
D-Penicillamine-Induced Myasthenia Gravis—A Probable Complication of Wilson’s Disease Treatment—A Case Report and Systematic Review of the Literature
 ,
, 
Abstract
:
1. Introduction
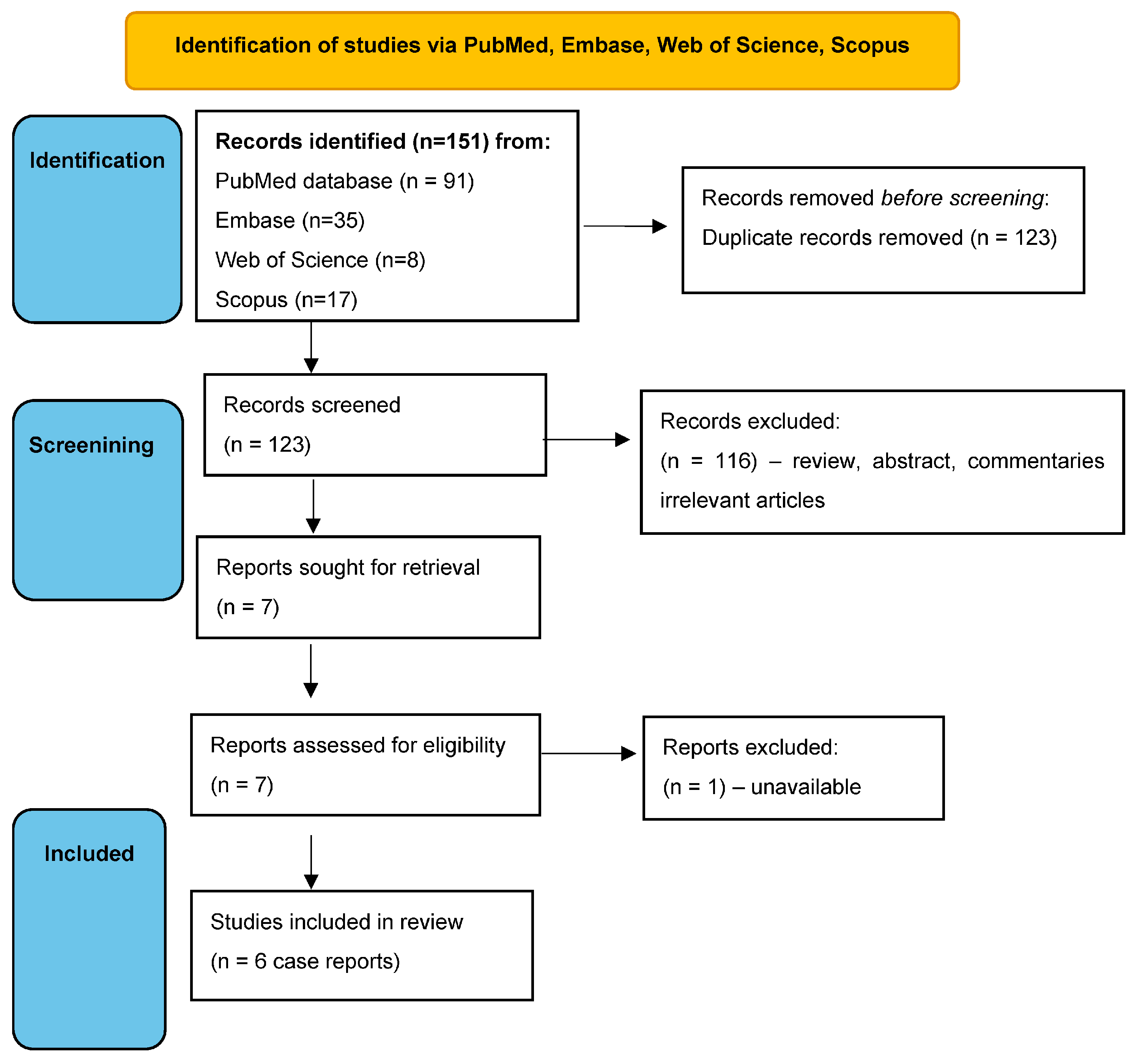
2. Materials and Methods
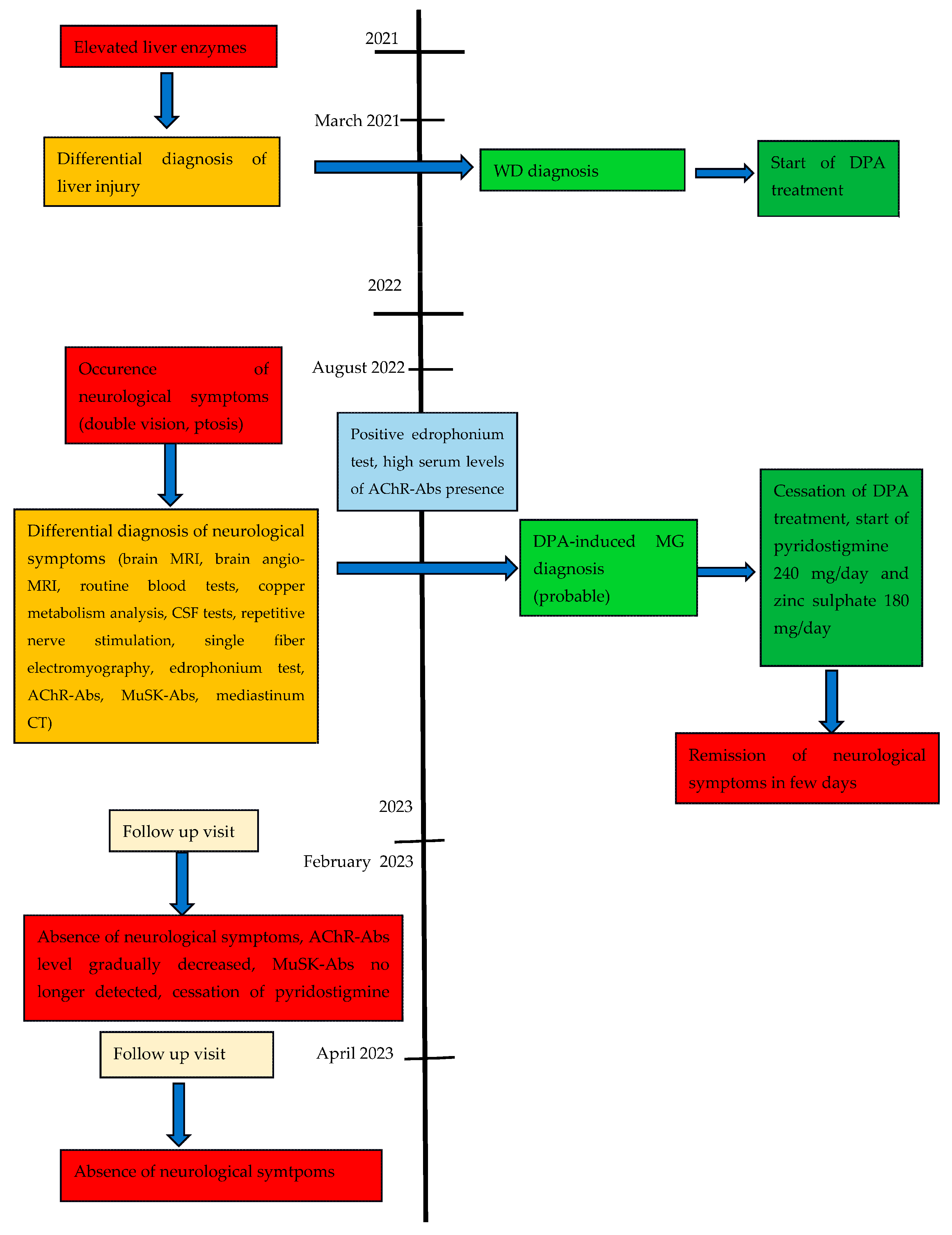
2.1. Case Report
2.2. Systematic Review of DPA-Induced MG in WD Patients
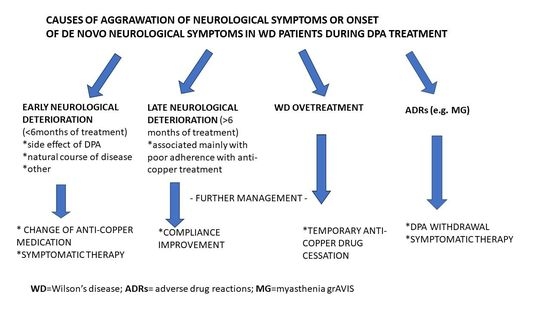
3. Discussion
4. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- European Association for The Study of The Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar] [CrossRef] [Green Version]
- Członkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Litwin, T. Wilson disease—Currently used anticopper therapy. Handb. Clin. Neurol. 2017, 142, 181–191. [Google Scholar]
- Weiss, K.H.; Askari, F.K.; Czlonkowska, A.; Ferenci, P.; Bronstein, J.M.; Bega, D.; Ala, A.; Nicholl, D.; Flint, S.; Olsson, L.; et al. Bis-choline tetrathiomolybdate in patients with Wilson’s disease: An open label, multicentre, phase 2 study. Lancet Gastroenterol. Hepatol. 2017, 2, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Dzieżyc, K.; Czlonkowska, A. Wilson disease—Treatment perspectives. Ann. Transl. Med. 2019, 7, S68. [Google Scholar] [CrossRef]
- Schilsky, M.L. Long-term outcome for Wilson disease: 85% good. Clin. Gastroenterol. Hepatol. 2014, 12, 690–691. [Google Scholar]
- Antos, A.; Członkowska, A.; Smolinski, L.; Bembenek, J.; Przybyłkowski, A.; Skowronska, M.; Kurkowska-Jastrzębska, I.; Litwin, T. Early neurological deterioraion in Wilson’s disease: A systematic literature review and meta-analysis. Neurol. Sci. 2023. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Antos, A.; Bembenek, J.; Przybyłkowski, A.; Kurkowska-Jastrzębska, I.; Skowrońska, M.; Członkowska, A. Copper deficeicny as Wilson’s disease overtreatment: A systematic review. Diagnostics 2023, 13, 2424. [Google Scholar] [CrossRef]
- Antos, A.J.; Litwin, T.; Przybylkowski, A.; Skowronska, M.; Kurkowska-Jastrzebska, I.; Czlonkowska, A. D-penicillamine-induced lupus erythematosus as an adverse reaction of treatment of Wilson’s disease. Neurol. Neurochir. Pol. 2021, 55, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Czlonkowska, A.; Socha, P. Oral chelator treatment of Wilson Disease: D-penicillamine. In Clinical and Translational Perspectives on Wilson Disease; Litwin, T., Czlonkowska, A., Socha, P., Eds.; Academic Press: Cambridge, UK, 2019; pp. 357–364. [Google Scholar]
