
Prebiotic Synthesis of Aspartate Using Life’s Metabolism as a Guide

Abstract
:1. Introduction
1.1. Life as a Guide to Protometabolism
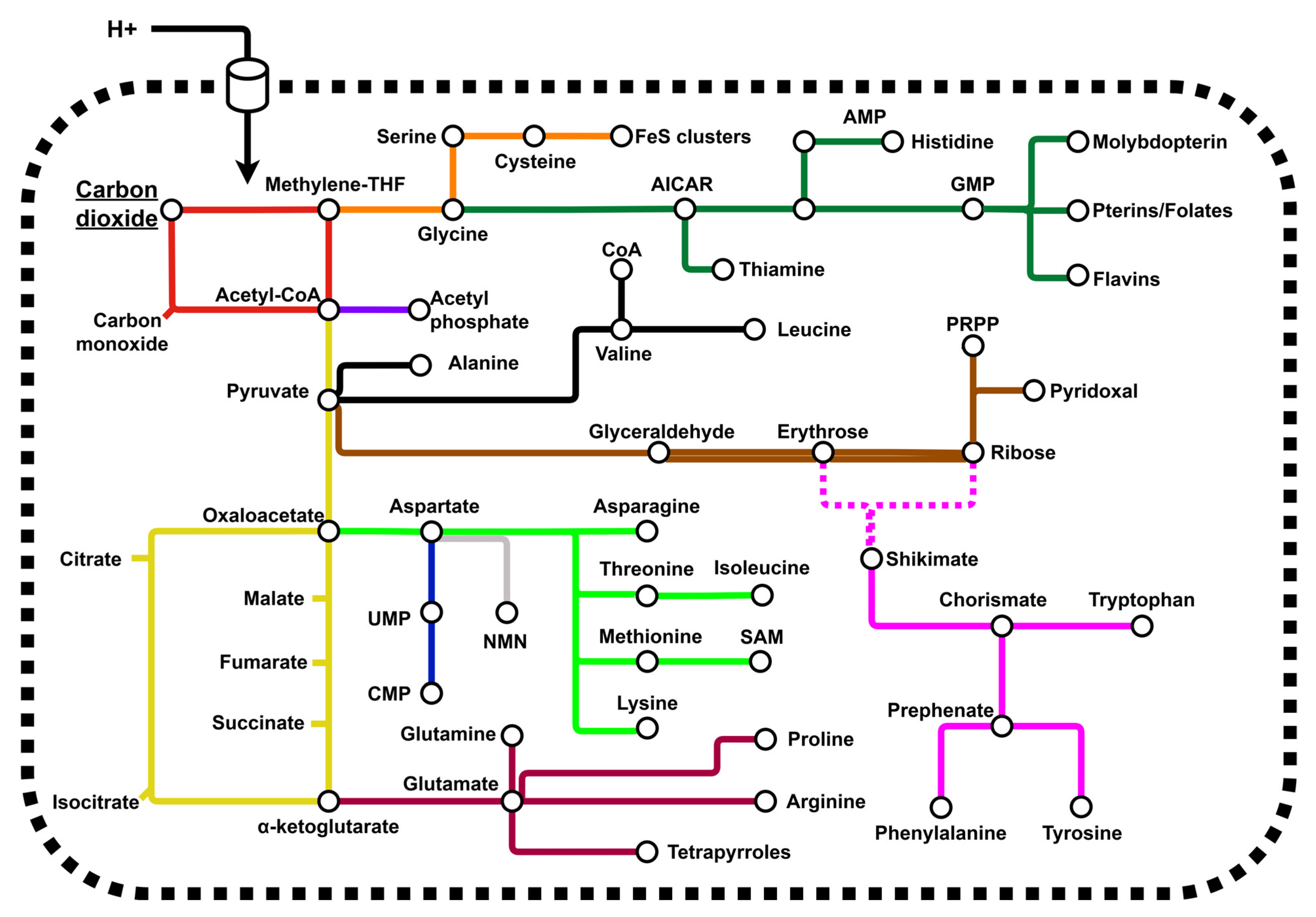
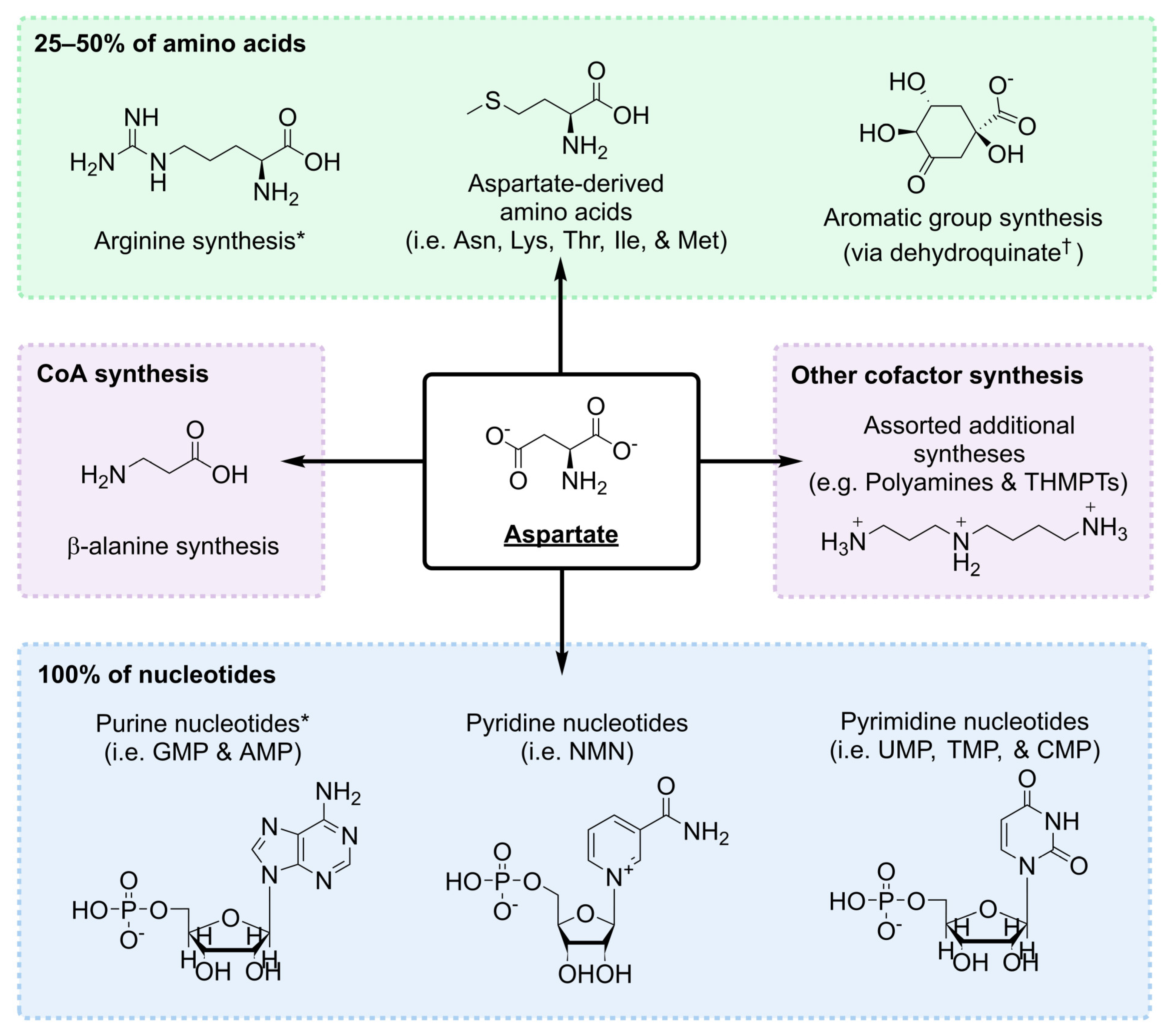
1.2. Aspartate Is a Nodal Metabolite
1.3. Prebiotic Synthesis of Aspartate
2. Materials and Methods
2.1. Reagents
2.2. General Reaction Conditions
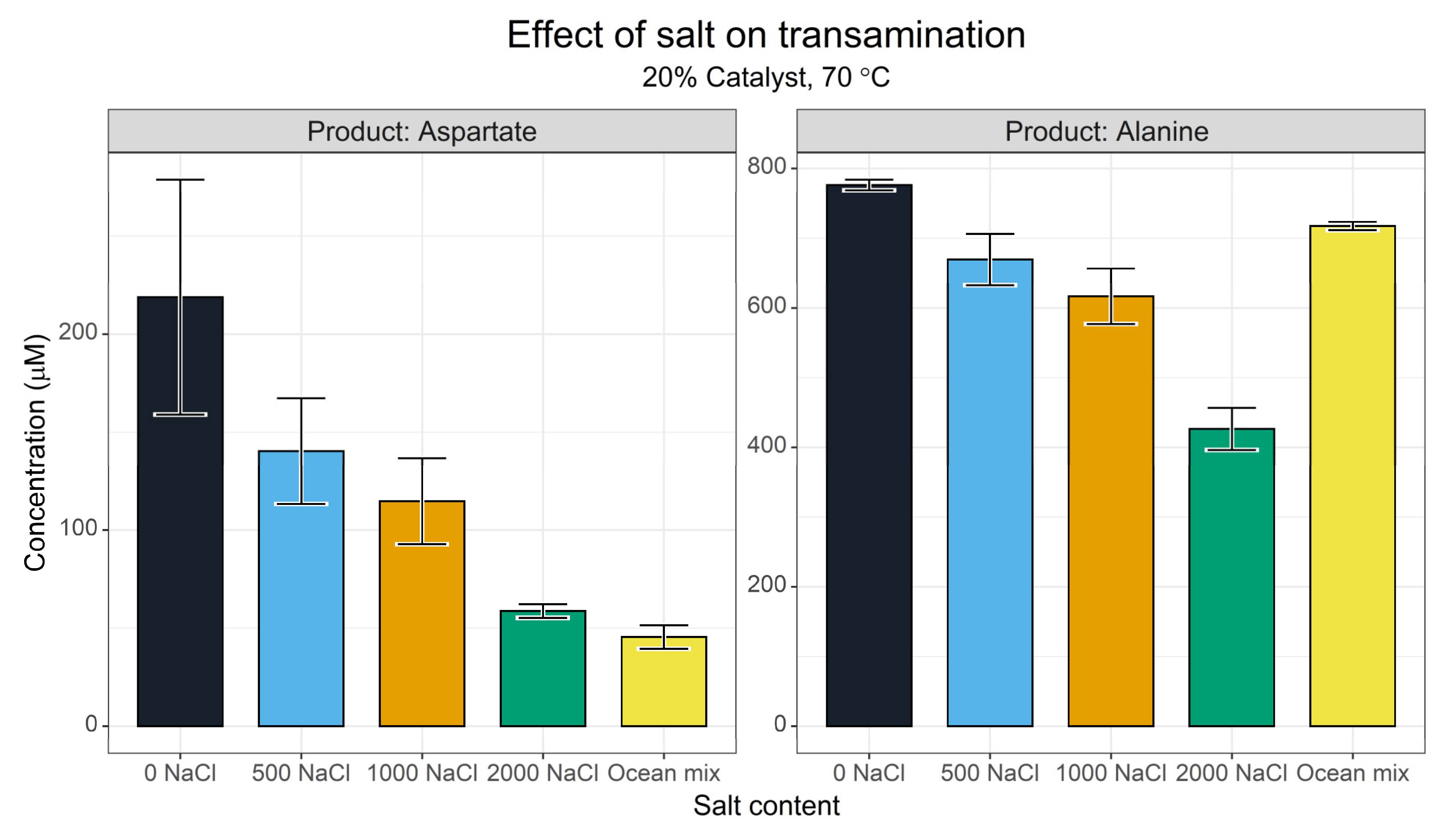
2.3. Ionic Strength Screens
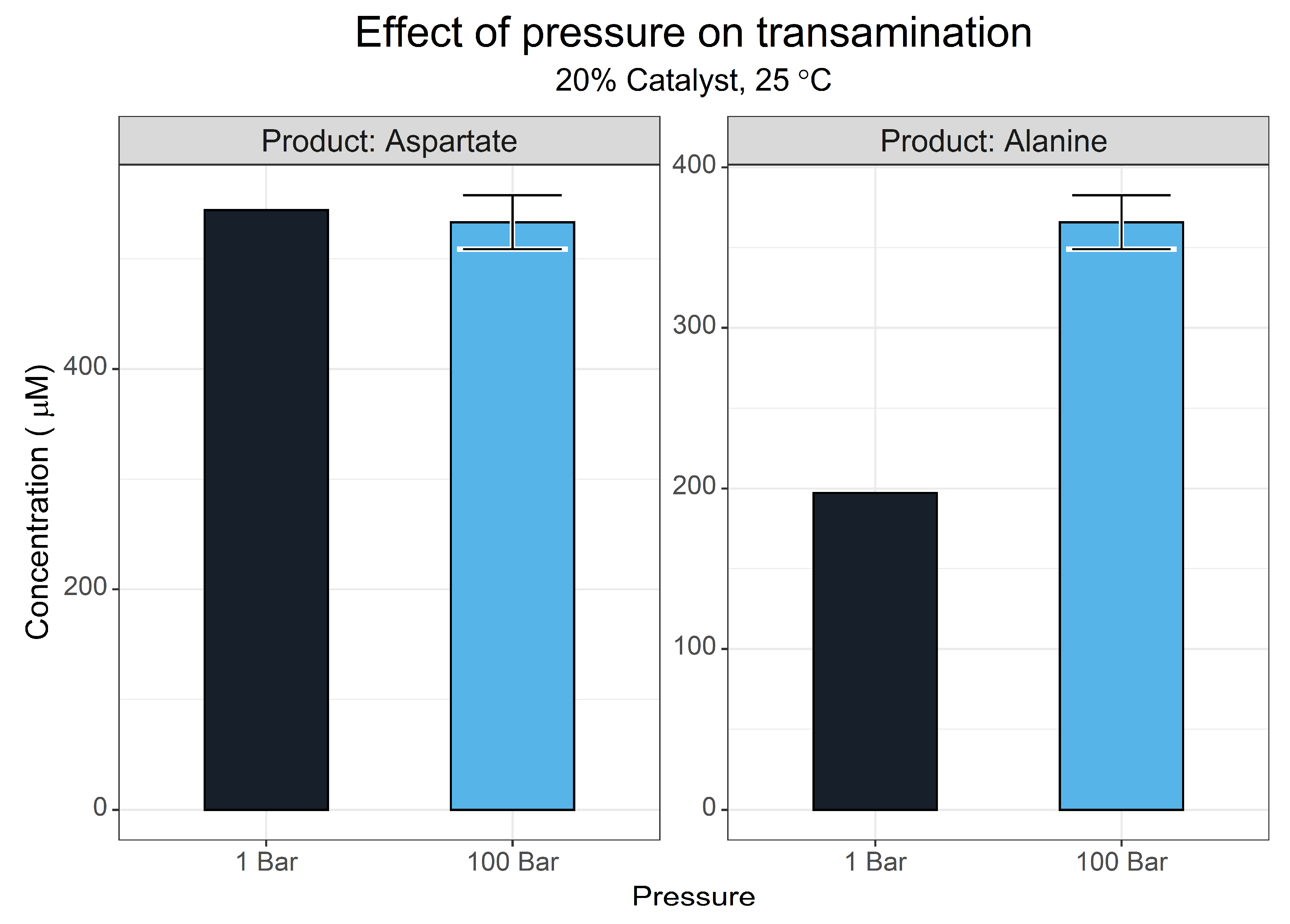
2.4. Pressure Screen
2.5. β-Alanine Synthesis
2.6. Complete Reactions
2.7. 9-Fluorenylmethylchloroformate (FMOC) Derivatisation
2.8. High-Performance Liquid Chromatography (HPLC) Analysis
2.9. Isobutylchloroformate (iBuCF) Derivatisation
2.10. Gas Chromatography Mass Spectrometry (GC-MS) Analysis
3. Results
3.1. Transamination of Oxaloacetate by Pyridoxamine
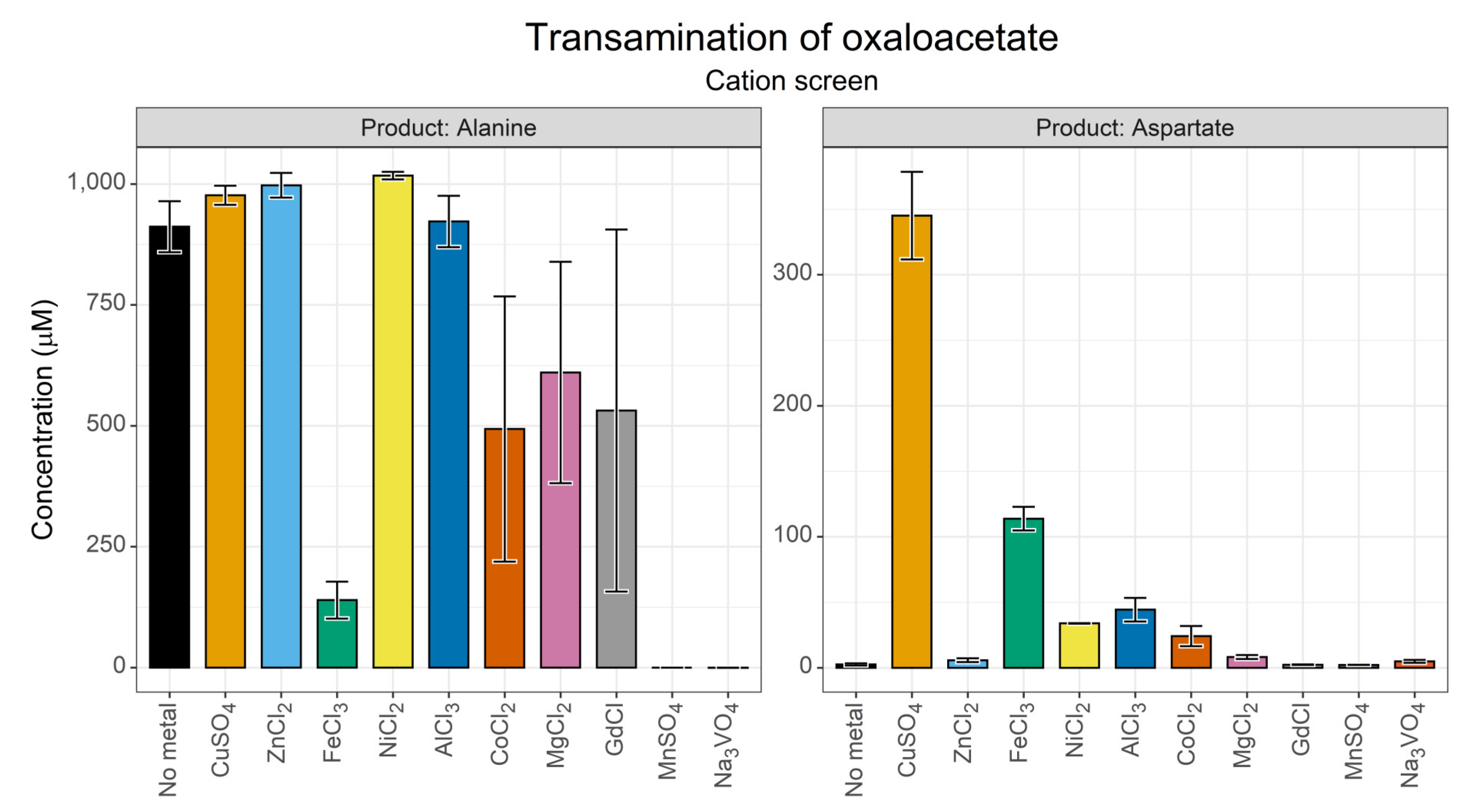
3.1.1. Effect of Transition Metal Ions
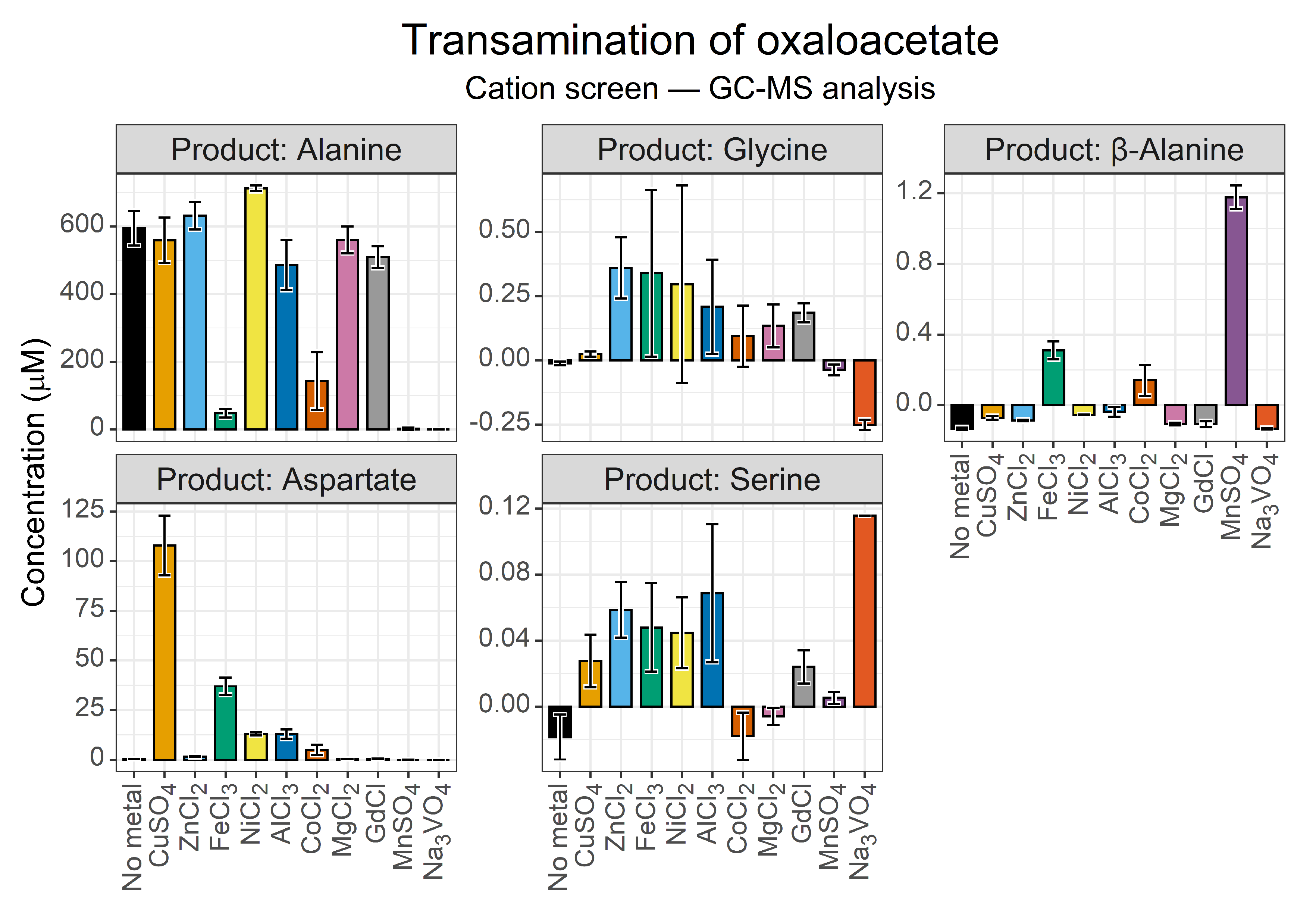
3.1.2. GC-MS Secondary Verification
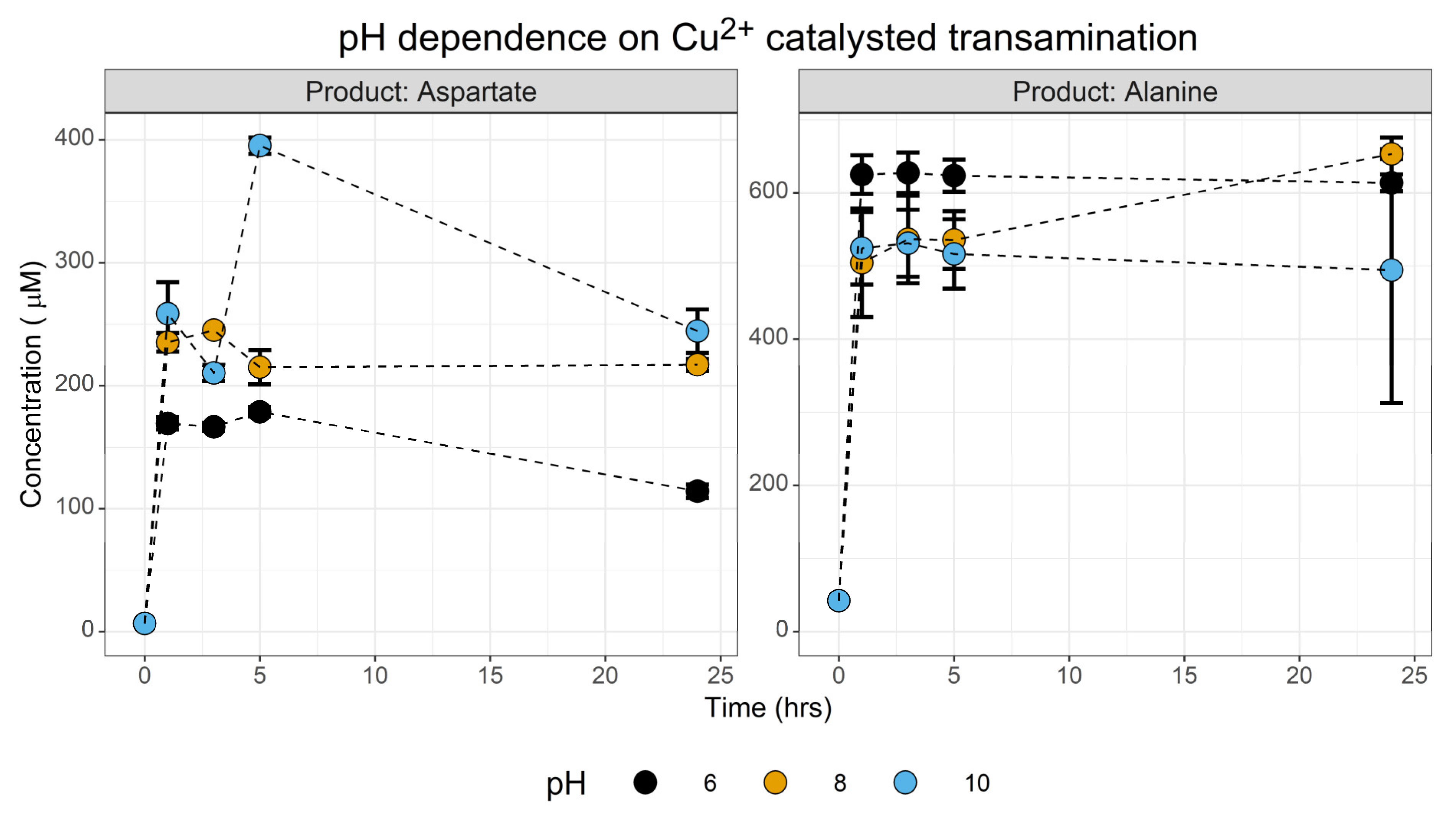
3.1.3. PH Screen
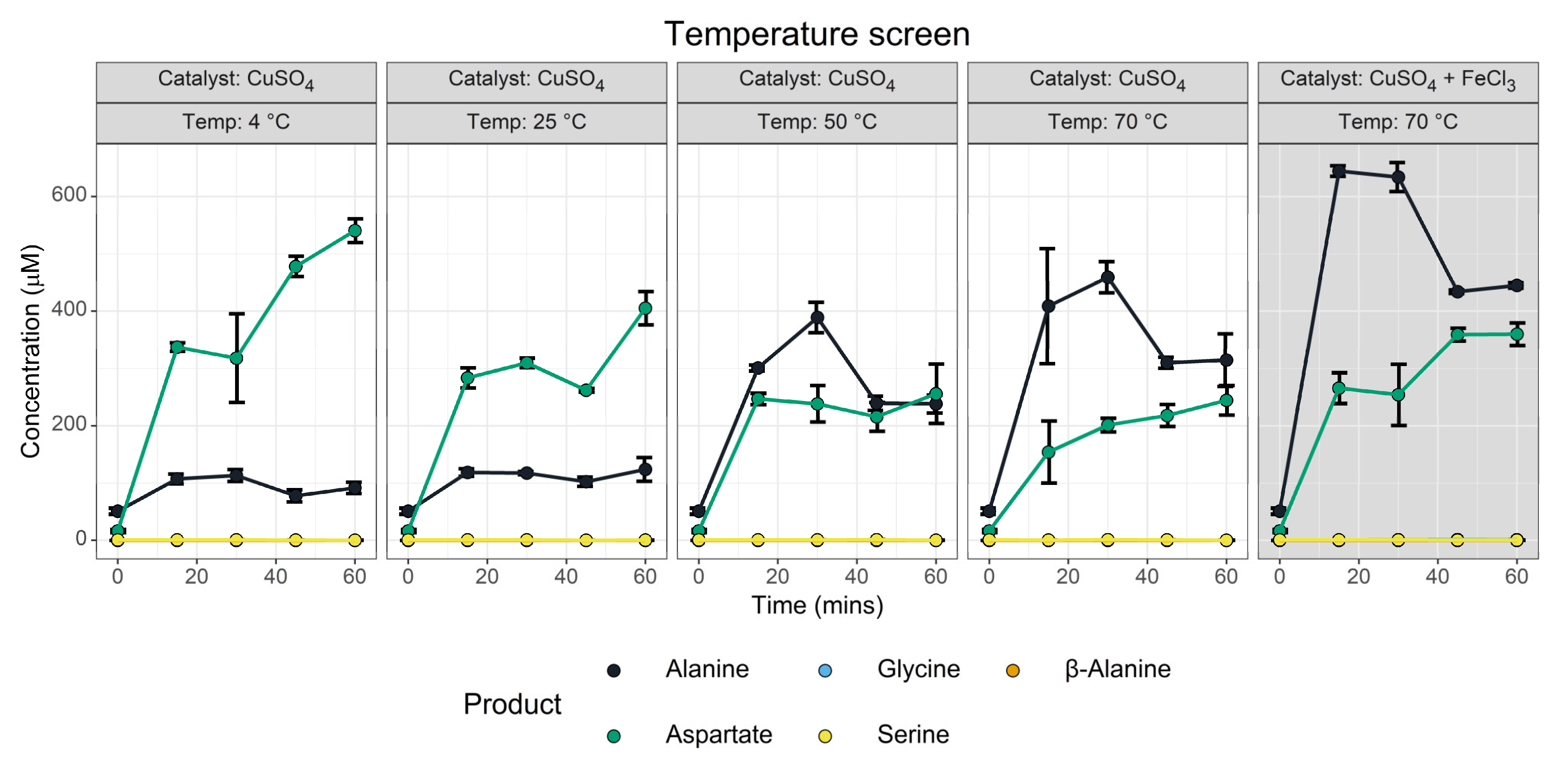
3.1.4. Temperature Screen
3.1.5. Ionic Strength
3.1.6. Pressure Effects
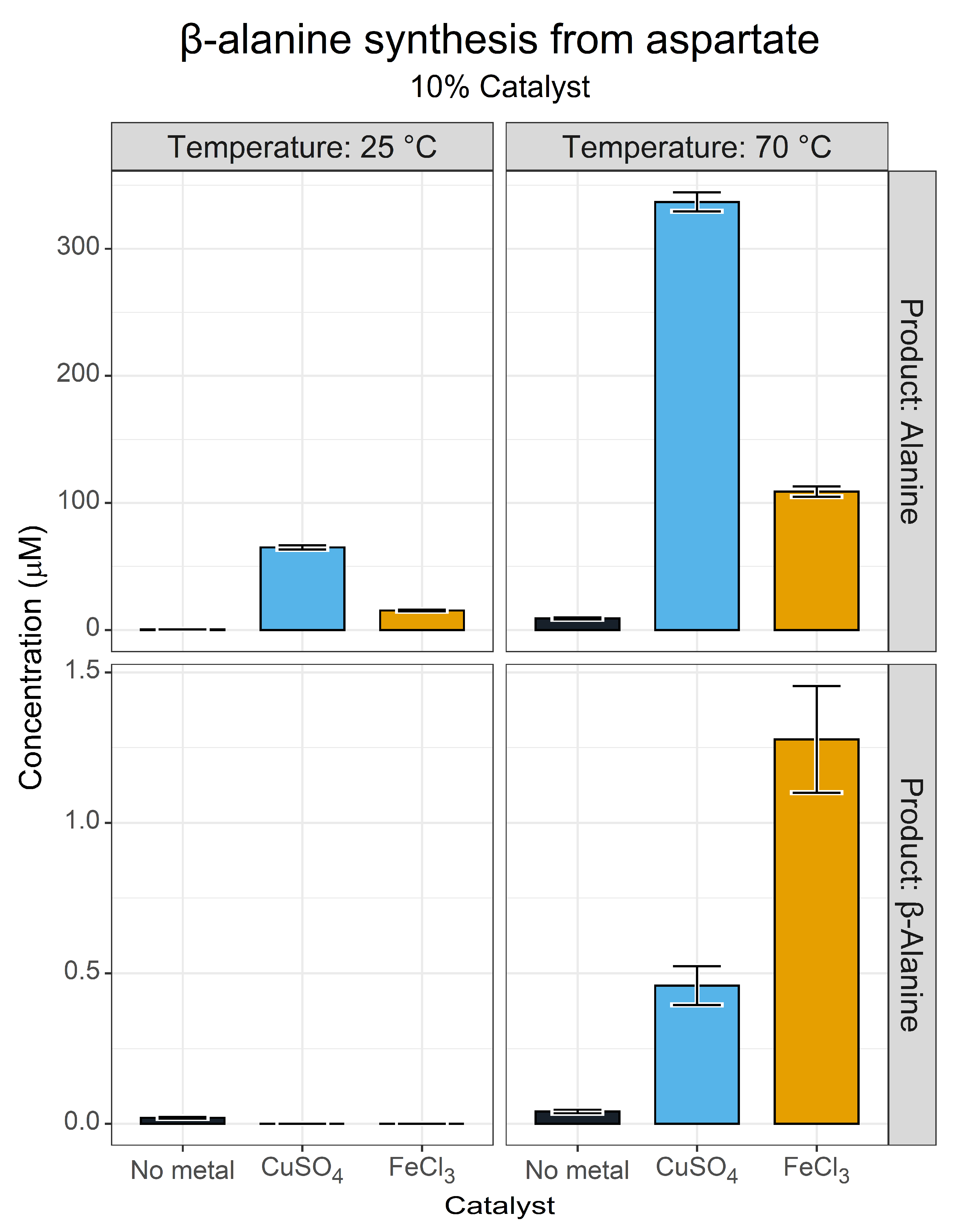
3.2. β-Alanine Synthesis
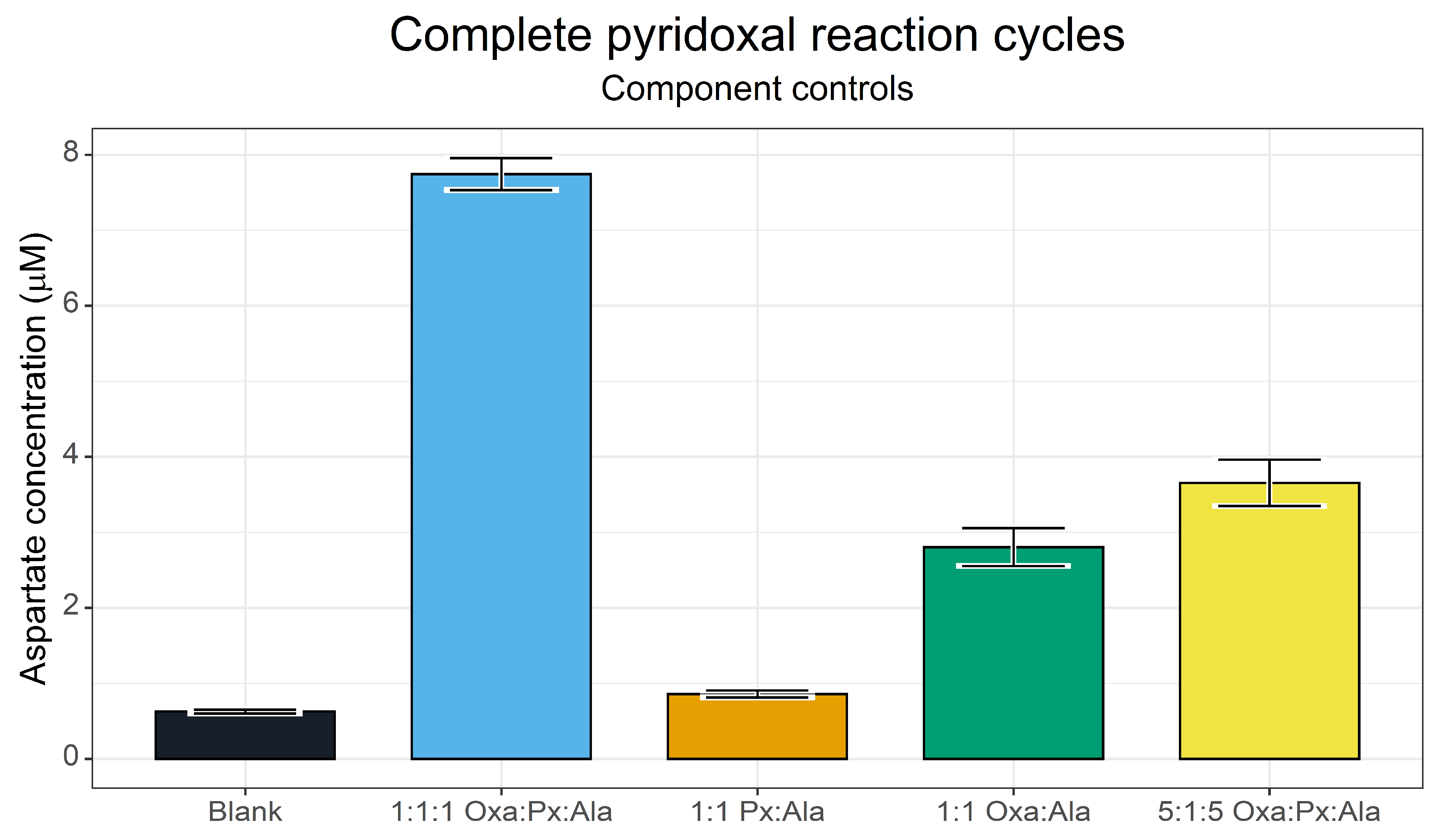
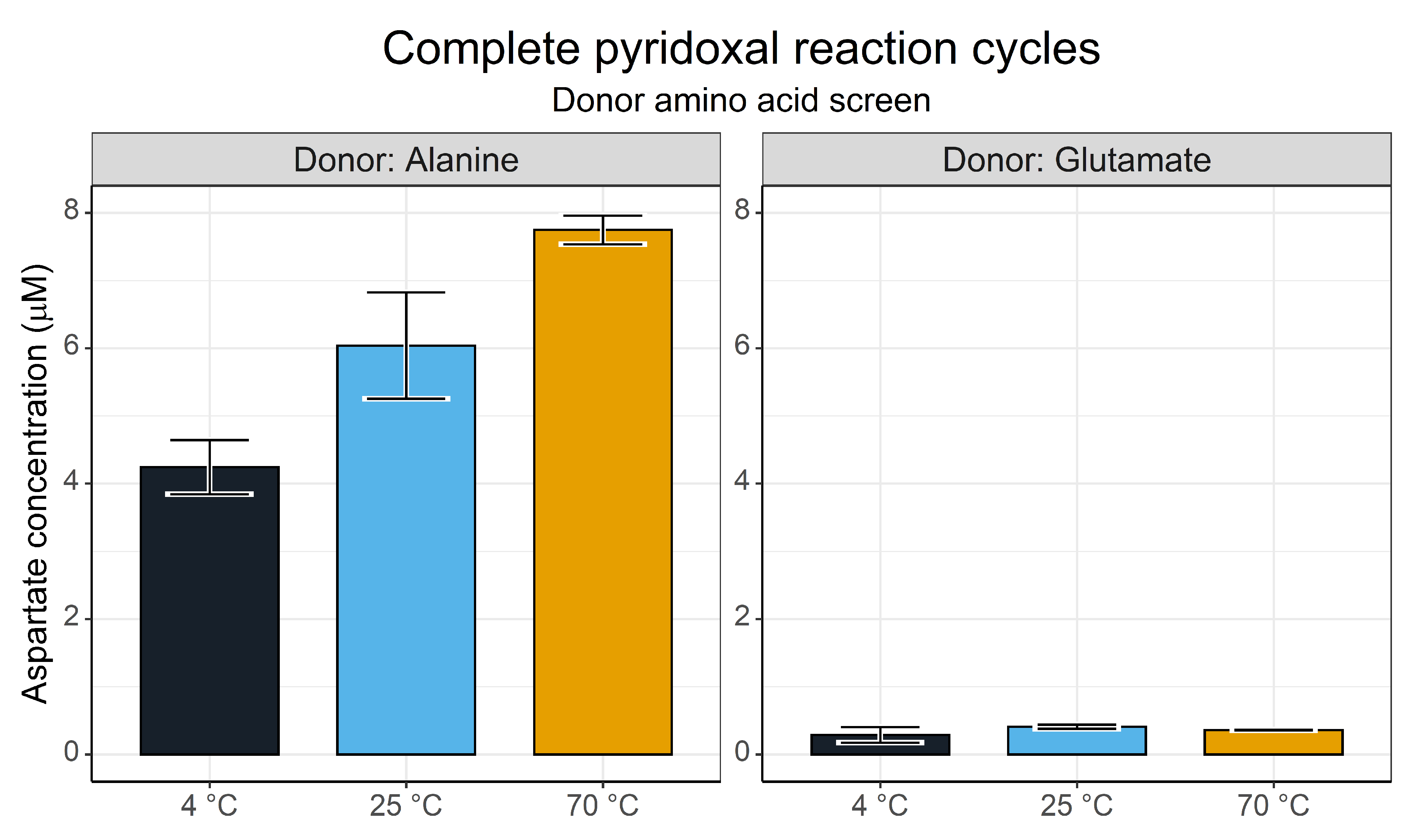
3.3. Complete Pyridoxal Cycles for the Synthesis of Aspartate
4. Discussion
4.1. Copper-Assisted Transamination Outcompetes Decarboxylation
4.2. Environmental Boundary Conditions
4.3. β-Alanine Synthesis at the Origins of Life
4.4. Complete Pyridoxal/Pyridoxamine Cycles
