
Homocysteine Editing, Thioester Chemistry, Coenzyme A, and the Origin of Coded Peptide Synthesis †

Abstract
:1. Introduction
2. Homocysteine (Hcy) is Edited by Class I and Class II Aminoacyl-tRNA Synthetases (AARSs)

2.1. Hcy Editing is Universal
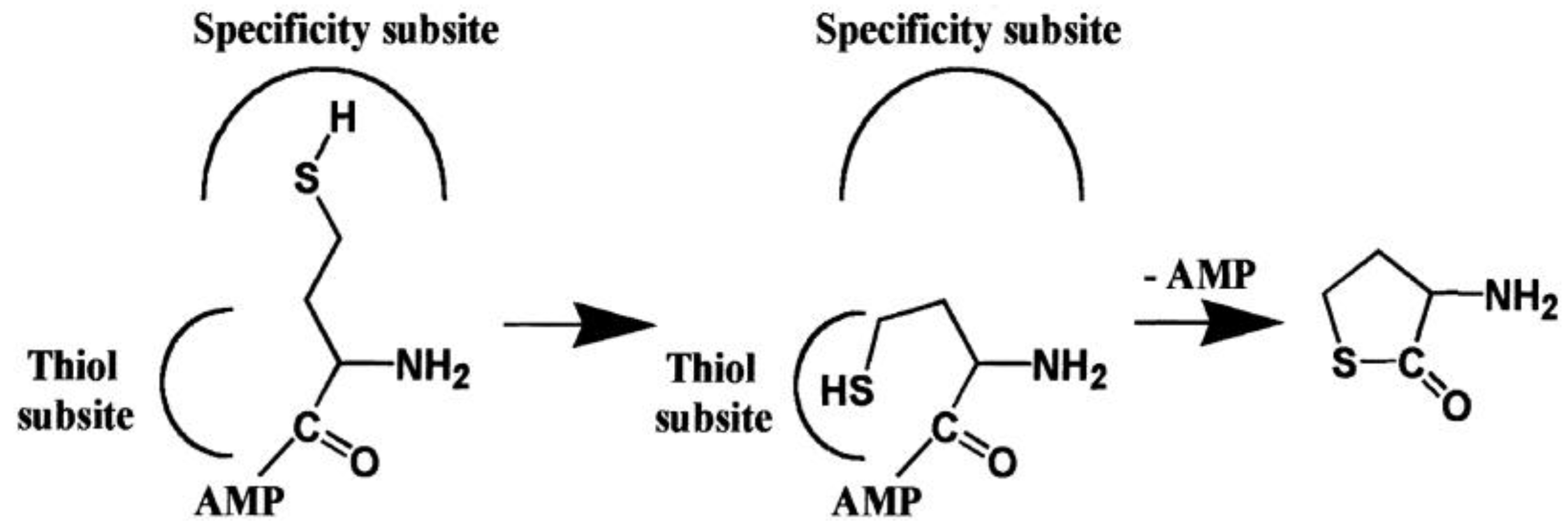
2.2. Mechanism of Hcy Editing
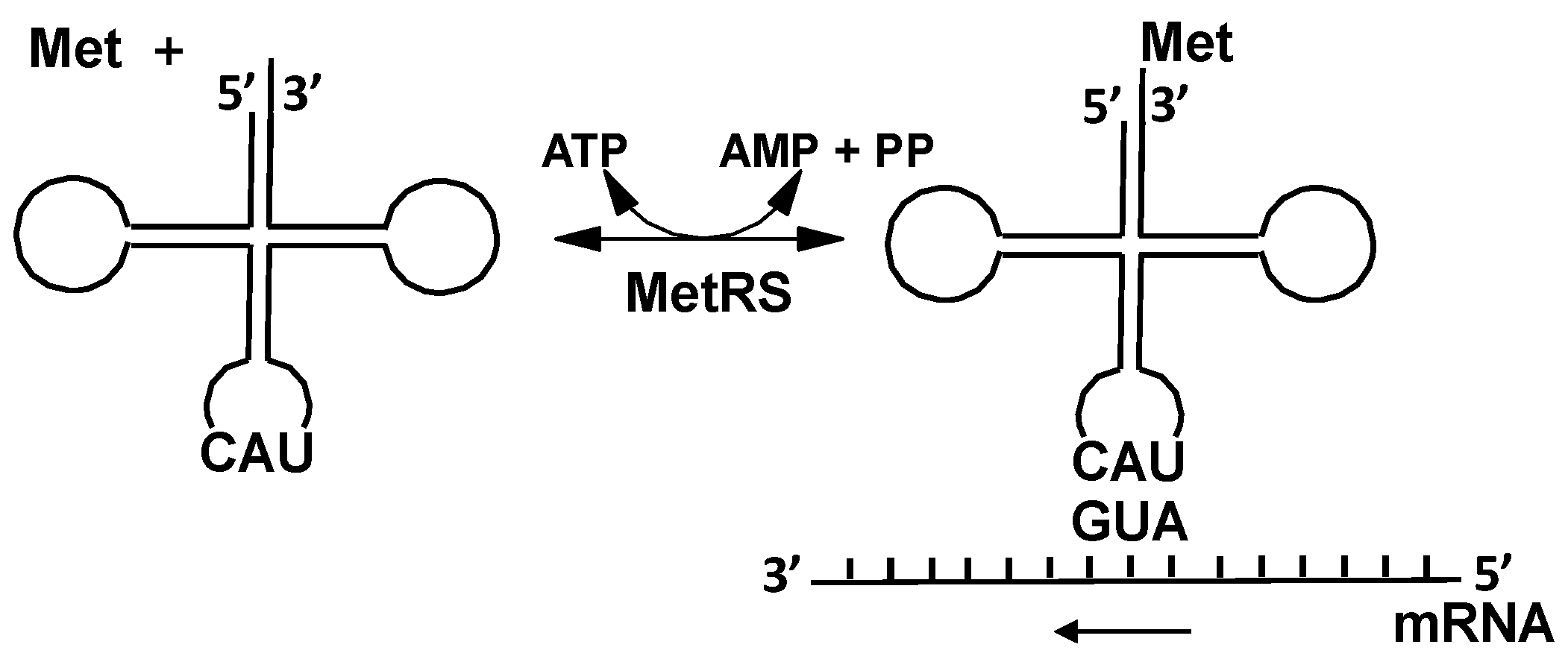
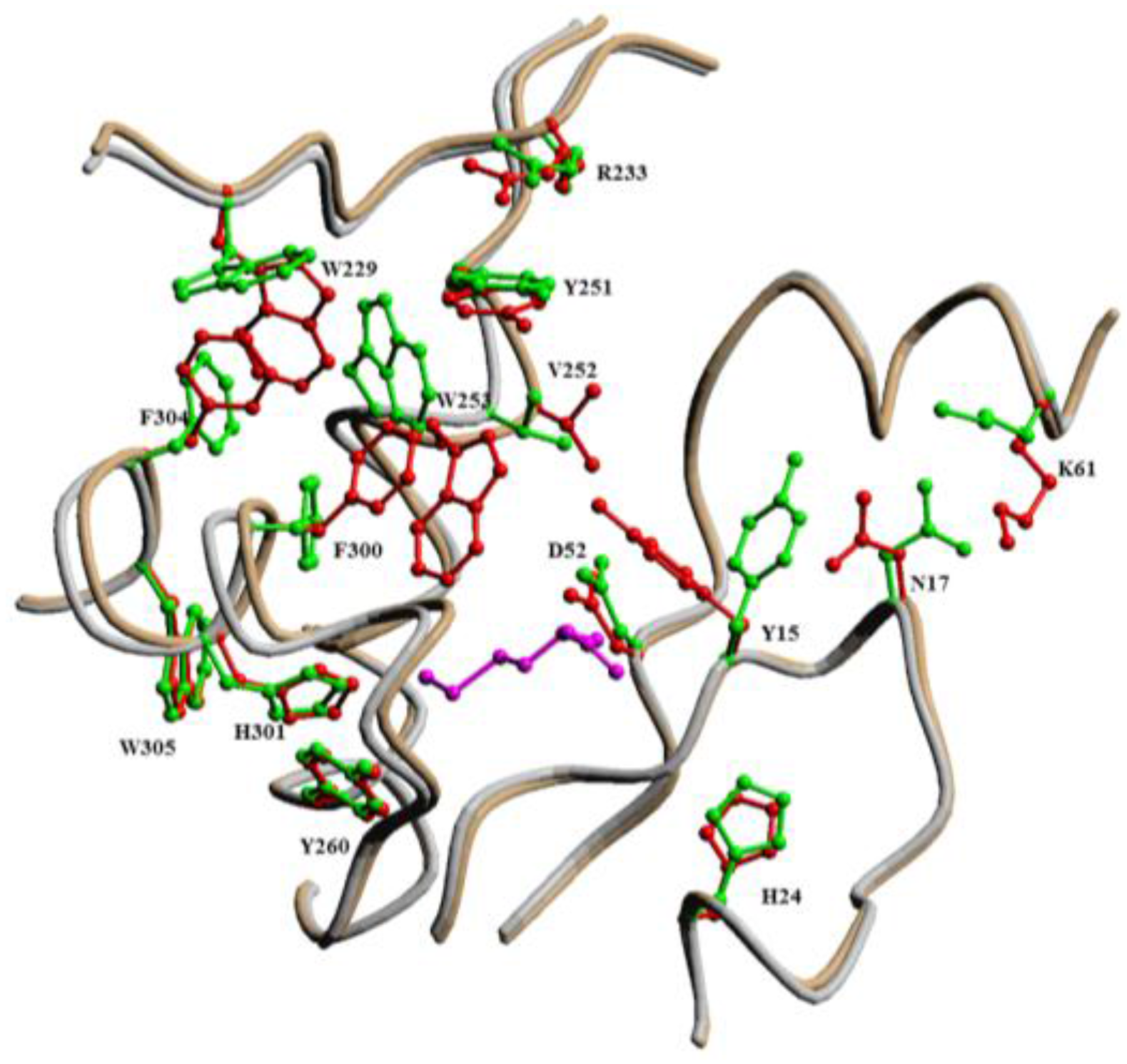
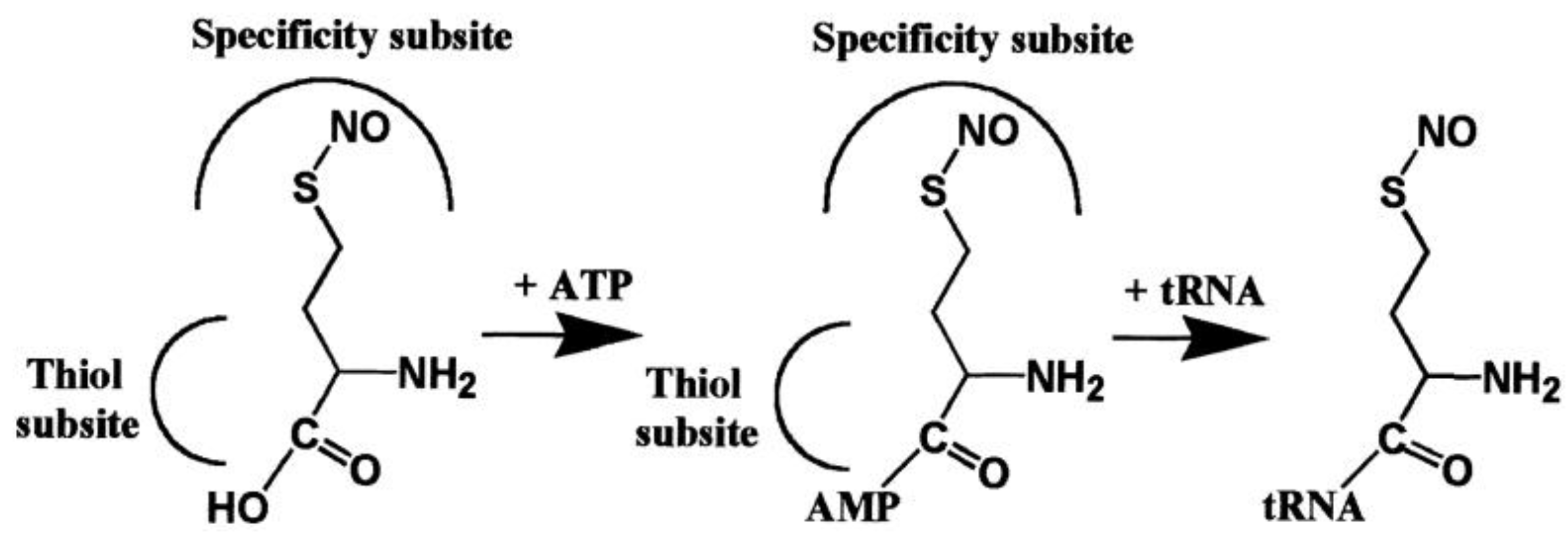
2.2.1. Methionyl-tRNA Synthetase (MetRS)
2.2.2. Leucyl-tRNA Synthetase (LeuRS), Isoleucyl-tRNA Synthetase (IleRS), Valyl-tRNA Synthetase (ValRS)
2.2.3. Lysyl-tRNA Synthetase (LysRS) Edits Homocysteine (Hcy), Ornithine (Orn), Homoserine (Hse), but Mischarges tRNALys with Proteinogenic Amino Acids
3. Expanding the Genetic Code: Decoding Methionine Codons by Homocysteine
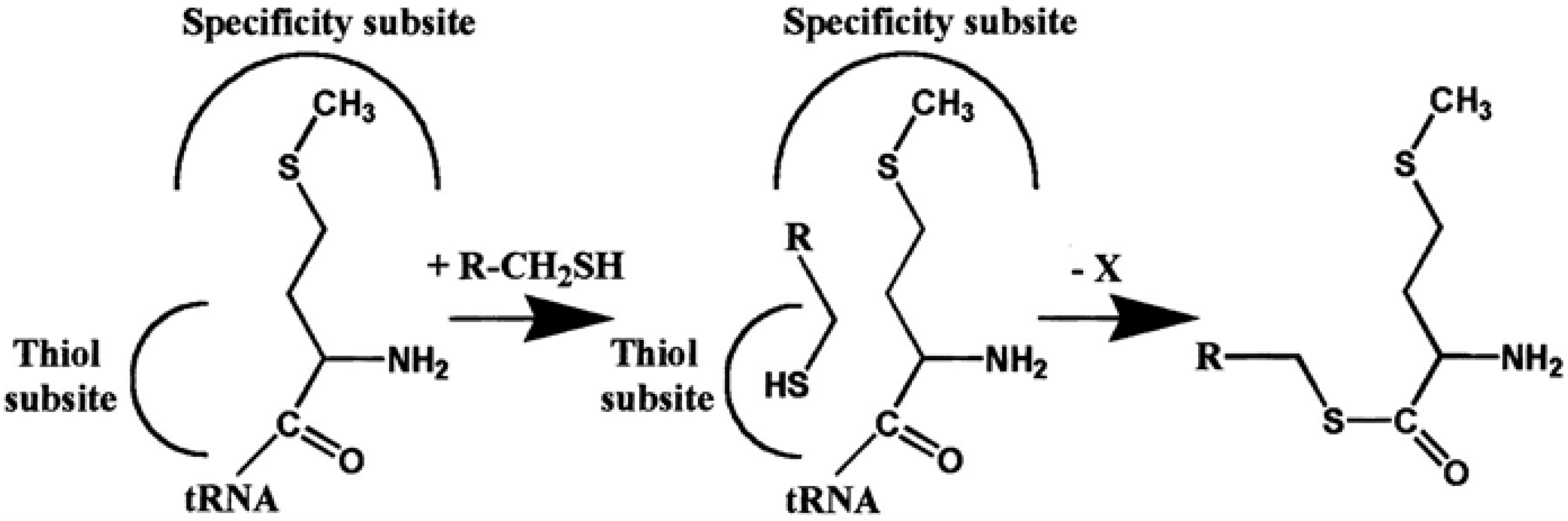
4. AARSs Support the Aminoacylation of Thiols and Peptide Bond Synthesis
4.1. Methionyl-tRNA Synthetase (MetRS)
4.2. IleRS, ValRS, LysRS
4.3. Arginyl-tRNA Synthease (ArgRS), Aspartyl-tRNA Synthetase (AspRS), Seryl-tRNA Synthetases (SerRS)
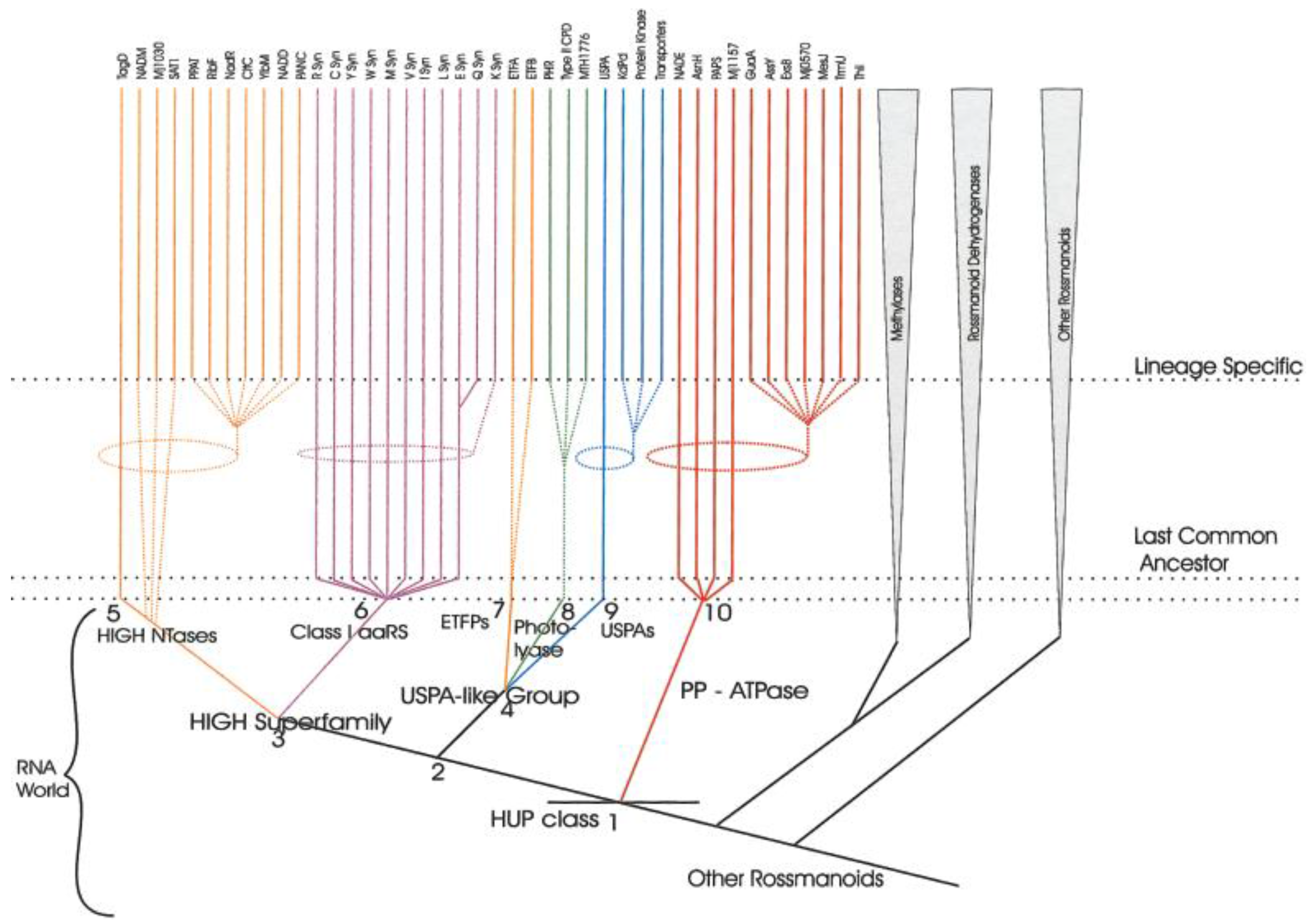
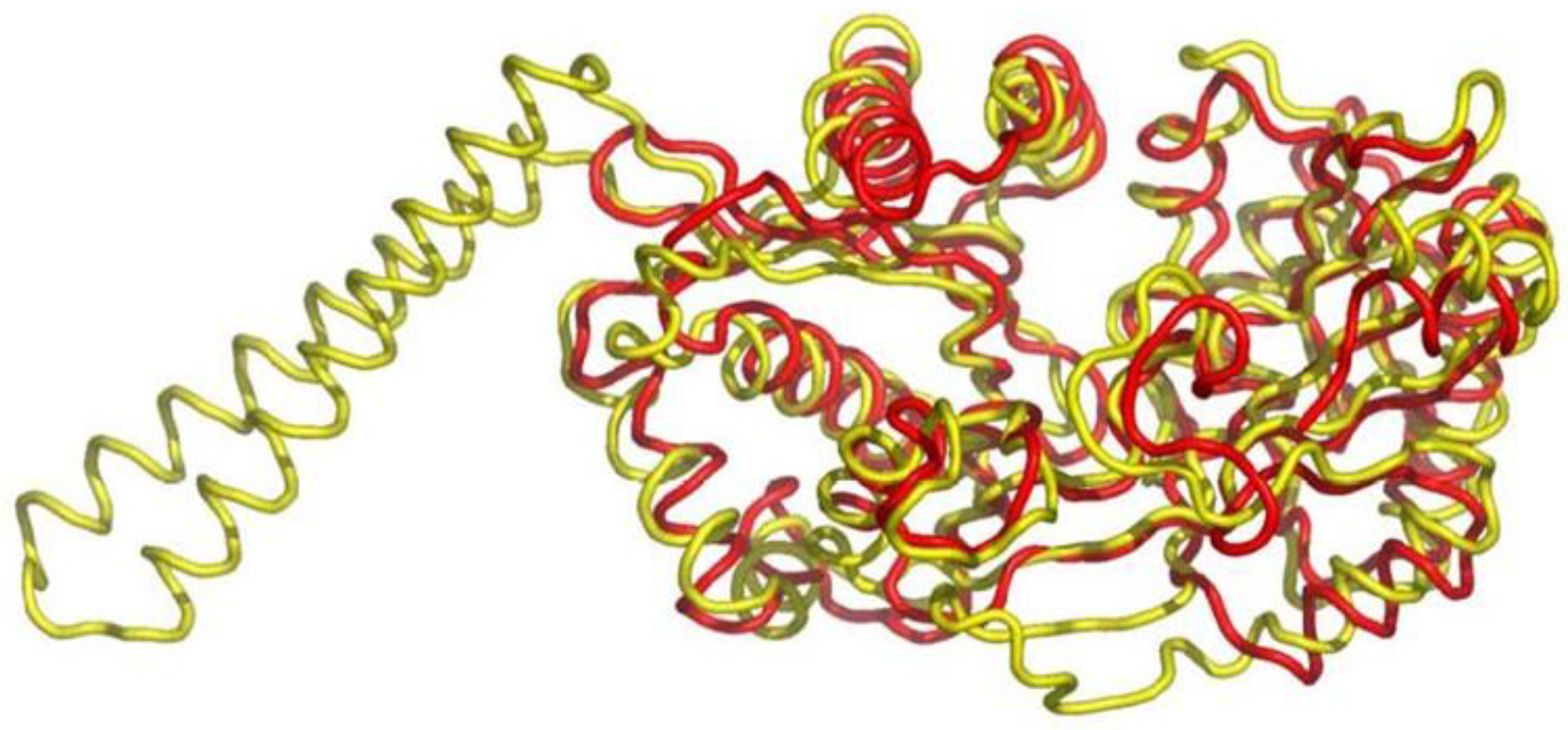
5. Class I AARS Are Related to Proteins Involved in Sulfur/CoA Metabolism
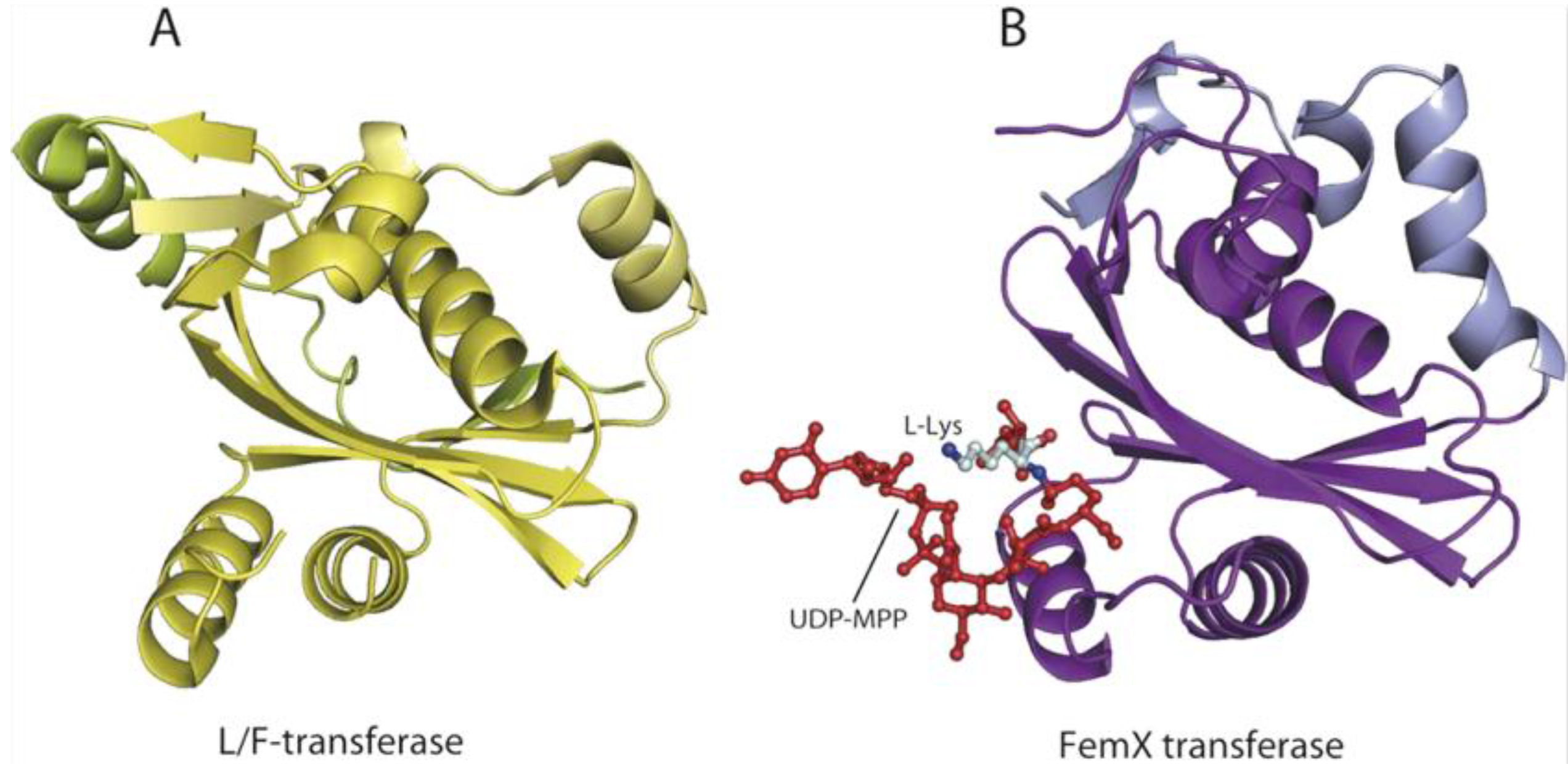
6. AARSs Are Related to Proteins Involved in Peptide Bond Synthesis
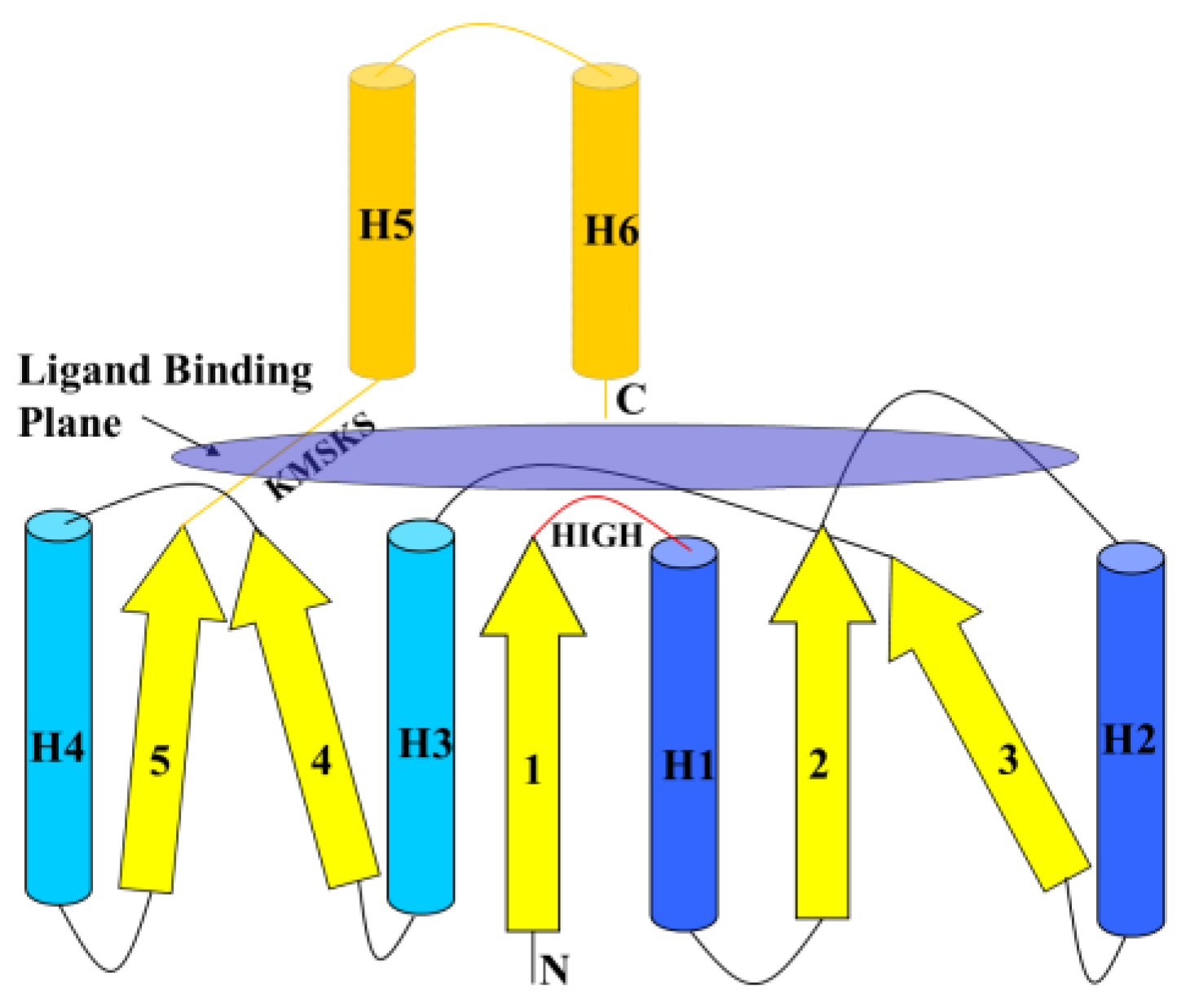
6.1. Class I AARSs
6.2. Class II AARSs
7. Thioester Chemistry of SerRS Homologues from Methanogenic Archaea
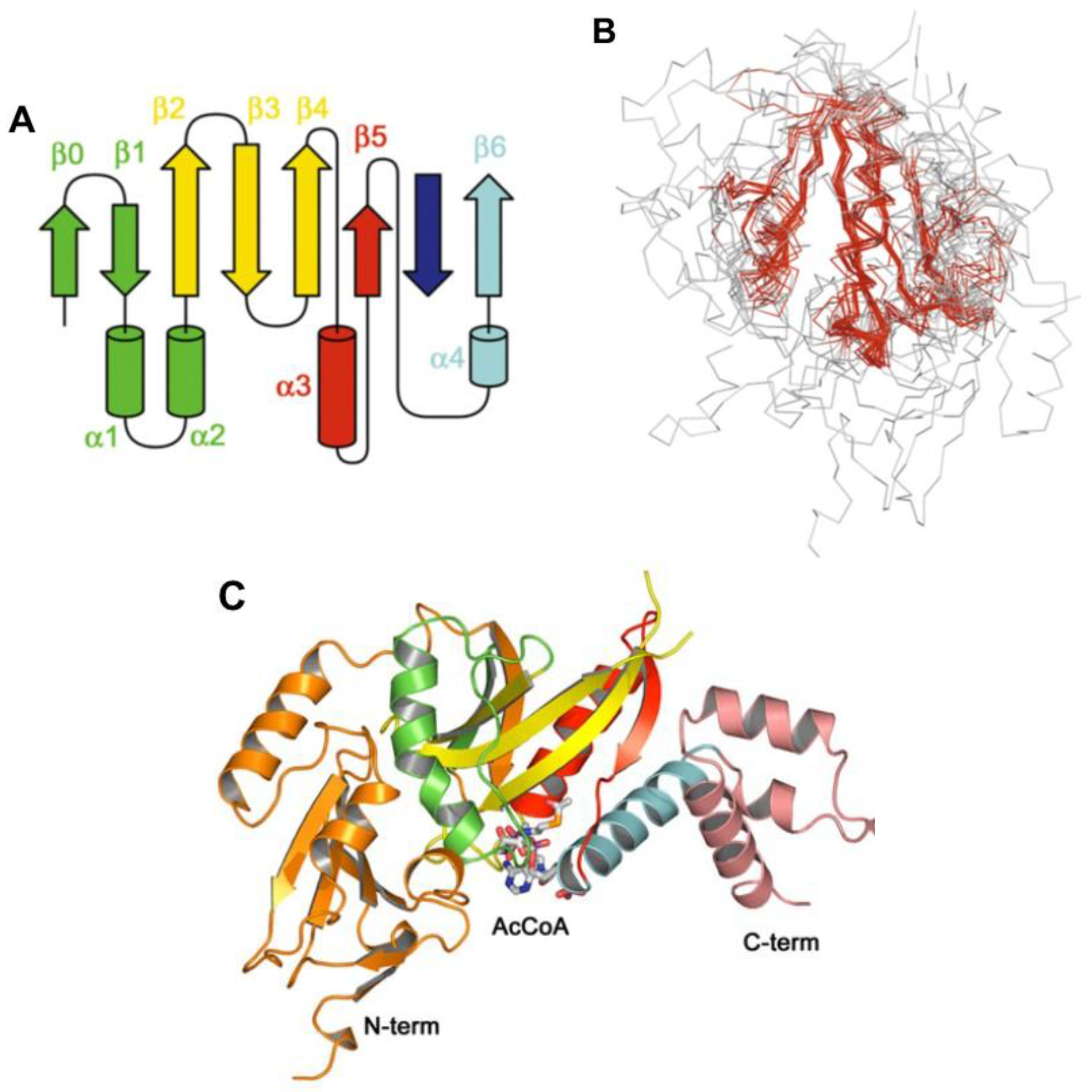
8. Acyl~S-CoA Thioesters and Aminoacyl~tRNA Esters are Used in Peptide Bond Synthesis Catalyzed the Gcn5-related N-acetyltransferases (GNAT) Fold Enzymes
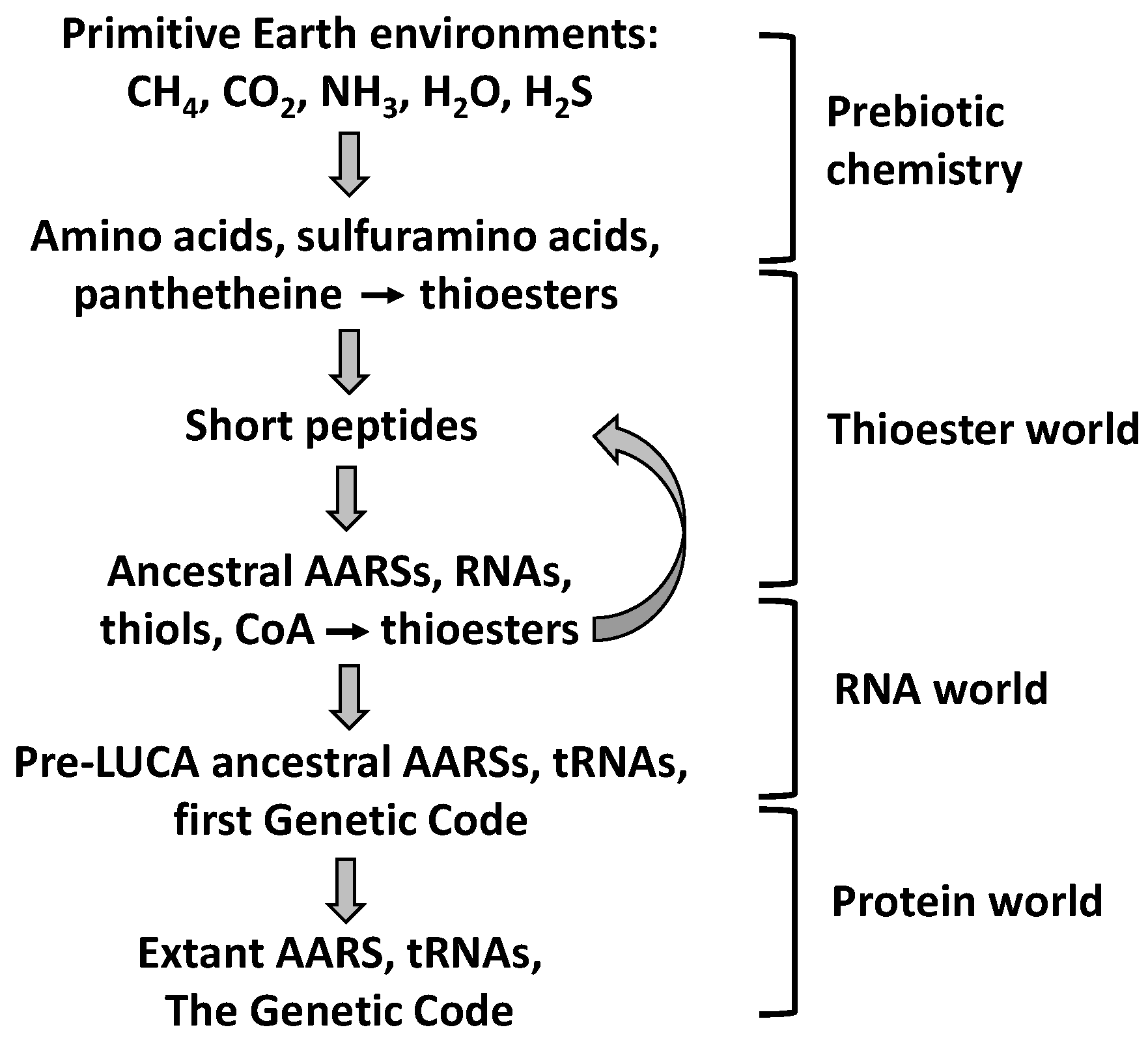
9. Evolutionary Implications
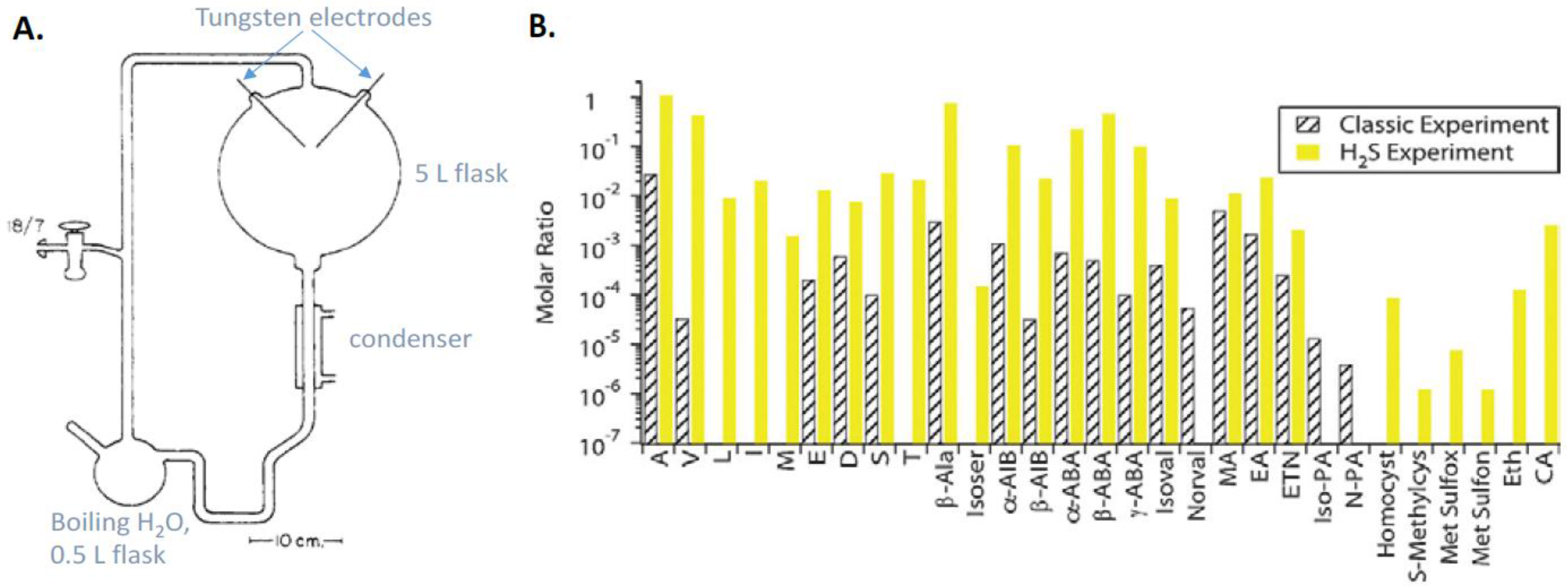
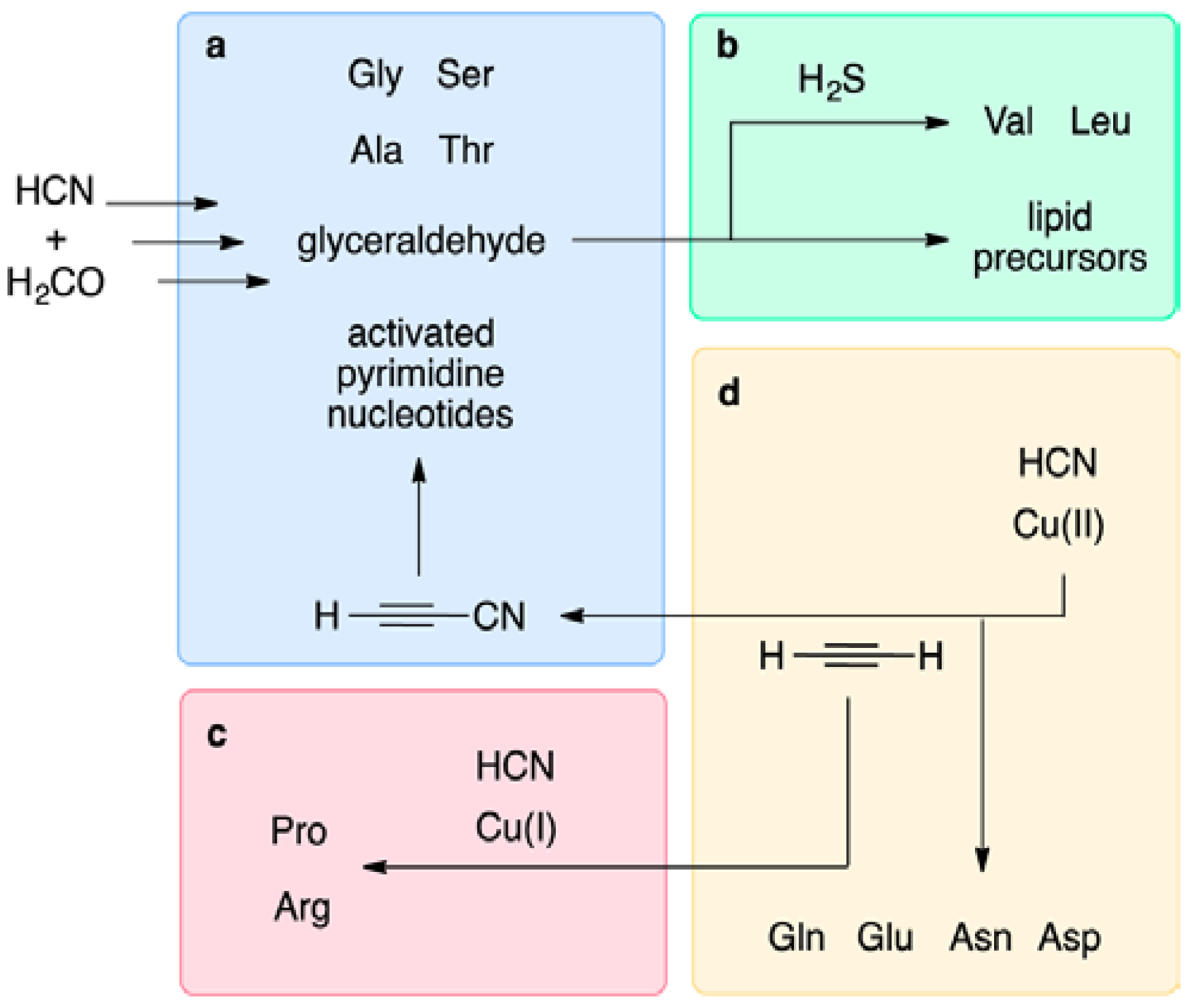
9.1. Prebiotic Synthesis of Amino Acids and Organosulfur Compounds
9.2. The Thioester World
10. Conclusions
- Coded peptide synthesis has been preceded by a prebiotic stage, a Thioester World, in which thioesters played key roles.
- Remnants of the Thioester World can be found in extant aminoacyl-tRNA synthetases (AARSs) and related proteins:
- A thiol-binding site at the catalytic domain of AARSs confers the amino acid:thiol ligase activity;
- AARSs are structurally related to proteins involved in sulfur/CoA-SH metabolism and peptide bond synthesis;
- Protein folds that bind pantetheine/CoA-SH can also bind tRNA.
Acknowledgments
Conflicts of Interest
References
- Woese, C.R.; Olsen, G.J.; Ibba, M.; Soll, D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 2000, 64, 202–236. [Google Scholar] [CrossRef] [PubMed]
- Giege, R.; Springer, M. Aminoacyl-tRNA Synthetases in the Bacterial World. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Plateau, P.; Mayaux, J.F.; Blanquet, S. Zinc(II)-dependent synthesis of diadenosine 5′,5′′′-P1,P4-tetraphosphate by Escherichia coli and yeast phenylalanyl transfer ribonucleic acid synthetases. Biochemistry 1981, 20, 4654–4662. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Synthesis of diadenosine 5′,5′′′-P1,P4-tetraphosphate and related compounds by plant (Lupinus luteus) seryl-tRNA and phenylalanyl-tRNA synthetases. Acta Biochim. Pol. 1983, 30, 51–69. [Google Scholar] [PubMed]
- Goerlich, O.; Foeckler, R.; Holler, E. Mechanism of synthesis of adenosine(5′)tetraphospho(5′)adenosine (AppppA) by aminoacyl-tRNA synthetases. Eur. J. Biochem. 1982, 126, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Schimmel, P. Essential nontranslational functions of tRNA synthetases. Nat. Chem Biol. 2013, 9, 145–153. [Google Scholar] [PubMed]