- Beinhardt, S.; Leiss, W.; Stättermayer, A.F.; Graziadei, I.; Zoller, H.; Stauber, R.; Maieron, A.; Datz, C.; Steindl-Munda, P.; Hofer, H.; et al. Long-term outcomes of patients with Wilson disease in a large Austrian cohort. Clin. Gastroenterol. Hepatol. 2014, 12, 683–689. [Google Scholar] [CrossRef]
- Bruha, R.; Marecek, Z.; Pospisilova, L.; Nevsimalova, S.; Vitek, L.; Martasek, P.; Nevoral, J.; Petrtyl, J.; Urbanek, P.; Jiraskova, A.; et al. Long-term follow-up of Wilson disease: Natural history, treatment, mutations analysis and phenotypic correlation. Liver Int. 2011, 31, 83–91. [Google Scholar] [PubMed]
- Poujois, A.; Trocello, J.-M.; Djebrani-Oussedik, N.; Poupon, J.; Collet, C.; Girardot-Tinant, N.; Sobesky, R.; Habès, D.; Debray, D.; Vanlemmens, C.; et al. Exchangeable copper: A reflection of the neurological severity in Wilson’s disease. Eur. J. Neurol. 2017, 24, 154–160. [Google Scholar] [PubMed] [Green Version]
- Członkowska, A.; Litwin, T.; Karliński, M.; Dziezyc, K.; Chabik, G.; Czerska, M. D-penicillamine versus zinc sulfate s first-line therapy for Wilson’s Disease. Eur. J. Neurol. 2014, 21, 599–606. [Google Scholar] [PubMed]
- Vincent, A.; Palace, J.; Hilton-Jones, D. Myasthenia gravis. Lancet 2001, 357, 2122–2128. [Google Scholar] [PubMed]
- Gilhus, N.E.; Skeie, G.O.; Romi, F.; Lazaridis, K.; Zisimopoulou, P.; Tzartos, S. Myasthenia gravis–autoantibody characteristics and their implications for therapy. Nat. Rev. Neurol. 2016, 12, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Hu, X.; Lu, Z.; Hackett, M.L. Prognostic factors of remission in myasthenia gravis after thymectomy. Eur. J. Cardiothorac. Surg. 2015, 48, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Naranjo, C.A.; Busto, U.; Sellers, E.M.; Sandor, P.; Ruiz, I.; Roberts, E.A.; Janecek, E.; Domecq, C.; Greenblatt, D.J. A method for estimating the probability of adverse drug reactions. Clin. Pharmacol. Ther. 1981, 30, 239–245. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gotzsche, P.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions. Explanation and elaboration. Ann. Intern. Med. 2009, 151, W65–W94. [Google Scholar] [CrossRef] [Green Version]