4.5. Future Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woese, C.R.; Dugre, D.H.; Saxinger, W.C.; Dugre, S.A. The molecular basis for the genetic code. Proc. Natl. Acad. Sci. USA 1966, 55, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Novozhilov, A.S. Origin and evolution of the genetic code: The universal enigma. IUBMB Life 2009, 61, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Palmeira, R.N.; Halpern, A.; Lane, N. A biophysical basis for the emergence of the genetic code in protocells. Biochim. Biophys. Acta Bioenerg. 2022, 1863, 148597. [Google Scholar] [CrossRef] [PubMed]
- Belousoff, M.; Davidovich, C.; Zimmerman, E.; Caspi, Y.; Wekselman, I.; Rozenszajn, L.; Shapira, T.; Sade-Falk, O.; Taha, L.; Bashan, A.; et al. Ancient machinery embedded in the contemporary ribosome. Biochem. Soc. Trans. 2010, 38, 422–427. [Google Scholar] [CrossRef]
- Bose, T.; Fridkin, G.; Davidovich, C.; Krupkin, M.; Dinger, N.; Falkovich, A.H.; Peleg, Y.; Agmon, I.; Bashan, A.; Yonath, A. Origin of life: Protoribosome forms peptide bonds and links RNA and protein dominated worlds. Nucleic Acids Res. 2022, 50, 1815–1828. [Google Scholar] [CrossRef]
- Anantharaman, V.; Koonin, E.V.; Aravind, L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002, 30, 1427–1464. [Google Scholar] [CrossRef]
- Koonin, E.V.; Krupovic, M.; Ishino, S.; Ishino, Y. The replication machinery of LUCA: Common origin of DNA replication and transcription. BMC Biol. 2020, 18, 61. [Google Scholar] [CrossRef]
- Wimmer, J.L.E.; Vieira, A.D.N.; Xavier, J.C.; Kleinermanns, K.; Martin, W.F.; Preiner, M. The Autotrophic Core: An Ancient Network of 404 Reactions Converts H2, CO2, and NH3 into Amino Acids, Bases, and Cofactors. Microorganisms 2021, 9, 458. [Google Scholar] [CrossRef]