- Ofir-Birin, Y.; Fang, P.; Bennett, S.P.; Zhang, H.M.; Wang, J.; Rachmin, I.; Shapiro, R.; Song, J.; Dagan, A.; Pozo, J.; et al. Structural switch of lysyl-tRNA synthetase between translation and transcription. Mol. Cell 2013, 49, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Brevet, A.; Chen, J.; Leveque, F.; Plateau, P.; Blanquet, S. In vivo synthesis of adenylylated bis(5′-nucleosidyl) tetraphosphates (Ap4N) by Escherichia coli aminoacyl-tRNA synthetases. Proc. Natl. Acad. Sci. USA 1989, 86, 8275–8279. [Google Scholar] [CrossRef] [PubMed]
- Yamane, T.; Hopfield, J.J. Experimental evidence for kinetic proofreading in the aminoacylation of tRNA by synthetase. Proc. Natl. Acad. Sci. USA 1977, 74, 2246–2250. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Quality control in tRNA charging—Editing of homocysteine. Acta Biochim. Pol. 2011, 58, 149–163. [Google Scholar] [PubMed]
- Jakubowski, H. Quality control in tRNA charging. Wiley Interdiscip. Rev. RNA 2012, 3, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Schulman, L.H. Recognition of tRNAs by aminoacyl-tRNA synthetases. Prog. Nucleic Acid Res. Mol. Biol. 1991, 41, 23–87. [Google Scholar] [PubMed]
- Netzer, N.; Goodenbour, J.M.; David, A.; Dittmar, K.A.; Jones, R.B.; Schneider, J.R.; Boone, D.; Eves, E.M.; Rosner, M.R.; Gibbs, J.S.; et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 2009, 462, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.H.; Waldbauer, J.R.; Zhang, L.; Pan, T. Global tRNA misacylation induced by anaerobiosis and antibiotic exposure broadly increases stress resistance in Escherichia coli. Nucleic Acids Res. 2016, 44, 10292–10303. [Google Scholar] [PubMed]
- Ling, J.; Soll, D. Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proc. Natl. Acad Sci. USA 2010, 107, 4028–4033. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H.; Goldman, E. Editing of errors in selection of amino acids for protein synthesis. Microbiol. Rev. 1992, 56, 412–429. [Google Scholar] [PubMed]
- Baldwin, A.N.; Berg, P. Transfer ribonucleic acid-induced hydrolysis of valyladenylate bound to isoleucyl ribonucleic acid synthetase. J. Biol. Chem. 1966, 241, 839–845. [Google Scholar] [PubMed]
- Jakubowski, H.; Fersht, A.R. Alternative pathways for editing non-cognate amino acids by aminoacyl-tRNA synthetases. Nucleic Acids Res. 1981, 9, 3105–3117. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Valyl-tRNA synthetase form yellow lupin seeds: Hydrolysis of the enzyme-bound noncognate aminoacyl adenylate as a possible mechanism of increasing specificity of the aminoacyl-tRNA synthetase. Biochemistry 1980, 19, 5071–5078. [Google Scholar] [CrossRef] [PubMed]
- Eldred, E.W.; Schimmel, P.R. Rapid deacylation by isoleucyl transfer ribonucleic acid synthetase of isoleucine-specific transfer ribonucleic acid aminoacylated with valine. J. Biol. Chem. 1972, 247, 2961–2964. [Google Scholar] [PubMed]
- Yarus, M. Phenylalanyl-tRNA synthetase and isoleucyl-tRNA Phe: A possible verification mechanism for aminoacyl-tRNA. Proc. Natl. Acad. Sci. USA 1972, 69, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Perona, J.J.; Gruic-Sovulj, I. Synthetic and editing mechanisms of aminoacyl-tRNA synthetases. Top. Curr. Chem. 2014, 344, 1–41. [Google Scholar] [PubMed]
- SternJohn, J.; Hati, S.; Siliciano, P.G.; Musier-Forsyth, K. Restoring species-specific posttransfer editing activity to a synthetase with a defunct editing domain. Proc. Natl. Acad. Sci. USA 2007, 104, 2127–2132. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Proofreading in vivo: Editing of homocysteine by methionyl-tRNA synthetase in Escherichia coli. Proc. Natl. Acad. Sci. USA 1990, 87, 4504–4508. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Proofreading in vivo. Editing of homocysteine by aminoacyl-tRNA synthetases in Escherichia coli. J. Biol. Chem. 1995, 270, 17672–17673. [Google Scholar] [PubMed]
- Sikora, M.; Jakubowski, H. Homocysteine editing and growth inhibition in Escherichia coli. Microbiology 2009, 155, 1858–1865. [Google Scholar] [CrossRef]
- Jakubowski, H. Proofreading in vivo: Editing of homocysteine by methionyl-tRNA synthetase in the yeast Saccharomyces cerevisiae. EMBO J. 1991, 10, 593–598. [Google Scholar] [PubMed]