- Thapa, L.; Thapa, M.; Bhattarai, S.; Shrestha, A.M.; Sharma, N.; Rai, N.; Pokharel, M.; Paudel, R. D-penicillamine induced myasthenia gravis in Wilson’s disease: A case report. JNMA J. Nepal. Med. Assoc. 2022, 60, 644–647. [Google Scholar]
- Reuner, U.; Stremmel, W.; Weiskirchen, R. The interesting case-orphan diseases-double trouble. Ann. Transl. Med. 2019, 7, S74. [Google Scholar] [CrossRef]
- Tan, S.S.; Latif, S.A.; Poh, W.Y. Concurrent massive breast enlargement, myasthenia gravis and dermopathy as manifestations of penicillamine toxicity in a Wilson’s disease patient. Med. J. Malaysia 2012, 67, 323–325. [Google Scholar]
- Varghese, T.; Ahmed, R.; Sankaran, J.D.; Al-Khusaiby, S.M. D-Penicillamine induced myasthenia gravis. Neurosciences 2002, 7, 293–295. [Google Scholar] [PubMed]
- Narayanan, C.S.; Behari, M. Generalized myasthenia gravis following use of D-pencillamine in Wilson’s disease. J. Assoc. Physician India 1999, 47, 648. [Google Scholar]
- Masters, C.L.; Dawkins, R.L.; Zilko, P.J.; Simpson, J.A.; Leedman, R.J. Penicillamine-associated myasthenia gravis, antiacetylcholine receptor and antistriational antibodies. Am. J. Med. 1977, 63, 689–694. [Google Scholar] [CrossRef]
- Czlonkowska, A. Myasthenia syndrome during penicillamine treatment. Br. Med. J. 1975, 28, 726–727. [Google Scholar] [CrossRef] [Green Version]
- Poulas, K.; Koutsouraki, E.; Kordas, G.; Kokla, A.; Tzartos, S.J. Anti-MuSK and anti-AChR-positive myasthenia gravis induced by d-penicillamine. J. Neuroimmunol. 2012, 250, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Adelman, H.M.; Winters, P.R.; Mahan, C.S.; Wallach, P.M. D-penicillamine-induced myasthenia gravis: Diagnosis obscured by coexisting chronic obstructive pulmonary disease. Am. J. Med. Sci. 1995, 309, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Marchiori, P.E.; Scaff, M.; Cossermelli, W.; De Assis, J.L. Myasthenia gravis induced by D-penicillamine in a patient with progressive systemic sclerosis. Arq. Neuropsiquiatr. 1984, 42, 380–383. [Google Scholar] [CrossRef]
- Essigman, W.K. Multiple side effects of penicillamine therapy in one patient with rheumatoid arthritis. Ann. Rheum. Dis. 1982, 41, 617–620. [Google Scholar] [CrossRef]
- Kimbrough, R.L.; Mewis, L.; Stewart, R.H. D-penicillamine and the ocular myasthenic syndrome. Ann. Ophthalmol. 1981, 13, 1171–1172. [Google Scholar]