- Braakman, R.; Smith, E. The compositional and evolutionary logic of metabolism. Phys. Biol. 2012, 10, 011001. [Google Scholar] [CrossRef]
- Braakman, R.; Smith, E. The Emergence and Early Evolution of Biological Carbon-Fixation. PLoS Comput. Biol. 2012, 8, e1002455. [Google Scholar] [CrossRef]
- Martin, W.F. Older Than Genes: The Acetyl CoA Pathway and Origins. Front. Microbiol. 2020, 11, 817. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Lane, N. Life as a guide to prebiotic nucleotide synthesis. Nat. Commun. 2018, 9, 5176. [Google Scholar] [CrossRef] [PubMed]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Nonenzymatic Metabolic Reactions and Life’s Origins. Chem. Rev. 2020, 120, 7708–7744. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M. An appeal to magic? The discovery of a non-enzymatic metabolism and its role in the origins of life. Biochem. J. 2018, 475, 2577–2592. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.D. Opinion: Studies on the origin of life—The end of the beginning. Nat. Rev. Chem. 2017, 1, 0012. [Google Scholar] [CrossRef]
- Xavier, J.C.; Hordijk, W.; Kauffman, S.; Steel, M.; Martin, W.F. Autocatalytic chemical networks at the origin of metabolism. Proc. R. Soc. B Boil. Sci. 2020, 287, 20192377. [Google Scholar] [CrossRef]
- Keller, M.A.; Turchyn, A.V.; Ralser, M. Non-enzymatic glycolysis and pentose phosphate pathway-like reactions in a plausibleArchean ocean. Mol. Syst. Biol. 2014, 10, 725. [Google Scholar] [CrossRef]