- Chwatko, G.; Boers, G.H.; Strauss, K.A.; Shih, D.M.; Jakubowski, H. Mutations in methylenetetrahydrofolate reductase or cystathionine beta-synthase gene, or a high-methionine diet, increase homocysteine thiolactone levels in humans and mice. FASEB J. 2007, 21, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Misacylation of tRNALys with noncognate amino acids by lysyl-tRNA synthetase. Biochemistry 1999, 38, 8088–8093. [Google Scholar] [CrossRef] [PubMed]
- Cvetesic, N.; Palencia, A.; Halasz, I.; Cusack, S.; Gruic-Sovulj, I. The physiological target for LeuRS translational quality control is norvaline. EMBO J. 2014, 33, 1639–1653. [Google Scholar] [CrossRef] [PubMed]
- Bullwinkle, T.; Lazazzera, B.; Ibba, M. Quality control and infiltration of translation by amino acids outside of the genetic code. Annu. Rev. Genet. 2014, 48, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S. The Non-Protein Amino Acids. In Chemistry and Biochemistry of the Amino Acids; Springer: Wien, Austria, 1985; pp. 55–138. [Google Scholar]
- Rosenthal, G.A. Plant Nonprotein Amino and Imino Acids; Academic Press: New York, NY, USA, 1982; p. 272. [Google Scholar]
- Caetano-Anolles, G.; Kim, K.M.; Caetano-Anolles, D. The phylogenomic roots of modern biochemistry: Origins of proteins, cofactors and protein biosynthesis. J. Mol. Evol. 2012, 74, 1–34. [Google Scholar] [CrossRef]
- Caetano-Anolles, D.; Caetano-Anolles, G. Piecemeal Buildup of the Genetic Code, Ribosomes, and Genomes from Primordial tRNA Building Blocks. Life 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Jiang, Y.Y.; Kim, K.M.; Qu, G.; Ji, H.F.; Mittenthal, J.E.; Zhang, H.Y.; Caetano-Anolles, G. A universal molecular clock of protein folds and its power in tracing the early history of aerobic metabolism and planet oxygenation. Mol. Biol. Evol. 2011, 28, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F.; Hartman, H. The evolution of Class II Aminoacyl-tRNA synthetases and the first code. FEBS Lett. 2015, 589, 3499–3507. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Aminoacyl thioester chemistry of class II aminoacyl-tRNA synthetases. Biochemistry 1997, 36, 11077–11085. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Metabolism of homocysteine thiolactone in human cell cultures. Possible mechanism for pathological consequences of elevated homocysteine levels. J. Biol. Chem. 1997, 272, 1935–1942. [Google Scholar] [PubMed]
- Jakubowski, H. Protein homocysteinylation: Possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999, 13, 2277–2283. [Google Scholar] [PubMed]
- Jakubowski, H. Homocysteine in Protein Structure/Function and Human Disease—Chemical Biology of Homocysteine-containing Proteins; Springer: Wien, Austria, 2013. [Google Scholar]
- Guzzo, M.B.; Nguyen, H.T.; Pham, T.H.; Wyszczelska-Rokiel, M.; Jakubowski, H.; Wolff, K.A.; Ogwang, S.; Timpona, J.L.; Gogula, S.; Jacobs, M.R.; et al. Methylfolate Trap Promotes Bacterial Thymineless Death by Sulfa Drugs. PLoS Pathog. 2016, 12, e1005949. [Google Scholar] [CrossRef] [PubMed]
- Rauch, B.J.; Perona, J.J. Efficient Sulfide Assimilation in Methanosarcina acetivorans Is Mediated by the MA1715 Protein. J. Bacteriol. 2016, 198, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. The determination of homocysteine-thiolactone in biological samples. Anal. Biochem. 2002, 308, 112–119. [Google Scholar] [CrossRef]
- Jakubowski, H.; Guranowski, A. Metabolism of homocysteine-thiolactone in plants. J. Biol. Chem. 2003, 278, 6765–6770. [Google Scholar] [CrossRef] [PubMed]
- Chwatko, G.; Jakubowski, H. Urinary excretion of homocysteine-thiolactone in humans. Clin. Chem. 2005, 51, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Senger, B.; Despons, L.; Walter, P.; Jakubowski, H.; Fasiolo, F. Yeast cytoplasmic and mitochondrial methionyl-tRNA synthetases: Two structural frameworks for identical functions. J. Mol. Biol. 2001, 311, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids 2015, 47, 1319–1339. [Google Scholar] [CrossRef] [PubMed]