- Hill, M.; Moss, P.; Wordsworth, P.; Newsom-Davis, J.; Willcox, N. T cell responses to D-penicillamine in drug-induced myasthenia gravis: Recognition of modified DR1:peptide complexes. J. Neuroimmunol. 1999, 97, 146–153. [Google Scholar] [CrossRef]
- Rodolico, C.; Bonanno, C.; Toscano, A.; Vita, G. Mu-SK-associated myasthenia gravis: Clinical features and management. Front. Neurol. 2020, 11, 660. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.; Alvi, U.; Soliven, B.; Rezania, K. Drugs that induce or cause deterioration of Myasthenia Gravis: An update. J. Clin. Med. 2021, 10, 1537. [Google Scholar] [CrossRef] [PubMed]
- Antczak-Kowalska, M.; Członkowska, A.; Eyileten, C.; Palejko, A.; Cudna, A.; Wolska, M.; Piechal, A.; Litwin, T. Autoantibodies in Wilson disease: Impact on clinical course. JIMD Rep. 2022, 63, 508–517. [Google Scholar] [CrossRef]
- Seessle, J.; Gotthardt, D.N.; Schäfer, M.; Gohdes, A.; Pfeiffenberger, J.; Ferenci, P.; Stremmel, W.; Weiss, K.H. Concomitant immune-related events in Wilson disease: Implications for monitoring chelator therapy. J. Inherit. Metab. Dis. 2016, 39, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Milewski, B. Immunological observations on patients with Wilson’s disease. J. Neurol. Sci. 1976, 29, 411–421. [Google Scholar] [CrossRef]
- Członkowska, A. The influence of prolonged treatment with D-penicillamine on the immune response in Wilson’s disease. Eur. J. Clin. Pharmacol. 1977, 12, 265–271. [Google Scholar] [CrossRef]
- Komal Kumar, R.N.; Patil, S.A.; Taly, A.B.; Nirmala, M.; Sinha, S.; Arunodaya, G.R. Effect of D-penicillamine on neuromuscular junction in patients with Wilson disease. Neurology 2004, 63, 935–936. [Google Scholar] [CrossRef]
- Antos, A.; Członkowska, A.; Bembenek, J.; Skowrońska, M.; Kurkowska-Jastrzębska, I.; Litwin, T. Blood based biomarkers of central nervous system involvement in Wilson’s disease. Diagnostics 2023, 13, 1554. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Reference | Patient Characteristics (Age, Gender, WD Symptoms) | Duration, Kind and Dose of WD Treatment | MG Clinical Presentation and Diagnostic Tests | MG Abs Induced by DPA | Treatment and Outcome |
|---|---|---|---|---|---|