- Muchowska, K.B.; Varma, S.J.; Chevallot-Beroux, E.; Lethuillier-Karl, L.; Li, G.; Moran, J. Metals promote sequences of the reverse Krebs cycle. Nat. Ecol. Evol. 2017, 1, 1716–1721. [Google Scholar] [CrossRef]
- Keller, M.A.; Kampjut, D.; Harrison, S.A.; Ralser, M. Sulfate radicals enable a non-enzymatic Krebs cycle precursor. Nat. Ecol. Evol. 2017, 1, 83. [Google Scholar] [CrossRef]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Synthesis and breakdown of universal metabolic precursors promoted by iron. Nature 2019, 569, 104–107. [Google Scholar] [CrossRef]
- Mayer, R.J.; Kaur, H.; Rauscher, S.A.; Moran, J. Mechanistic Insight into Metal Ion-Catalyzed Transamination. J. Am. Chem. Soc. 2021, 143, 19099–19111. [Google Scholar] [CrossRef]
- Yi, J.; Kaur, H.; Kazöne, W.; Rauscher, S.A.; Gravillier, L.; Muchowska, K.B.; Moran, J. A Nonenzymatic Analog of Pyrimidine Nucleobase Biosynthesis. Angew. Chem. Int. Ed. 2022, 61, e202117211. [Google Scholar] [CrossRef]
- Whicher, A.; Camprubi, E.; Pinna, S.; Herschy, B.; Lane, N. Acetyl Phosphate as a Primordial Energy Currency at the Origin of Life. Orig. Life Evol. Biosph. 2018, 48, 159–179. [Google Scholar] [CrossRef]
- Pinna, S.; Kunz, C.; Halpern, A.; Harrison, S.A.; Jordan, S.F.; Ward, J.; Werner, F.; Lane, N. A prebiotic basis for ATP as the universal energy currency. PLoS Biol. 2022, 20, e3001437. [Google Scholar] [CrossRef]
- Varma, S.J.; Muchowska, K.B.; Chatelain, P.; Moran, J. Native iron reduces CO2 to intermediates and end-products of the acetyl-CoA pathway. Nat. Ecol. Evol. 2018, 2, 1019–1024. [Google Scholar] [CrossRef]
