
First Report of a Low-Frequency Mosaic Mutation in the Hydroxymethylbilane Synthase Gene Causing Acute Intermittent Porphyria
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Informed Consent
2.2. Biochemical Analyses
2.3. Sanger Sequencing
2.4. Long-Range PCR
2.5. Nanopore Sequencing
2.6. Sequence-Specific Primer PCR
2.7. Analysis of Short Tandem Repeats
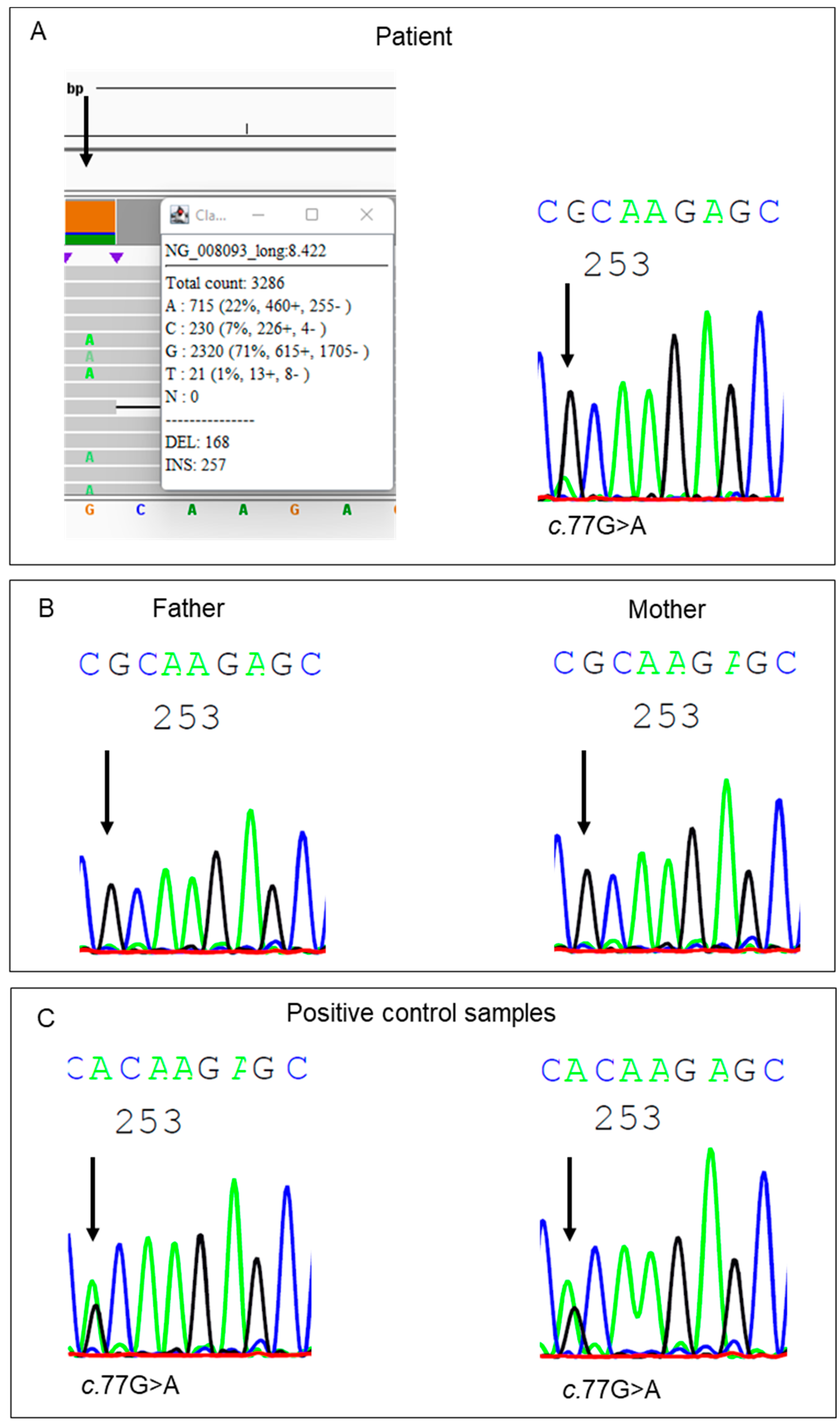
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stölzel, U.; Doss, M.O.; Schuppan, D. Clinical Guide and Update on Porphyrias. Gastroenterology 2019, 157, 365–381.e4. [Google Scholar] [CrossRef] [PubMed]
- Schulenburg-Brand, D.; Stewart, F.; Stein, P.; Rees, D.; Badminton, M. Update on the diagnosis and management of the autosomal dominant acute hepatic porphyrias. J. Clin. Pathol. 2022, 75, 537–543. [Google Scholar] [CrossRef]
- Stein, P.E.; Edel, Y.; Mansour, R.; Mustafa, R.A.; Sandberg, S.; Members of the Acute Porphyria Expert Panel. Key terms and definitions in acute porphyrias: Results of an international Delphi consensus led by the European porphyria network. J. Inherit. Metab. Dis. 2023, 46, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Kazamel, M.; Pischik, E.; Desnick, R.J. Pain in acute hepatic porphyrias: Updates on pathophysiology and management. Front. Neurol. 2022, 13, 1004125. [Google Scholar] [CrossRef]
- Schmitt, C.; Lenglet, H.; Yu, A.; Delaby, C.; Benecke, A.; Lefebvre, T.; Letteron, P.; Paradis, V.; Wahlin, S.; Sandberg, S.; et al. Recurrent attacks of acute hepatic porphyria: Major role of the chronic inflammatory response in the liver. J. Intern. Med. 2018, 284, 78–91. [Google Scholar] [CrossRef]
- Ricci, A.; Sandri, G.; Marcacci, M.; Di Pierro, E.; Granata, F.; Cuoghi, C.; Marchini, S.; Pietrangelo, A.; Ventura, P. Endothelial Dysfunction in Acute Hepatic Porphyrias. Diagnostics 2022, 12, 1303. [Google Scholar] [CrossRef] [PubMed]
- Pischik, E.; Baumann, K.; Karpenko, A.; Kauppinen, R. Pathogenesis of acute encephalopathy in acute hepatic porphyria. J. Neurol. 2023, 270, 2613–2630. [Google Scholar] [CrossRef]
- Mustajoki, P.; Nordmann, Y. Early administration of heme arginate for acute porphyric attacks. Arch. Intern. Med. 1993, 153, 2004–2008. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Neeleman, R.A.; Wensink, D.; Wagenmakers MA, E.M.; Mijnhout, G.S.; Friesema EC, H.; Langendonk, J.G. Diagnostic and therapeutic strategies for porphyrias. Neth. J. Med. 2020, 78, 149–160. [Google Scholar]