| Thapa et al. 2022 [20] | 15-year-old male with neurological WD (generalized weakness, spastic limbs, hand tremor) | Duration of WD treatment with DPA: 6 years Since diagnosis aged 9, patient was treated with DPA 500 mg/day and zinc 60 mg/day | Bilateral facial weakness (dropping jaw and both upper eyelids) and dysphagia RNS from the facial nerve (orbicularis oculi muscle) showed >10% decremental response Normal chest CT | Positive serum AChR-Abs (14.3 nmol/L; normal < 0.4) Negative for MuSK-Abs | Withdrawal of DPA Pyridostigmine introduced (dose not known) Remarkable improvement within 2 months |
| Reuner et al. 2019 [21] | 17-year-old female with hepatic WD (moderately elevated transaminases) | Duration of WD treatment with DPA: 6 years Since diagnosis aged 11, patient was treated with DPA 300 mg/day with pyridoxine supplementation 20 mg/week | Exercise-induced speech difficulties, mouth and tongue motility and dysphagia RNS showed significant decrement in the orbicularis oris (38%) and trapezius (25%) muscle Chest magnetic resonance imaging visualized thymus hyperplasia | Positive serum AChR-Abs (>5 nM) MuSK-Abs not tested | Continuation of DPA Pyridostigmine introduced (180 mg/day) Robot-assisted endoscopic thymectomy showed marked lymphofolicular hyperplasia (no thymoma) Remarkable improvement, 2 years after cessation of antimyasthenic therapy patient remains asymptomatic |
| Tan et al. 2012 [22] | 16-year-old female with WD (clinically asymptomatic—diagnosed during family screening) | Duration of WD treatment with DPA: 4 months Since diagnosis aged 15 (10 months earlier), patient was treated with DPA 750 mg/day | Diplopia, bilateral ptosis, jaw weakness with difficulties in smiling and chewing, dysarthria and dysphagia, proximal limb weakness (with massive bilateral breast enlargement, galactorrhea, and dermopathy) RNS showed a decremental response of the right deltoid and orbicularis muscles Chest CT not done | AChR-Abs and MuSK-Abs not tested | Withdrawal of DPA; trientine 900 mg/day introduced with clobetasone butyrate for skin lesions (indication dermopathy) One month later disappearance of the visual and skin complaints At follow-up at 7 months, complete resolution of all neurological and dermatological DPA-induced side effects was observed |