- Preiner, M.; Igarashi, K.; Muchowska, K.B.; Yu, M.; Varma, S.J.; Kleinermanns, K.; Nobu, M.K.; Kamagata, Y.; Tüysüz, H.; Moran, J.; et al. A hydrogen-dependent geochemical analogue of primordial carbon and energy metabolism. Nat. Ecol. Evol. 2020, 4, 534–542. [Google Scholar] [CrossRef]
- Beyazay, T.; Belthle, K.S.; Farès, C.; Preiner, M.; Moran, J.; Martin, W.F.; Tüysüz, H. Ambient temperature CO2 fixation to pyruvate and subsequently to citramalate over iron and nickel nanoparticles. Nat. Commun. 2023, 14, 570. [Google Scholar] [CrossRef]
- Rauscher, S.A.; Moran, J. Hydrogen Drives Part of the Reverse Krebs Cycle under Metal or Meteorite Catalysis. Angew. Chem. Int. Ed. 2022, 61, e202212932. [Google Scholar] [CrossRef]
- Beyazay, T.; Ochoa-Hernández, C.; Song, Y.; Belthle, K.S.; Martin, W.F.; Tüysüz, H. Influence of Composition of Nickel-Iron Nanoparticles for Abiotic CO2 Conversion to Early Prebiotic Organics. Angew. Chem. 2023, e202218189. [Google Scholar] [CrossRef]
- West, T.O.; Sojo, V.; Pomiankowski, A.; Lane, N. The origin of heredity in protocells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160419. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, R.N.; Colnaghi, M.; Harrison, S.A.; Pomiankowski, A.; Lane, N. The limits of metabolic heredity in protocells. Proc. R. Soc. B Biol. Sci. 2022, 289, 20221469. [Google Scholar] [CrossRef] [PubMed]
- Aulakh, S.K.; Varma, S.J.; Ralser, M. Metal ion availability and homeostasis as drivers of metabolic evolution and enzyme function. Curr. Opin. Genet. Dev. 2022, 77, 101987. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.L.; Butcher, A.; Valas, R.E.; Bourne, P.E.; Caetano-Anollés, G. History of biological metal utilization inferred through phylogenomic analysis of protein structures. Proc. Natl. Acad. Sci. USA 2010, 107, 10567–10572. