Pathogenic PSEN1 Thr119Ile Mutation in Two Korean Patients with Early-Onset Alzheimer’s Disease

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Findings
2.2. Molecular Genetic Analysis
2.3. In Silico Analyses and Protein Structure Prediction
3. Results
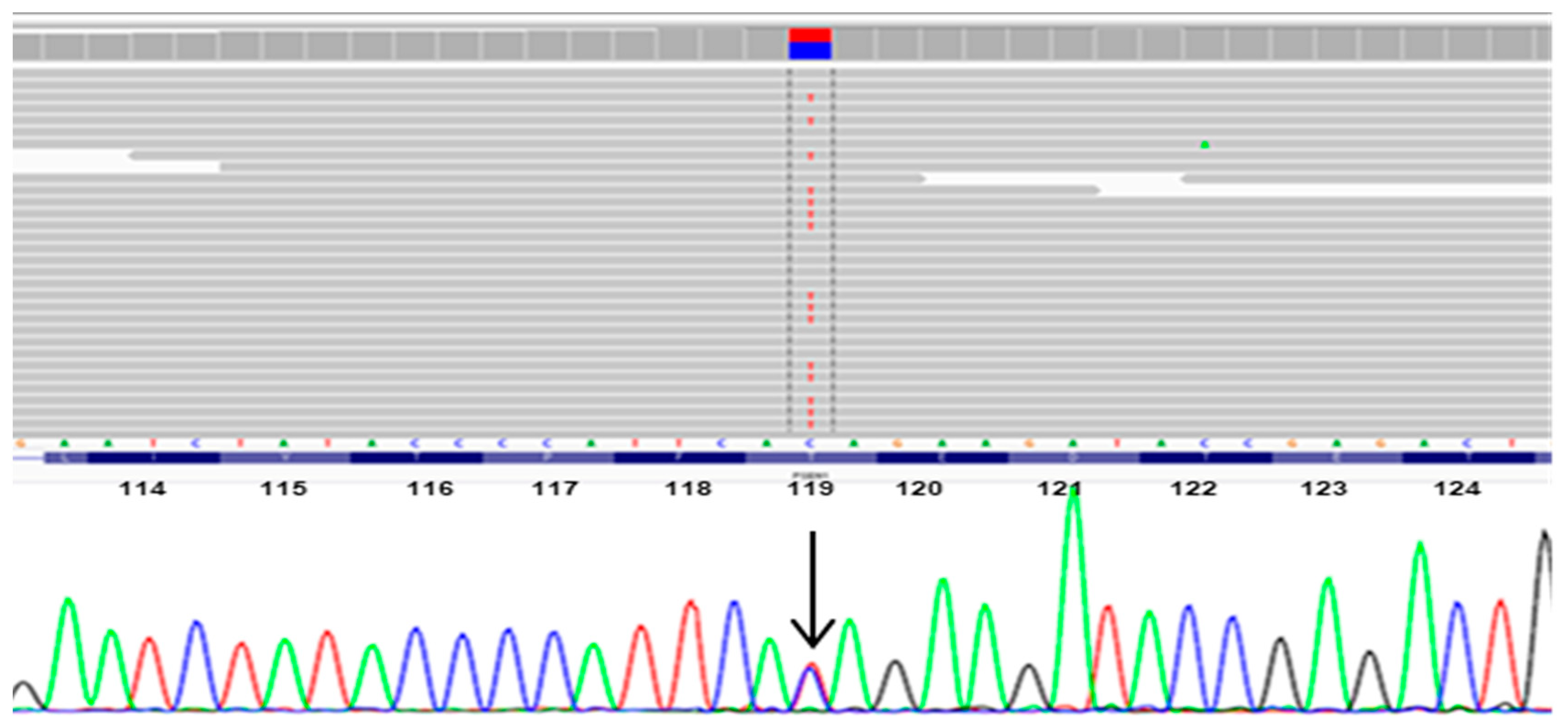
3.1. Mutation Analysis
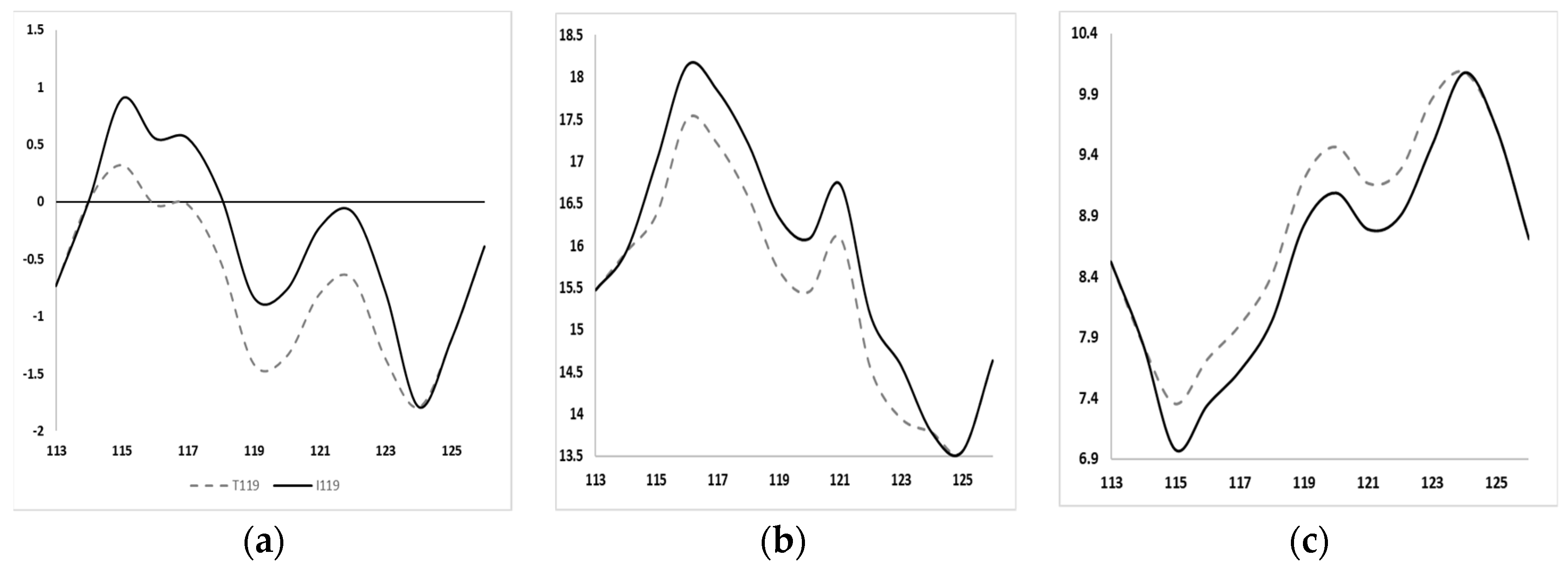
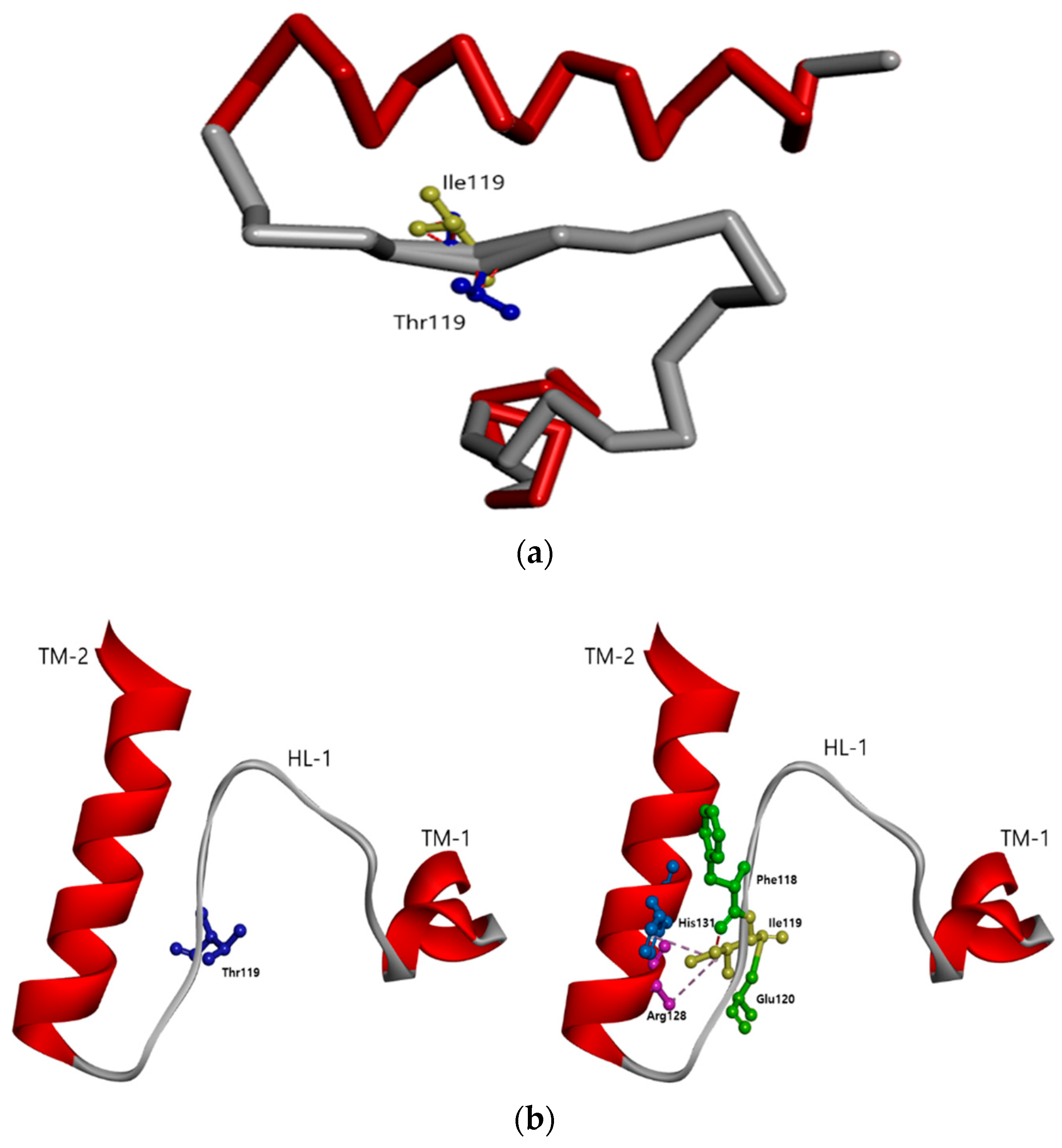
3.2. In Silico Predictions
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Lublin, A.L.; Gandy, S. Amyloid-β Oligomers: Possible Roles as Key Neurotoxins in Alzheimer’s Disease. Mt. Sinai J. Med. A J. Transl. Pers. Med. 2010, 77, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A Recessive Mutation in the APP Gene with Dominant-Negative Effect on Amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S.S. Trafficking and Proteolytic Processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Campion, D.; Brice, A.; Dumanchin, C.; Puel, M.; Baulac, S.; De La Sayette, V.; Mannequin, D.; Duyckaerts, C.; Michon, A.; Martin, C.; et al. A novel presenilin 1 mutation resulting in familial Alzheimer’s disease with an onset age of 29 years. NeuroReport 1996, 7, 1582–1584. [Google Scholar] [CrossRef]
- Ataka, S.; Tomiyama, T.; Takuma, H.; Yamashita, T.; Shimada, H.; Tsutada, T.; Kawabata, K.; Mori, H.; Miki, T. A Novel Presenilin-1 Mutation (Leu85Pro) in Early-Onset Alzheimer Disease With Spastic Paraparesis. Arch. Neurol. 2004, 61, 1773–1776. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bagyinszky, E.; Chang, Y.H.; Choe, G.; Choi, B.-O.; An, S.S.A.; Kim, S. A novel PSEN1 H163P mutation in a patient with early-onset Alzheimer’s disease: Clinical, neuroimaging, and neuropathological findings. Neurosci. Lett. 2012, 530, 109–114. [Google Scholar] [CrossRef]