| Varghese et al. 2002 [23] | 12-year-old female with hepatic WD (prior hepatomegaly and jaundice) | Duration of WD treatment with DPA: 4 years Since diagnosis aged 8, patient was treated with DPA 1000 mg/day and pyridoxine 1 mg/day | Left-sided ptosis (cranial nerves normal, systemic examination unremarkable) RNS showed decremental response of more than 20% in both deltoid muscles and the left orbicularis oculi Positive edrophonium test Normal chest CT | Positive serum AChR-Abs (29.1 nmol/L; normal < 0.4) MuSK-Abs not tested | Withdrawal of DPA and trientine 1500 mg/day introduced Remarkable improvement within 3 months, all symptoms disappeared After 1 year, AChR-Abs became negative |
| Masters et al. 1976 [25] | 18-year-old female with hepatic WD | Duration of WD treatment with DPA: 8 years Since diagnosis aged 10, patient was treated with DPA 1000 mg/day | Progressive muscular weakness with bilateral ptosis, diplopia, bilateral facial and palatal weakness, dysphagia, dysarthria and marked fatigability RNS showed a decremental response of the right deltoid and orbicularis muscles Positive edrophonium test Normal chest CT | Serum AChR-Abs titer 19 U (normal 1 U) MuSK-Abs not tested | DPA was continued Pyridostigmine (120 mg/day) and neostigmine (15 mg as required) was introduced Thymectomy (enlarged thymus, with thymic hyperplasia) but no thymoma MG symptoms disappeared over 5 months; however, patient was diagnosed as MG and treated with pyridostigmine as increased serum AChR-Abs were observed during follow-up |
| Czlonkowska et al. 1975 [26] | 14-year-old male with WD (clinically asymptomatic—family screening) | Duration of WD treatment with DPA: 13 months Since diagnosis aged 13, patient was treated with DPA 1000 mg/day and pyridoxine 1 mg/day | Initial right-sided ptosis (cranial nerves normal, systemic examination unremarkable). After 10 days, left ptosis also occurred RNS not determined Positive edrophonium test Chest CT not done | Serum AChR-Abs positive MuSK-Abs not tested | Withdrawal of DPA Neostigmine was introduced (dose not known) Remarkable improvement with cessation of all symptoms within 6 weeks After 8 months, DPA 750 mg/day was reintroduced without adverse events during next 10 months |
| Affected System | Symptoms and Their Estimated Frequency | Type of ADR (Early/Late) |
|---|---|---|