- Minder, E.; Schneider-Yin, X. Porphyrins, Porphobilinogen, and δ-Aminolevulinic Acid. In Laboratory Guide to the Methods in Biochemical Genetics; Blau, N., Duran, M., Gibson, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar] [CrossRef]
- Kauppinen, R.; von und zu Fraunberg, M. Molecular and biochemical studies of acute intermittent porphyria in 196 patients and their families. Clin. Chem. 2002, 48, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Enriquez de Salamanca, R.; Sepulveda, P.; Moran, M.J.; Santos, J.L.; Fontanellas, A.; Hernández, A. Clinical utility of fluorometric scanning of plasma porphyrins for the diagnosis and typing of porphyrias. Clin. Exp. Dermatol. 1993, 18, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Gregor, A.; Kostrzewska, E.; Tarczyńska-Nosal, S.; Stachurska, H. Fluorescencja porfiryn w osoczu w róznych typach porfirii [Porphyrin fluorescence in plasma of various types of porphyria]. Pol. Tyg. Lek. 1994, 49, 284–286. [Google Scholar]
- Peoc’h, K.; Manceau, H.; Karim, Z.; Wahlin, S.; Gouya, L.; Puy, H.; Deybach, J.C. Hepatocellular carcinoma in acute hepatic porphyrias: A Damocles Sword. Mol. Genet. Metab. 2019, 128, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Badminton, M.N.; Rees, D.C. Update review of the acute porphyrias. Br. J. Haematol. 2017, 176, 527–538. [Google Scholar] [CrossRef]
- Saberi, B.; Naik, H.; Overbey, J.R.; Erwin, A.L.; Anderson, K.E.; Bissell, D.M.; Bonkovsky, H.L.; Phillips, J.D.; Wang, B.; K.Singal, A.; et al. Hepatocellular Carcinoma in Acute Hepatic Porphyrias: Results from the Longitudinal Study of the U.S. Porphyrias Consortium. Hepatology 2021, 73, 1736–1746. [Google Scholar] [CrossRef]
- Lissing, M.; Vassiliou, D.; Floderus, Y.; Harper, P.; Bottai, M.; Kotopouli, M.; Hagström, H.; Sardh, E.; Wahlin, S. Risk of primary liver cancer in acute hepatic porphyria patients: A matched cohort study of 1244 individuals. J. Intern. Med. 2022, 291, 824–836. [Google Scholar] [CrossRef]
- Schneider-Yin, X.; Harms, J.; Minder, E.I. Porphyria in Switzerland, 15 years experience. Swiss Med. Wkly. 2009, 139, 198–206. [Google Scholar] [CrossRef]
- Anderson, K.E.; Sassa, S.; Peterson, C.M.; Kappas, A. Increased erythrocyte uroporphyrinogen-l-synthetase, delta-aminolevulinic acid dehydratase and protoporphyrin in hemolytic anemias. Am. J. Med. 1977, 63, 359–364. [Google Scholar] [CrossRef]
- Whatley, S.D.; Roberts, A.G.; Llewellyn, D.H.; Bennett, C.P.; Garrett, C.; Elder, G.H. Non-erythroid form of acute intermittent porphyria caused by promoter and frameshift mutations distant from the coding sequence of exon 1 of the HMBS gene. Hum. Genet. 2020, 107, 243–248. [Google Scholar] [CrossRef]