- Paoli, P.; Sbrana, F.; Tiribilli, B.; Caselli, A.; Pantera, B.; Cirri, P.; De Donatis, A.; Formigli, L.; Nosi, D.; Manao, G.; et al. Protein N-homocysteinylation induces the formation of toxic amyloid-like protofibrils. J. Mol. Biol. 2010, 400, 889–907. [Google Scholar] [CrossRef] [PubMed]
- Khayati, K.; Antikainen, H.; Bonder, E.M.; Weber, G.F.; Kruger, W.D.; Jakubowski, H.; Dobrowolski, R. The amino acid metabolite homocysteine activates mTORC1 to inhibit autophagy and form abnormal proteins in human neurons and mice. FASEB J. 2017, 31, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Keating, A.K.; Freehauf, C.; Jiang, H.; Brodsky, G.L.; Stabler, S.P.; Allen, R.H.; Graham, D.K.; Thomas, J.A.; van Hove, J.L.; Maclean, K.N. Constitutive induction of pro-inflammatory and chemotactic cytokines in cystathionine beta-synthase deficient homocystinuria. Mol. Genet. Metab. 2011, 103, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Perla, J.; Lacinski, M.; Trzeciak, W.; Kazmierski, R.; Jakubowski, H. Autoantibodies against N-homocysteinylated proteins in humans: Implications for atherosclerosis. Stroke 2004, 35, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Zhang, D.; Cheng, Z.; Yan, C.; Jiang, X.; Kruger, W.D.; Meng, S.; Arning, E.; Bottiglieri, T.; Choi, E.T.; et al. Hyperhomocysteinemia potentiates hyperglycemia-induced inflammatory monocyte differentiation and atherosclerosis. Diabetes 2014, 63, 4275–4290. [Google Scholar] [CrossRef] [PubMed]
- Crepin, T.; Schmitt, E.; Blanquet, S.; Mechulam, Y. Three-dimensional structure of methionyl-tRNA synthetase from Pyrococcus abyssi. Biochemistry 2004, 43, 2635–2644. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. The synthetic/editing active site of an aminoacyl-tRNA synthetase: Evidence for binding of thiols in the editing subsite. Biochemistry 1996, 35, 8252–8259. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Ghosh, G.; Schulman, L.H.; Brunie, S.; Jakubowski, H. The relationship between synthetic and editing functions of the active site of an aminoacyl-tRNA synthetase. Proc. Natl. Acad. Sci. USA 1993, 90, 11553–11557. [Google Scholar] [CrossRef] [PubMed]
- Serre, L.; Verdon, G.; Choinowski, T.; Hervouet, N.; Risler, J.L.; Zelwer, C. How methionyl-tRNA synthetase creates its amino acid recognition pocket upon l-methionine binding. J. Mol. Biol. 2001, 306, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Fortowsky, G.B.; Simard, D.J.; Aboelnga, M.M.; Gauld, J.W. Substrate-Assisted and Enzymatic Pretransfer Editing of Nonstandard Amino Acids by Methionyl-tRNA Synthetase. Biochemistry 2015, 54, 5757–5765. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Translational incorporation of S-nitrosohomocysteine into protein. J. Biol. Chem. 2000, 275, 21813–21816. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Translational accuracy of aminoacyl-tRNA synthetases: Implications for atherosclerosis. J. Nutr. 2001, 131, 2983S–2987S. [Google Scholar] [PubMed]
- Jakubowski, H.; Zhang, L.; Bardeguez, A.; Aviv, A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: Implications for atherosclerosis. Circ. Res. 2000, 87, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Accuracy of Aminoacyl-tRNA Synthetases: Proofreading of Amino Acids. In The Aminoacyl-tRNA Synthetases; Ibba, M., Francklyn, C., Cusack, S., Eds.; Landes Bioscience/Eurekah.com: Georgetown, TX, USA, 2005; pp. 384–396. [Google Scholar]
