Pro152Leu | 3.3 | 62 | 5 | 23 | WT | P | ND |
| c.845G > A, p.Arg282Gln | 3.9 | P | C | |||||
| T/2 | c.557C > G; p.His193Asp | 35.6 | 71 | 60 | 155 | M | ND | LP |
| c.586C > T; p. Arg196* | 5.6 | P | P | |||||
| T/3 | c.820G > T; p.Val274Phe | 33.3 | 50 | 90 | 230 | M | N | LP |
| T/4 | c.743G > A; p.Arg248Gln | 3.1 | 62 | 15 | 25 | WT | P | P |
| M1/1 | c.726C > A; p.Cys242* | 10.8 | 57 | 70 | 150 | M | P | ND |
| c.769G > A; p.Gly266Arg | 7.4 | ND | C | |||||
| c.328C > T; p.Arg110Cys | 8.3 | ND | US | |||||
| c.747G > T; p.Arg249Ser | 4.0 | ND | C | |||||
| c.820G > T, p.Val274Phe | 23.1 | N | LP | |||||
| M1/2 | c.827C > T; p.Ala276Val | 15.7 | 64 | 100 | 300 | M | ND | ND |
| c.917G > A; p.Arg306Gln | 28.1 | P | C | |||||
| c.820G > T; p.Val274Phe | 6.1 | N | LP | |||||
| M2 | c.820G > T; p.Val274Phe | 7.1 | 31 | 90 | 225 | M | N | LP |
| M3 | c.820G > T; p.Val274Phe | 3.14 | 30 | 80 | 195 | M | N | LP |
| M4/1 | c.820G > T; p.Val274Phe | 37.9 | 62 | 80 | 195 | M | N | LP |
| M4/2 | c.820G > T; p.Val274Phe | 72.8 | 91 | 100 | 300 | Mutant | N | LP |
| M4/3 | c.820G > T; p.Val274Phe | 25.2 | 46 | 100 | 300 | Mutant | N | LP |
| M5 | c.820G > T; p.Val274Phe | 45.6 | 24 | 100 | 300 | Mutant | N | LP |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bádon, E.S.; Mokánszki, A.; Mónus, A.; András, C.; Damjanovich, L.; Méhes, G. Quadruplicate Synchronous Adenocarcinoma of the Colon with Distant Metastases—Long-Term Molecular Follow-Up by KRAS and TP53 Mutational Profiling. Diagnostics 2020, 10, 407. https://doi.org/10.3390/diagnostics10060407
Bádon ES, Mokánszki A, Mónus A, András C, Damjanovich L, Méhes G. Quadruplicate Synchronous Adenocarcinoma of the Colon with Distant Metastases—Long-Term Molecular Follow-Up by KRAS and TP53 Mutational Profiling. Diagnostics. 2020; 10(6):407. https://doi.org/10.3390/diagnostics10060407
Chicago/Turabian StyleBádon, Emese Sarolta, Attila Mokánszki, Anikó Mónus, Csilla András, László Damjanovich, and Gábor Méhes. 2020. "Quadruplicate Synchronous Adenocarcinoma of the Colon with Distant Metastases—Long-Term Molecular Follow-Up by KRAS and TP53 Mutational Profiling" Diagnostics 10, no. 6: 407. https://doi.org/10.3390/diagnostics10060407