- Brancaleoni, V.; Granata, F.; Colancecco, A.; Tavazzi, D.; Cappellini, M.D.; Di Pierro, E. Seven novel genetic mutations within the 5’UTR and the housekeeping promoter of HMBS gene responsible for the non-erythroid form of acute intermittent porphyria. Blood Cells Mol. Dis. 2012, 49, 147–151. [Google Scholar] [CrossRef]
- Barman-Aksözen, J.; Suter, L.; Wegmann, F.; Meienberg, J.; Minder, A.E.; Beer, M.; Komminoth, P.; Minder, E.I.; Schneider-Yin, X. A next-generation-sequencing panel for mutational analysis of dominant acute hepatic porphyrias. Scand J. Clin. Lab. Investig. 2019, 79, 305–313. [Google Scholar] [CrossRef]
- Gueuning, M.; Thun, G.A.; Wittig, M.; Galati, A.L.; Meyer, S.; Trost, N.; Gourri, E.; Fuss, J.; Sigurdardottir, S.; Merki, Y.; et al. Haplotype sequence collection of ABO blood group alleles by long-read sequencing reveals putative A1-diagnostic variants. Blood Adv. 2023, 7, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Barman-Aksözen, J.; Minder, E.I.; Schubiger, C.; Biolcati, G.; Schneider-Yin, X. In ferrochelatase-deficient protoporphyria patients, ALAS2 expression is enhanced and erythrocytic protoporphyrin concentration correlates with iron availability. Blood Cells Mol. Dis. 2015, 54, 71–77. [Google Scholar] [CrossRef]
- Llewellyn, D.H.; Whatley, S.; Elder, G.H. Acute intermittent porphyria caused by an arginine to histidine substitution (R26H) in the cofactor-binding cleft of porphobilinogen deaminase. Hum. Mol. Genet. 1993, 2, 1315–1316. [Google Scholar] [CrossRef]
- Johnson, B.N.; Ehli, E.A.; Davies, G.E.; Boomsma, D.I. Chimerism in health and potential implications on behavior: A systematic review. Am. J. Med. Genet. Part A 2020, 182, 1513–1529. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Hidalgo, R.; Veltman, J.A.; Hoischen, A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016, 17, 241. [Google Scholar] [CrossRef]
- Pacot, L.; Pelletier, V.; Chansavang, A.; Briand-Suleau, A.; Burin des Roziers, C.; Coustier, A.; Maillard, T.; Vaucouleur, N.; Orhant, L.; Barbance, C.; et al. Contribution of whole genome sequencing in the molecular diagnosis of mosaic partial deletion of the NF1 gene in neurofibromatosis type 1. Hum. Genet. 2023, 142, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.M.; Shaw, C.A.; Stankiewicz, P.; Lupski, J.R. Somatic mosaicism: Implications for disease and transmission genetics. Trends Genet. 2015, 31, 382–392, Erratum in Trends Genet. 2016, 32, 138. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| An Acute Porphyria Attack Is Characterised by: | |