- Jakubowski, H. Aminoacylation of coenzyme A and pantetheine by aminoacyl-tRNA synthetases: Possible link between noncoded and coded peptide synthesis. Biochemistry 1998, 37, 5147–5153. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Amino acid selectivity in the aminoacylation of coenzyme A and RNA minihelices by aminoacyl-tRNA synthetases. J. Biol. Chem. 2000, 275, 34845–34848. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Synthesis of cysteine-containing dipeptides by aminoacyl-tRNA synthetases. Nucleic Acids Res. 1995, 23, 4608–4615. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Proofreading in trans by an aminoacyl-tRNA synthetase: A model for single site editing by isoleucyl-tRNA synthetase. Nucleic Acids Res. 1996, 24, 2505–2510. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H.; Goldman, E. Synthesis of homocysteine thiolactone by methionyl-tRNA synthetase in cultured mammalian cells. FEBS Lett. 1993, 317, 237–240. [Google Scholar] [CrossRef]
- Jakubowski, H. Editing function of Escherichia coli cysteinyl-tRNA synthetase: Cyclization of cysteine to cysteine thiolactone. Nucleic Acids Res. 1994, 22, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Gulick, A.M. Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 2009, 4, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Finking, R.; Marahiel, M.A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef] [PubMed]
- Hartman, H.; Smith, T.F. The evolution of the ribosome and the genetic code. Life 2014, 4, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Aravind, L.; Grishin, N.V.; Koonin, E.V. Evolution of aminoacyl-tRNA synthetases—Analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999, 9, 689–710. [Google Scholar] [PubMed]
- Aravind, L.; Anantharaman, V.; Koonin, E.V. Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: Implications for protein evolution in the RNA. Proteins 2002, 48, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fournier, G.P.; Alm, E.J. Ancestral Reconstruction of a Pre-LUCA Aminoacyl-tRNA Synthetase Ancestor Supports the Late Addition of Trp to the Genetic Code. J. Mol. Evol. 2015, 80, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Von Delft, F.; Lewendon, A.; Dhanaraj, V.; Blundell, T.L.; Abell, C.; Smith, A.G. The crystal structure of E. coli pantothenate synthetase confirms it as a member of the cytidylyltransferase superfamily. Structure 2001, 9, 439–450. [Google Scholar] [CrossRef]
- Izard, T. A novel adenylate binding site confers phosphopantetheine adenylyltransferase interactions with coenzyme A. J. Bacteriol. 2003, 185, 4074–4080. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Lansdon, E.B.; Segel, I.H.; Fisher, A.J. Crystal structure of the bifunctional ATP sulfurylase-APS kinase from the chemolithotrophic thermophile Aquifex aeolicus. J. Mol. Biol. 2007, 365, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Lemongello, D.; Segel, I.H.; Fisher, A.J. Crystal structure of Saccharomyces cerevisiae 3′-phosphoadenosine-5′-phosphosulfate reductase complexed with adenosine 3′,5′-bisphosphate. Biochemistry 2008, 47, 12777–12786. [Google Scholar] [CrossRef] [PubMed]
- Sareen, D.; Steffek, M.; Newton, G.L.; Fahey, R.C. ATP-dependent l-cysteine:1D-myo-inosityl 2-amino-2-deoxy-alpha-d-glucopyranoside ligase, mycothiol biosynthesis enzyme MshC, is related to class I cysteinyl-tRNA synthetases. Biochemistry 2002, 41, 6885–6890. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, L.W.; Fan, F.; Vetting, M.W.; Blanchard, J.S. The 1.6 A crystal structure of Mycobacterium smegmatis MshC: The penultimate enzyme in the mycothiol biosynthetic pathway. Biochemistry 2008, 47, 13326–13335. [Google Scholar] [CrossRef] [PubMed]
- Newberry, K.J.; Hou, Y.M.; Perona, J.J. Structural origins of amino acid selection without editing by cysteinyl-tRNA synthetase. EMBO J. 2002, 21, 2778–2787. [Google Scholar] [CrossRef] [PubMed]
- Gondry, M.; Sauguet, L.; Belin, P.; Thai, R.; Amouroux, R.; Tellier, C.; Tuphile, K.; Jacquet, M.; Braud, S.; Courcon, M.; et al. Cyclodipeptide synthases are a family of tRNA-dependent peptide bond-forming enzymes. Nat. Chem. Biol. 2009, 5, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Belin, P.; Moutiez, M.; Lautru, S.; Seguin, J.; Pernodet, J.L.; Gondry, M. The nonribosomal synthesis of diketopiperazines in tRNA-dependent cyclodipeptide synthase pathways. Nat. Prod. Rep. 2012, 29, 961–979. [Google Scholar] [CrossRef] [PubMed]