- Park, J.; An, S.S.A.; Van Giau, V.; Shim, K.; Youn, Y.C.; Bagyinszky, E.; Kim, S. Identification of a novel PSEN1 mutation (Leu232Pro) in a Korean patient with early-onset Alzheimer’s disease and a family history of dementia. Neurobiol. Aging 2017, 56, 212.e11–212.e17. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Van Giau, V.; An, S.S.A.; Bagyinszky, E.; Kim, S. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol. Cell. Toxicol. 2015, 11, 89–143. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Park, Y.H.; Wang, M.J.; Park, S.Y.; Jang, J.W.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Novel, PSEN1 T119I mutation in an early-onset Alzheimer’s disease patient. Alzheimer Dement. J. Alzheimer Assoc. 2016, 12, P645. [Google Scholar] [CrossRef]
- Van Giau, V.; Bagyinszky, E.; Yang, Y.S.; Youn, Y.C.; An, S.S.A.; Kim, S. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9, 8368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lü, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzcovich, T.; Chrem-Méndez, P.; Vázquez, S.; Barbieri-Kennedy, M.; Niikado, M.; Martinettoa, H.; Allegri, R.; Sevlever, G.; Suracead, E.I. A novel mutation in PSEN1 (p.T119I) in an Argentine family with early and late-onset Alzheimer’s disease. Neurobiol. Aging 2020, 85, 155.e9–155.e12. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Zhang, L.; Meng, X.; Ma, L.; Zhang, G.; Wu, H.; Liang, L.; Cao, M.; Mei, F. Identification of a Rare PSEN1 Mutation (Thr119Ile) in Late-Onset Alzheimer’s Disease With Early Presentation of Behavioral Disturbance. Front. Psychol. 2020, 11. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Lee, H.M.; Vo, V.G.; Koh, S.-B.; Jeong, J.H.; An, S.S.A.; Kim, S. PSEN1 p.Thr116Ile Variant in Two Korean Families with Young Onset Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 2604. [Google Scholar] [CrossRef] [Green Version]