|---|---|
| Two or More of the Following Clinical Manifestations, Typically Persisting > 24 h: | AND Increased Urinary Porphobilinogen (PBG): |
| Urinary PBG * to creatinine ratio of typically more than 10 times the upper limit of normal |
| |
| |
| |
| |
| |
| |
| Forward primer (5′-3′) | GCAAAGGAAGCGCCATAGAAG |
| Reverse primer (3′-5′) | AGGCAAGGCAGTCATCAAGG |
| Sequence with reference nucleotide (5′-3′) | ACCCGCAAGAGCCAGGTGGGTGCAGGAG |
| Sequence with alternative nucleotide (5′-3′) | ACCCACAAGAGCCAGGTGGGTGCAGGAG |
| Reverse primer with mismatch (3′-5′) | TGATCTCGGTCCACCCACGT |
| (A) | |||||
| Aminolevulinic Acid (urine) | Porphobilinogen (urine) | Plasma Fluorescence Scan | Faecal Coproporphyrin III (1) | Faecal Protoporphyrin (2) | |
| Reference range | <2.5 [μmol/mmol creatinine] | <1.25 [μmol/mmol creatinine] | Pos/neg [nm] | <12.0 [mmol/g dry weight] | <80 [mmol/g dry weight] |
| During the acute attack | 76.4 | 35.2 | |||
| After the acute attack | 15.5 (a) 12.5 (b) | 22.4 (a) 25.0 (b) | 619 (a) (c) | 3.57 (c) | 16.9 (c) |
| (B) | |||||
| Reference Range | Healthy Controls (n = 20), Mean (SD) (3) | AIP Patients (n = 16), Mean (SD) (3) | Patient 1st Measurement | Patient 2nd Measurement | |
| HMBS enzymatic activity | 66–126 [pmol/mgHb/h] | 99.6 (±27.8) | 44.9 (±9.0) | 85 (c) | 58 (88% LLN) (a) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosevic, A.; Minder, A.-E.; Gueuning, M.; van Breemen, F.; Thun, G.A.; Mattle-Greminger, M.P.; Meyer, S.; Baumer, A.; Minder, E.I.; Schneider-Yin, X.; et al. First Report of a Low-Frequency Mosaic Mutation in the Hydroxymethylbilane Synthase Gene Causing Acute Intermittent Porphyria. Life 2023, 13, 1889. https://doi.org/10.3390/life13091889
Belosevic A, Minder A-E, Gueuning M, van Breemen F, Thun GA, Mattle-Greminger MP, Meyer S, Baumer A, Minder EI, Schneider-Yin X, et al. First Report of a Low-Frequency Mosaic Mutation in the Hydroxymethylbilane Synthase Gene Causing Acute Intermittent Porphyria. Life. 2023; 13(9):1889. https://doi.org/10.3390/life13091889
Chicago/Turabian StyleBelosevic, Adrian, Anna-Elisabeth Minder, Morgan Gueuning, Franziska van Breemen, Gian Andri Thun, Maja P. Mattle-Greminger, Stefan Meyer, Alessandra Baumer, Elisabeth I. Minder, Xiaoye Schneider-Yin, and et al. 2023. "First Report of a Low-Frequency Mosaic Mutation in the Hydroxymethylbilane Synthase Gene Causing Acute Intermittent Porphyria" Life 13, no. 9: 1889. https://doi.org/10.3390/life13091889