- Moutiez, M.; Schmitt, E.; Seguin, J.; Thai, R.; Favry, E.; Belin, P.; Mechulam, Y.; Gondry, M. Unravelling the mechanism of non-ribosomal peptide synthesis by cyclodipeptide synthases. Nat. Commun. 2014, 5, 5141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravind, L.; de Souza, R.F.; Iyer, L.M. Predicted class-I aminoacyl tRNA synthetase-like proteins in non-ribosomal peptide synthesis. Biol. Direct 2010, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.P.; Qian, X.L.; Alemany, L.B.; Moran, S.; Parry, R.J. Investigations of valanimycin biosynthesis: Elucidation of the role of seryl-tRNA. Proc. Natl. Acad. Sci. USA 2008, 105, 6543–6547. [Google Scholar] [CrossRef] [PubMed]
- Artymiuk, P.J.; Rice, D.W.; Poirrette, A.R.; Willet, P. A tale of two synthetases. Nat. Struct. Biol. 1994, 1, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Cusack, S.; Berthet-Colominas, C.; Hartlein, M.; Nassar, N.; Leberman, R. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 A. Nature 1990, 347, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Sumida, T.; Ishii, R.; Takemoto, C.; Yokoyama, S. A paralog of lysyl-tRNA synthetase aminoacylates a conserved lysine residue in translation elongation factor P. Nat. Struct. Mol. Biol. 2010, 17, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.B.; Roy, H.; Ibba, M.; Navarre, W.W. Elongation factor P mediates a novel post-transcriptional regulatory pathway critical for bacterial virulence. Virulence 2011, 2, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Blaise, M.; Frechin, M.; Olieric, V.; Charron, C.; Sauter, C.; Lorber, B.; Roy, H.; Kern, D. Crystal structure of the archaeal asparagine synthetase: Interrelation with aspartyl-tRNA and asparaginyl-tRNA synthetases. J. Mol. Biol. 2011, 412, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Mocibob, M.; Ivic, N.; Bilokapic, S.; Maier, T.; Luic, M.; Ban, N.; Weygand-Durasevic, I. Homologs of aminoacyl-tRNA synthetases acylate carrier proteins and provide a link between ribosomal and nonribosomal peptide synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 14585–14590. [Google Scholar] [CrossRef] [PubMed]
- Mocibob, M.; Ivic, N.; Luic, M.; Weygand-Durasevic, I. Adaptation of aminoacyl-tRNA synthetase catalytic core to carrier protein aminoacylation. Structure 2013, 21, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; LP, S.d.C.; Yu, M.; Hegde, S.S.; Magnet, S.; Roderick, S.L.; Blanchard, J.S. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 2005, 433, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Roderick, S.L.; Yu, M.; Blanchard, J.S. Crystal structure of mycothiol synthase (Rv0819) from Mycobacterium tuberculosis shows structural homology to the GNAT family of N-acetyltransferases. Protein Sci. 2003, 12, 1954–1959. [Google Scholar] [CrossRef] [PubMed]
- Benson, T.E.; Prince, D.B.; Mutchler, V.T.; Curry, K.A.; Ho, A.M.; Sarver, R.W.; Hagadorn, J.C.; Choi, G.H.; Garlick, R.L. X-ray crystal structure of Staphylococcus aureus FemA. Structure 2002, 10, 1107–1115. [Google Scholar] [CrossRef]
- Biarrotte-Sorin, S.; Maillard, A.P.; Delettre, J.; Sougakoff, W.; Arthur, M.; Mayer, C. Crystal structures of Weissella viridescens FemX and its complex with UDP-MurNAc-pentapeptide: Insights into FemABX family substrates recognition. Structure 2004, 12, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Toh, Y.; Suto, K.; Shimizu, Y.; Oka, N.; Wada, T.; Tomita, K. Protein-based peptide-bond formation by aminoacyl-tRNA protein transferase. Nature 2007, 449, 867–871. [Google Scholar] [CrossRef]
- Dong, X.; Kato-Murayama, M.; Muramatsu, T.; Mori, H.; Shirouzu, M.; Bessho, Y.; Yokoyama, S. The crystal structure of leucyl/phenylalanyl-tRNA-protein transferase from Escherichia coli. Protein Sci. 2007, 16, 528–534. [Google Scholar] [PubMed]