- Vardarajan, B.N.; Zhang, Y.; Lee, J.H.; Cheng, R.; Bohm, C.; Ghani, M.; Reitz, C.; Reyes-Dumeyer, L.; Shen, Y.; Rogaeva, E.; et al. Coding mutations in SORL1 and Alzheimer disease. Ann. Neurol. 2015, 77, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Cuccaro, M.L.; Carney, R.M.; Zhang, Y.; Bohm, C.; Kunkle, B.W.; Vardarajan, B.N.; Whitehead, P.L.; Cukier, H.N.; Mayeux, R.; George-Hyslop, P.S.; et al. SORL1 mutations in early- and late-onset Alzheimer disease. Neurol. Genet. 2016, 2, e116. [Google Scholar] [CrossRef] [Green Version]
- Thonberg, H.; Chiang, H.-H.; Lilius, L.; Forsell, C.; Lindström, A.-K.; Johansson, C.; Björkström, J.; Thordardottir, S.; Sleegers, K.; Van Broeckhoven, C.; et al. Identification and description of three families with familial Alzheimer disease that segregate variants in the SORL1 gene. Acta Neuropathol. Commun. 2017, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019, 138, 201–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, K.; Abe-Dohmae, S.; Yokoyama, S.; George-Hyslop, P.S.; Fraser, P.E. ATP-binding Cassette Transporter A7 (ABCA7) Loss of Function Alters Alzheimer Amyloid Processing*. J. Biol. Chem. 2015, 290, 24152–24165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raux, G.; Guyant-Maréchal, L.; Martin, C.; Bou, J.; Penet, C.; Brice, A.; Hannequin, D.; Frebourg, T.; Campion, D. Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: An update. J. Med. Genet. 