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, L.; Mansy, S. Metal Catalysts and the Origin of Life. Elements 2016, 12, 413–418. [Google Scholar] [CrossRef]
- Martin, W.F. Carbon–Metal Bonds: Rare and Primordial in Metabolism. Trends Biochem. Sci. 2019, 44, 807–818. [Google Scholar] [CrossRef]
- Paulus, H. Biosynthesis of the Aspartate Family of Amino Acids. In Bacillus Subtilis and Other Gram-Positive Bacteria; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; pp. 237–267. [Google Scholar] [CrossRef]
- Jander, G.; Joshi, V. Aspartate-Derived Amino Acid Biosynthesis in Arabidopsis thaliana. Arab. Book 2009, 7, e0121. [Google Scholar] [CrossRef]
- White, R.H. l-Aspartate Semialdehyde and a 6-Deoxy-5-ketohexose 1-Phosphate Are the Precursors to the Aromatic Amino Acids in Methanocaldococcus Jannaschii. Biochemistry 2004, 43, 7618–7627. [Google Scholar] [CrossRef]
- Pittard, J.; Yang, J. Biosynthesis of the Aromatic Amino Acids. EcoSal Plus 2008, 3. [Google Scholar] [CrossRef]
- Haines, R.J.; Pendleton, L.C.; Eichler, D.C. Argininosuccinate synthase: At the center of arginine metabolism. Int. J. Biochem. Mol. Biol. 2010, 2, 8–23. [Google Scholar]
- Cunin, R.; Glansdorff, N.; Piérard, A.; Stalon, V. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 1986, 50, 314–352. [Google Scholar] [CrossRef]
- Ouchi, T.; Tomita, T.; Horie, A.; Yoshida, A.; Takahashi, K.; Nishida, H.; Lassak, K.; Taka, H.; Mineki, R.; Fujimura, T.; et al. Lysine and arginine biosyntheses mediated by a common carrier protein in Sulfolobus. Nat. Chem. Biol. 2013, 9, 277–283. [Google Scholar] [CrossRef]