- Linne, U.; Schafer, A.; Stubbs, M.T.; Marahiel, M.A. Aminoacyl-coenzyme A synthesis catalyzed by adenylation domains. FEBS Lett. 2007, 581, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Reanney, D.C. Aminoacyl thiol esters and the origins of genetic specificity. J. Theor. Biol. 1977, 65, 555–569. [Google Scholar] [CrossRef]
- Miller, S.L. A production of amino acids under possible primitive earth conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Bada, J.L. New insights into prebiotic chemistry from Stanley Miller’s spark discharge experiments. Chem. Soc. Rev. 2013, 42, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Van Trump, J.E.; Miller, S.L. Prebiotic synthesis of methionine. Science 1972, 178, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Khare, B.N.; Sagan, C. Synthesis of cystine in simulated primitive conditions. Nature 1971, 232, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Eisenreich, W.; Wachtershauser, G. Synthesis of α-amino and α-hydroxy acids under volcanic conditions: Implications for the origin of life. Tetrahedron Lett. 2010, 51, 1069–1071. [Google Scholar] [CrossRef]
- Parker, E.T.; Cleaves, H.J.; Callahan, M.P.; Dworkin, J.P.; Glavin, D.P.; Lazcano, A.; Bada, J.L. Prebiotic synthesis of methionine and other sulfur-containing organic compounds on the primitive Earth: A contemporary reassessment based on an unpublished 1958 Stanley Miller experiment. Orig. Life Evol. Biosph. 2011, 41, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L. Production of some organic compounds under possible promitive Earth conditions. J. Am. Chem. Soc. 1955, 77, 2351–2361. [Google Scholar] [CrossRef]
- Parker, E.T.; Cleaves, H.J.; Dworkin, J.P.; Glavin, D.P.; Callahan, M.; Aubrey, A.; Lazcano, A.; Bada, J.L. Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment. Proc. Natl. Acad. Sci. USA 2011, 108, 5526–5531. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Newton, G.L.; Miller, S.L. A possible prebiotic synthesis of pantetheine, a precursor to coenzyme A. Nature 1995, 373, 683–685. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. Vital Dust; Basic Books: New York, NY, USA, 1994. [Google Scholar]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Blackmond, D.G. The Future of Prebiotic Chemistry. ACS Cent. Sci. 2016, 2, 775–777. [Google Scholar] [CrossRef] [PubMed]
- White, H.B., 3rd. Coenzymes as fossils of an earlier metabolic state. J. Mol. Evol. 1976, 7, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Kowtoniuk, W.E.; Shen, Y.; Heemstra, J.M.; Agarwal, I.; Liu, D.R. A chemical screen for biological small molecule-RNA conjugates reveals CoA-linked RNA. Proc. Natl. Acad. Sci. USA 2009, 106, 7768–7773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubowski, H.; Goldman, E. Quantities of individual aminoacyl-tRNA families and their turnover in Escherichia coli. J. Bacteriol. 1984, 158, 769–776. [Google Scholar] [PubMed]
- Benner, S.A.; Ellington, A.D.; Tauer, A. Modern metabolism as a palimpsest of the RNA world. Proc. Natl. Acad. Sci. USA 1989, 86, 7054–7058. [Google Scholar] [CrossRef] [PubMed]
- Lipmann, F. Attempts to map a process evolution of peptide biosynthesis. Science 1971, 173, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Wieland, T.; Bokelmann, E.; Bauer, L.; Lang, H.U.; Lau, H. Uber Peptidsynthesen. 8. Mitteilung. Bildung von S-haltigen Peptiden durch intramolekulare Wanderung Von Aminoacylresten. Eur. J. Org. Chem. 1953, 583, 129–149. [Google Scholar]