2005, 42, 793–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Bella, V.; Liguori, M.; Cittadella, R.; Settipani, N.; Piccoli, T.; Manna, I.; Quattrone, A.; Piccoli, F. A novel mutation (Thr116Ile) in the presenilin 1 gene in a patient with early-onset Alzheimer’s disease. Eur. J. Neurol. 2004, 11, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. Mutations, associated with early-onset Alzheimer’s disease, discovered in Asian countries. Clin. Interv. Aging 2016, 11, 1467–1488. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, T.; Dowjat, W.K.; Buxbaum, J.D.; Khorkova, O.; Efthimiopoulos, S.; Kulczycki, J.; Lojkowska, W.; Wegiel, J.; Wisniewski, H.M.; Frangione, B. A novel Polish presenilin-1 mutation (P117L) is associated with familial Alzheimer’s disease and leads to death as early as the age of 28 years. NeuroReport 1998, 9, 217–221. [Google Scholar] [CrossRef]
- Romero, I.; Jørgensen, P.; Bolwig, G.; Fraser, P.E.; Rogaeva, E.; Mann, D.; Havsager, A.-M.; Jørgensen, A.L. A presenilin-1 Thr116Asn substitution in a family with early-onset Alzheimer’s disease. NeuroReport 1999, 10, 2255–2260. [Google Scholar] [CrossRef]
- Finckh, U.; Kuschel, C.; Anagnostouli, M.; Patsouris, E.; Pantes, G.V.; Gatzonis, S.; Kapaki, E.; Davaki, P.; Lamszus, K.; Stavrou, D.; et al. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics 2005, 6, 85–89. [Google Scholar] [CrossRef]
- Dobričić, V.; Stefanova, E.; Janković, M.; Gurunlian, N.; Novakovic, I.; Hardy, J.; Kostic, V.; Guerreiro, R. Genetic testing in familial and young-onset Alzheimer’s disease: Mutation spectrum in a Serbian cohort. Neurobiol. Aging 2012, 33, 1481.e7–1481.e12. [Google Scholar] [CrossRef]