- Zhang, Y.; Morar, M.; Ealick, S.E. Structural biology of the purine biosynthetic pathway. Cell. Mol. Life Sci. 2008, 65, 3699–3724. [Google Scholar] [CrossRef]
- Kappock, T.J.; Ealick, S.E.; Stubbe, J. Modular evolution of the purine biosynthetic pathway. Curr. Opin. Chem. Biol. 2000, 4, 567–572. [Google Scholar] [CrossRef]
- Nasu, S.; Wicks, F.; Gholson, R. L-Aspartate oxidase, a newly discovered enzyme of Escherichia coli, is the B protein of quinolinate synthetase. J. Biol. Chem. 1982, 257, 626–632. [Google Scholar] [CrossRef]
- Chandler, J.; Gholson, R.; Scott, T. Studies on the de novo biosynthesis of NAD in Escherichia coli I. Labelling patterns from precursors. Biochim. Biophys. Acta Gen. Subj. 1970, 222, 523–526. [Google Scholar] [CrossRef]
- Gazzaniga, F.; Stebbins, R.; Chang, S.Z.; McPeek, M.A.; Brenner, C. Microbial NAD Metabolism: Lessons from Comparative Genomics. Microbiol. Mol. Biol. Rev. 2009, 73, 529–541. [Google Scholar] [CrossRef]
- Bossi, R.T.; Negri, A.; Tedeschi, G.; Mattevi, A. Structure of FAD-Bound l-Aspartate Oxidase: Insight into Substrate Specificity and Catalysis. Biochemistry 2002, 41, 3018–3024. [Google Scholar] [CrossRef]
- Goldford, J.E.; Smith, H.B.; Longo, L.M.; Wing, B.A.; McGlynn, S.E. Continuity between ancient geochemistry and modern metabolism enabled by non-autocatalytic purine biosynthesis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lee, J.; Sperandio, V.; Frantz, D.E.; Longgood, J.; Camilli, A.; Phillips, M.A.; Michael, A.J. An Alternative Polyamine Biosynthetic Pathway Is Widespread in Bacteria and Essential for Biofilm Formation in Vibrio cholerae. J. Biol. Chem. 2009, 284, 9899–9907. [Google Scholar] [CrossRef]