- Jakubowski, H. Aminoacyl-tRNA synthetases and the evolution of coded peptide synthesis: The Thioester World. FEBS Lett. 2016, 590, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Baggott, J.E.; Tamura, T. Iron-dependent formation of homocysteine from methionine and other thioethers. Eur. J. Clin. Nutr. 2007, 61, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, M.; Kunishi, A.T. Ethylene production from methionine. Biochem. J. 1965, 97, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Facile syntheses of [35S]homocysteine-thiolactone, [35S]homocystine, [35S]homocysteine, and [S-nitroso-35S]homocysteine. Anal. Biochem. 2007, 370, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wachtershauser, G. α-Hydroxy and α-amino acids under possible Hadean, volcanic origin-of-life conditions. Science 2006, 314, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Racker, E. Metabolism of thiolesters of glutathione. Fed. Proc. 1953, 12, 711–714. [Google Scholar] [PubMed]
- Huber, C.; Eisenreich, W.; Hecht, S.; Wachtershauser, G. A possible primordial peptide cycle. Science 2003, 301, 938–940. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.T.; Zhou, M.; Burton, A.S.; Glavin, D.P.; Dworkin, J.P.; Krishnamurthy, R.; Fernandez, F.M.; Bada, J.L. A plausible simultaneous synthesis of amino acids and simple peptides on the primordial Earth. Angew. Chem. Int. Ed. Engl. 2014, 53, 8132–8136. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Novikova, O.; Topilina, N.; Belfort, M. Enigmatic distribution, evolution, and function of inteins. J. Biol. Chem. 2014, 289, 14490–14497. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, P.; Kelley, S.O. Exiting an RNA world. Nat. Struct. Biol. 2000, 7, 5–7. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
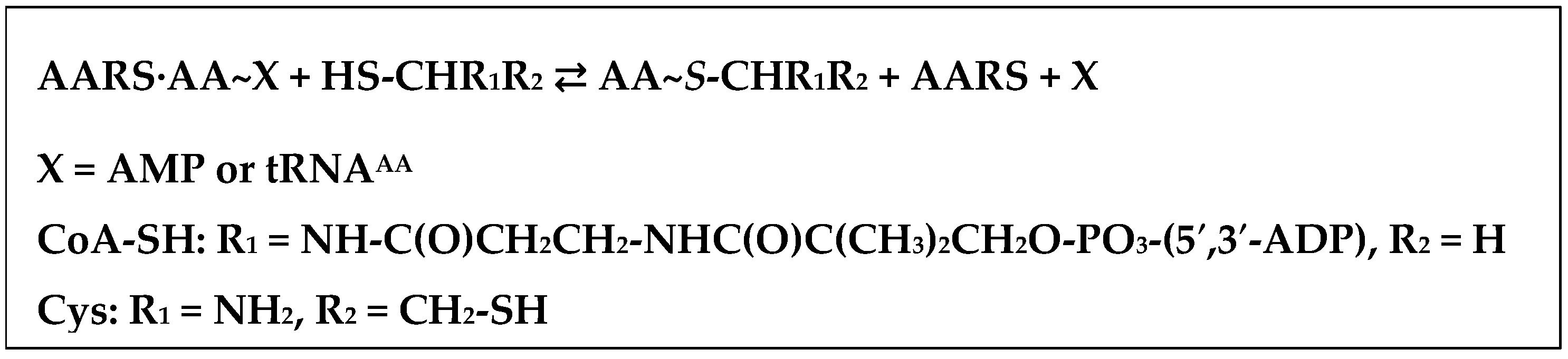{kind=link}
{kind=link}
{kind=link}
| Average Distance, Å | ||||
|---|---|---|---|---|
| MetRS | LeuRS | ValRS | IleRS | |
| SHcy···OAsp/Glu | 5.30 | 5.51 | 4.94 | 4.70 |
| SHcy···Ccarb | 3.91 | 5.19 | 4.14 | 4.66 |
| Ccarb···OAsp/Glu | 4.25 | 4.39 | 5.26 | 4.10 |
| SHcy···Ophos | 6.99 | 7.78 | 7.01 | 4.25 |
| Cell Labeling Conditions | Translational | Post-Translational | |
|---|---|---|---|
| [35S]Hcy-Protein | [35S]Met-Protein | εN-[35S]Hcy-Lys-Protein | |
| % | % | % | |
| [35S]Hcy (10 μM, 50 μCi/mL) | 37 | 25 | 38 |
| [35S]Hcy + folic acid, 10 μM | <1 | >98 | <1 |
| [35S]Hcy + HDL, 1 mg/mL | 68 | 25 | 7 |
| [35S]Hcy + Met, 20 μM | 12 | 76 | 12 |
| Control, εN-[35S]Hcy-Lys-protein | <4 | 0 | >96 |
| AARS | Hcy Editing | Thiol-Binding Site Residue | Thiol Aminoacylation | Peptide Synthesis | References |
|---|---|---|---|---|---|
| Class I | |||||
| MetRS | Yes | Asp359 | Yes | Yes | [24,25,26,27,47,55,56,58,63,67] |
| LeuRS | Yes | Glu532 | ? | ? | [25,26,58] |
| IleRS | Yes | Glu550 | Yes | Yes | [25,26,58,63,64,66] |
| ValRS | Yes | Asp490 | Yes | Yes | [58,64,65] |
| CysRS | No | ? | Yes | ? | [68] |
| ArgRS | No | ? | Yes | Yes | [65] |
| Class II | |||||
| SerRS | No | ? | Yes | Yes | [38,63] |
| AspRS | No | ? | Yes | Yes | [38,63] |
| LysRS | Yes | ? | Yes | Yes | [29,38] |
| Amino Acid | IleRS, kcat/KM (min−1·M−1) | ValRS, kcat/KM (min−1·M−1) | LysRS, v (µM/h) |
|---|---|---|---|
| Isoleucine | 440,000 | 115 | 0.24 |
| Valine | 45,000 | 3300 | 0.67 |
| Leucine | 7700 | <5 | 2.7 |
| Threonine | 1200 | 1400 | 2.7 |
| Serine | 170 | <0.1 | |
| Alanine | 1300 | 500 | 1.7 |
| Lysine | 1.9 |
| GlnRS | GluRS | TyrRS | PPAT | |
|---|---|---|---|---|
| Sequence identity to Pan C (%) * | 10.8 | 14.5 | 11.2 | 14.5 |
| RMSD of Cα atoms (Å) | 1.8 | 1.5 | 2.2 | 1.8 |
| Number of Cα aligned for RMSD | 65 | 62 | 68 | 76 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakubowski, H. Homocysteine Editing, Thioester Chemistry, Coenzyme A, and the Origin of Coded Peptide Synthesis †. Life 2017, 7, 6. https://doi.org/10.3390/life7010006
Jakubowski H. Homocysteine Editing, Thioester Chemistry, Coenzyme A, and the Origin of Coded Peptide Synthesis †. Life. 2017; 7(1):6. https://doi.org/10.3390/life7010006
Chicago/Turabian StyleJakubowski, Hieronim. 2017. "Homocysteine Editing, Thioester Chemistry, Coenzyme A, and the Origin of Coded Peptide Synthesis †" Life 7, no. 1: 6. https://doi.org/10.3390/life7010006