- Campion, D.; Flaman, J.-M.; Brice, A.; Hannequin, D.; Dubois, B.; Martin, C.; Moreau, V.; Charbonnier, F.; Didierjean, O.; Tardieu, S.; et al. Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Hum. Mol. Genet. 1995, 4, 2373–2377. [Google Scholar] [CrossRef]
- Hutton, M.; Busfield, F.; Wragg, M.; Crook, R.; Tur, J.P.; Clark, R.F.; Prihar, G.; Phillips, H.M.; Wright, K.; Baker, M.; et al. Complete analysis of the presenilin 1 gene in early onset Alzheimer’s disease. NeuroReport 1996, 7, 801–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, P.; Vetrivel, K.S.; Nguyen, P.D.; Meckler, X.; Cheng, H.; Kounnas, M.Z.; Wagner, S.L.; Parent, A.T.; Thinakaran, G. Mutation Analysis of the Presenilin 1 N-terminal Domain Reveals a Broad Spectrum of γ-Secretase Activity toward Amyloid Precursor Protein and Other Substrates*. J. Biol. Chem. 2010, 285, 38042–38052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi-Niidome, S.; Sasaki, T.; Osawa, S.; Sato, T.; Morishima, K.; Cai, T.; Iwatsubo, T.; Tomita, T. Cooperative Roles of Hydrophilic Loop 1 and the C-Terminus of Presenilin 1 in the Substrate-Gating Mechanism of γ-Secretase. J. Neurosci. 2015, 35, 2646–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denvir, J.; Neitch, S.; Fan, J.; Niles, R.M.; Boskovic, G.; Schreurs, B.G.; Primerano, D.A.; Alkon, D.L. Identification of the PS1 Thr147Ile Variant in a Family with Very Early Onset Dementia and Expressive Aphasia. J. Alzheimer’s Dis. 2015, 46, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhou, J.; Li, H.-L.; Chen, Y.-G.; Cheng, H.-R.; Ye, L.-Q.; Liu, D.-S.; Chen, D.-F.; Tao, Q.-Q.; Wu, Z.-Y. Mutation screening in Chinese patients with familial Alzheimer’s disease by whole-exome sequencing. Neurobiol. Aging 2019, 76, 215.e15–215.e21. [Google Scholar] [CrossRef] [PubMed]