| Skin | Degenerative dermatoses (elastosis perforans serpiginosa a, cutis laxa (skin laxity) a, anterodermax, pseudo-pseudoxanthoma elasticum a) Bullous dermatosesx (pemphigus and bullous disease) Miscellaneous cutaneous conditions (lichen planus-like eruptionsx, aphtous stomatitis or glossitis a, oral ulcerations a, alopecia a, psoriasiform dermatitisx, seborrheic dermatitis-like picturex, yellow-nail syndromex) | late early late |
| Nervous system | Paradoxical neurological deterioration b, myasthenia-like syndromesx, peripheral sensory-motor neuropathiesx, optic nerve neuropathyx, serous retinitisx, hypogeusia (diminution in taste perception)x, deafnessx | early/late late |
| Connective tissue disease | Lupus-like syndrome, rheumatoid arthritisx, polymyositisx, arthralgiax, | late early/late |
| Renal | Proteinuria b, hematuria a, Goodpasture syndromex, severe fatal glomerulonephritis associated with intra-alveolar hemorrhagex, renal vasculitisx | early/late late |
| Respiratory | Bronchiolitisx, pulmonary fibrosisx, pneumonitisx, pleural effusionx, dyspneax | late |
| Gastrointestinal | Nauseax, vomitingx, diarrheax, cholestatic jaundicex, liver siderosisx | early/late late |
| Hematologic | Thrombocytopenia c, neutropeniax, hemolytic anemiax Agranulocytosisx, aplastic anemiax, | early/late late |
| Immunologic | Immunoglobulin deficiencyx | late |
| Reproductive system and breast disorders | Breast enlargement a | late |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antos, A.; Członkowska, A.; Bembenek, J.; Kurkowska-Jastrzębska, I.; Litwin, T. D-Penicillamine-Induced Myasthenia Gravis—A Probable Complication of Wilson’s Disease Treatment—A Case Report and Systematic Review of the Literature. Life 2023, 13, 1715. https://doi.org/10.3390/life13081715
Antos A, Członkowska A, Bembenek J, Kurkowska-Jastrzębska I, Litwin T. D-Penicillamine-Induced Myasthenia Gravis—A Probable Complication of Wilson’s Disease Treatment—A Case Report and Systematic Review of the Literature. Life. 2023; 13(8):1715. https://doi.org/10.3390/life13081715
Chicago/Turabian StyleAntos, Agnieszka, Anna Członkowska, Jan Bembenek, Iwona Kurkowska-Jastrzębska, and Tomasz Litwin. 2023. "D-Penicillamine-Induced Myasthenia Gravis—A Probable Complication of Wilson’s Disease Treatment—A Case Report and Systematic Review of the Literature" Life 13, no. 8: 1715. https://doi.org/10.3390/life13081715