- Rogaeva, E.A.; Fafel, K.; Song, Y.-Q.; Medeiros, H.; Sato, C.; Liang, Y.; Richard, E.; I Rogaev, E.; Frommelt, P.; Sadovnick, A.D.; et al. Screening for PS1 mutations in a referral-based series of AD cases: 21 novel mutations. Neurology 2001, 57, 621–625. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosome | Mutation | Patient 1 | Patient 2 | Rs ID | 1000g | ExAC | SIFT | Polyphen2 |
|---|---|---|---|---|---|---|---|---|---|
| ABCA7 | 19 | E188G | found | found | rs3764645 | 0.399561 | 0.4838 | 0.647, T | 0.358, B |
| R463H | NA | found | rs3752233 | 0.060703 | 0.0478 | 0.254, T | 0.997, D | ||
| N718T | found | found | rs3752239 | 0.059105 | 0.0703 | 0.239, T | 0.529, P | ||
| G1527A | found | found | rs3752246 | 0.825479 | 0.8405 | 0.877, T | 0.0, B | ||
| Q1686R | found | found | rs4147918 | 0.05651 | 0.0479 | 0.234, T | 0.001, B | ||
| V1729M | NA | found | NA | NA | NA | 0.151, T | 0.1, B | ||
| A2045S | found | found | rs4147934 | 0.605032 | 0.7317 | 0.962, T | 0.057, B | ||
| ALS2 | 2 | V368M | found | found | rs3219156 | 0.896565 | 0.9106 | 0.191, T | 0.006, B |
| BACE1 | 11 | C412R | found | found | rs539765 | 1 | 0.9997 | 1.0, T | 0.0, B |
| BIN1 | 2 | R263Q | NA | found | rs117721706 | 0.004593 | 0.0015 | 0.349, T | 0.997, D |
| CR1 | 1 | T1858M | found | found | rs3737002 | 0.248802 | 0.275 | 0.021, D | 1.0, D |
| T2060S | found | found | rs4844609 | 0.995008 | 0.9853 | 1.0, T | 0.003, B | ||
| T2419A | found | found | rs2296160 | 0.828075 | 0.8159 | 1.0, T | 0.0, B | ||
| CTNNA3 | 10 | S596N | found | found | rs4548513 | 0.485024 | 0.412 | 1.0, T | 0.0, B |
| LRP6 | 12 | V1062I | found | found | rs2302685 | 0.885583 | 0.8474 | 1.0, T | 0.0, B |
| LRRK2 | 12 | R50H | found | found | rs2256408 | 0.969249 | 0.9911 | 1.0, T | 0.0, B |
| N551K | found | found | rs7308720 | 0.099441 | 0.0861 | 0.009, D | 1.0, D | ||
| R1398H | found | found | rs7133914 | 0.100439 | 0.0841 | 0.1, T | 0.992, D | ||
| M2397T | found | found | rs3761863 | 0.551717 | 0.624 | 0.466, T | 0.0, B | ||
| MAPT | 17 | Y441H | found | found | rs2258689 | 0.312899 | 0.2752 | 0.978, T | 0.001, B |
| NOTCH3 | 19 | A2223V | found | found | rs1044009 | 0.629393 | 0.7591 | 0.175, T | 0.001, B |
| OPTN | 10 | K322E | found | found | rs523747 | 0.993411 | 0.9973 | 1.0, T | 0.0, B |
| PARK2 | 6 | S167N | NA | found | rs1801474 | 0.117412 | 0.0676 | 0.229, T | 0.027, B |
| PSEN1 | 14 | T119I | found | found | NA | NA | NA | 0.385, T | 0.998, D |
| SIGMAR1 | 9 | Q2P | found | found | rs1800866 | 0.217252 | 0.184 | 0.513, T | 0.0, B |
| SORL1 | 11 | A528T | NA | found | rs2298813 | 0.103235 | 0.0722 | 0.306, T | 0.962, D |
| N828S | NA | found | rs377222446 | 0.0002 | 0.0002 | 0.745, T | 0.0, B | ||
| Q1074E | found | found | rs1699107 | 0.984824 | 0.9949 | 1.0, T | 0.0, B | ||
| G1524R | NA | found | rs201415809 | 0.0008 | 0.0002 | 0.008, D | 1.0, D | ||
| V1967I | found | found | rs1792120 | 0.979433 | 0.9953 | 1.0, T | 0.0, B | ||
| SPG11 | 15 | F463S | found | found | rs3759871 | 0.47484 | 0.4659 | 0.343, T | 0.066, B |
| Argentinian Family | Chinese Family | Korean-1 | Korean-2 | |
|---|---|---|---|---|
| Age of onset (years) | 49–71 years | Late 60s | 64 years | 49 years |
| Family history | Positive | Positive | Probably de novo | Positive |
| Disease | EOAD | LOAD, behavioral variant | EOAD | EOAD |
| CDR | 0.5, later 3 | 2 | 0.5 | 1.5 |
| MMSE | 28/30 | 16/30 | 28/30 | 14/30 |
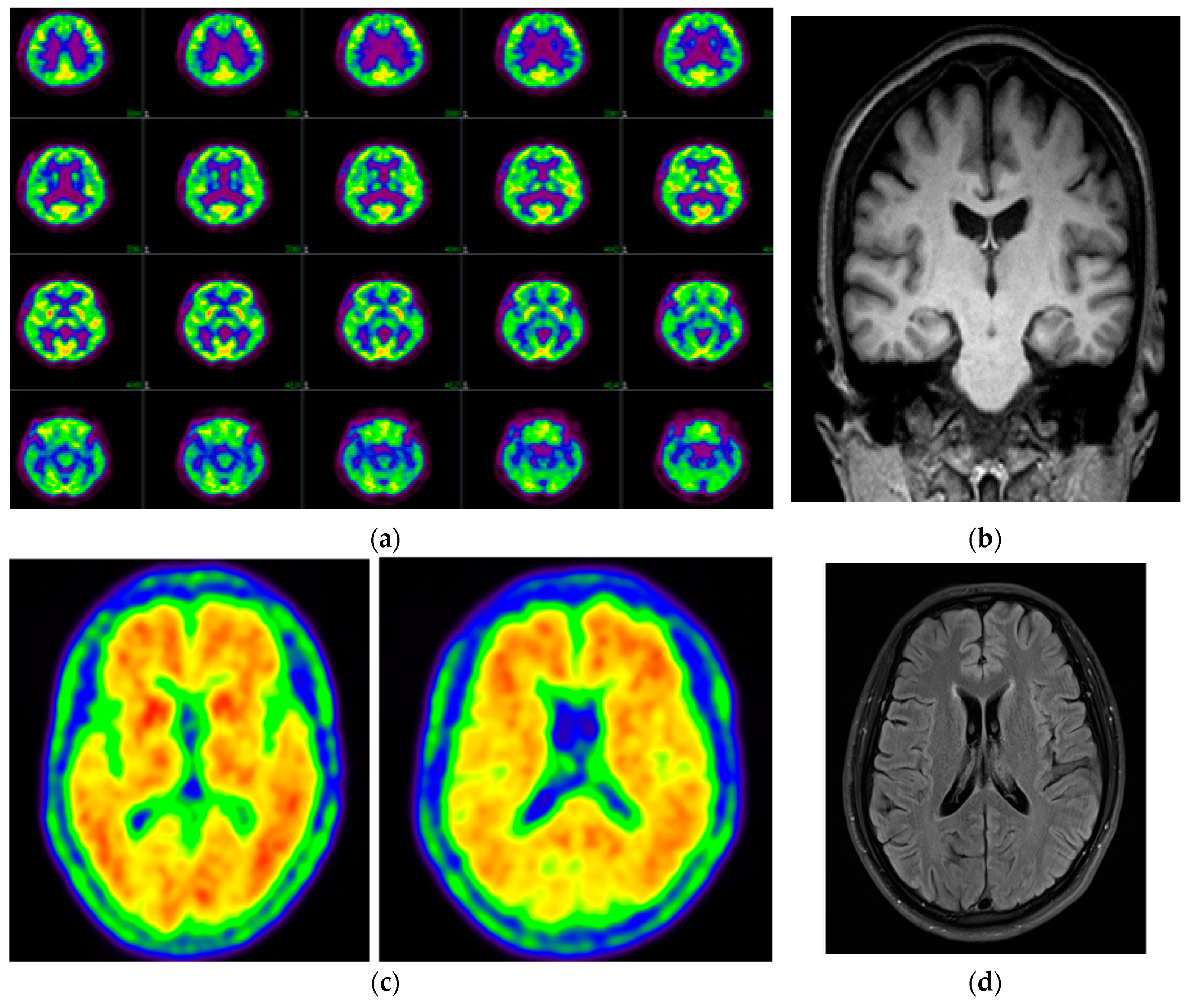
| Amyloid PET | Positive, frontal, parietal, precuneus/posterior cingulate, lateral temporal, and striatum | Diffuse amyloid retention in bilateral-parietal and temporal cortex | NA | Positive, posterior cingulate, and precuneus |
| FDG-PET | Mild bilateral hypometabolism in parietal lobe, precuneus, anterior cingulate, dorsal frontal lobe, and lateral temporal lobe with left predominance | NA | Bilateral hypometabolism in parietal and temporal cortices | NA |
| MRI | NA | Atrophy in frontal-temporal lobe, shrinkage of hippocampus | Global atrophy | No significant changes |
| CSF biomarkers | Reduced Aβ or elevated Tau | NA | NA | NA |
| References | [15] | [16] | [11,12] recent finding | Recent finding |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagyinszky, E.; Lee, H.; Pyun, J.M.; Suh, J.; Kang, M.J.; Vo, V.G.; An, S.S.A.; Park, K.H.; Kim, S. Pathogenic PSEN1 Thr119Ile Mutation in Two Korean Patients with Early-Onset Alzheimer’s Disease. Diagnostics 2020, 10, 405. https://doi.org/10.3390/diagnostics10060405
Bagyinszky E, Lee H, Pyun JM, Suh J, Kang MJ, Vo VG, An SSA, Park KH, Kim S. Pathogenic PSEN1 Thr119Ile Mutation in Two Korean Patients with Early-Onset Alzheimer’s Disease. Diagnostics. 2020; 10(6):405. https://doi.org/10.3390/diagnostics10060405
Chicago/Turabian StyleBagyinszky, Eva, Hyon Lee, Jung Min Pyun, Jeewon Suh, Min Ju Kang, Van Giau Vo, Seong Soo A. An, Kee Hyung Park, and SangYun Kim. 2020. "Pathogenic PSEN1 Thr119Ile Mutation in Two Korean Patients with Early-Onset Alzheimer’s Disease" Diagnostics 10, no. 6: 405. https://doi.org/10.3390/diagnostics10060405