- White, R.H. The Conversion of a Phenol to an Aniline Occurs in the Biochemical Formation of the 1-(4-Aminophenyl)-1-deoxy-d-ribitol Moiety in Methanopterin. Biochemistry 2011, 50, 6041–6052. [Google Scholar] [CrossRef]
- Cronan, J.E.; Littel, K.J.; Jackowski, S. Genetic and biochemical analyses of pantothenate biosynthesis in Escherichia coli and Salmonella typhimurium. J. Bacteriol. 1982, 149, 916–922. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.; White, R.H. β-Alanine Biosynthesis in Methanocaldococcus jannaschii. J. Bacteriol. 2014, 196, 2869–2875. [Google Scholar] [CrossRef]
- van Poelje, P.D.; Snell, E.E. Pyruvoyl-Dependent Enzymes. Annu. Rev. Biochem. 1990, 59, 29–59. [Google Scholar] [CrossRef]
- Diaz, E.; Anton, D.L. Alkylation of an active-site cysteinyl residue during substrate-dependent inactivation of Escherichia coli S-adenosylmethionine decarboxylase. Biochemistry 1991, 30, 4078–4081. [Google Scholar] [CrossRef]
- Kezmarsky, N.D.; Xu, H.; Graham, D.; White, R.H. Identification and characterization of a l-tyrosine decarboxylase in Methanocaldococcus jannaschii. Biochim. Biophys. Acta Gen. Subj. 2005, 1722, 175–182. [Google Scholar] [CrossRef]
- Yokooji, Y.; Tomita, H.; Atomi, H.; Imanaka, T. Pantoate Kinase and Phosphopantothenate Synthetase, Two Novel Enzymes Necessary for CoA Biosynthesis in the Archaea. J. Biol. Chem. 2009, 284, 28137–28145. [Google Scholar] [CrossRef]
- von Delft, F.; Lewendon, A.; Dhanaraj, V.; Blundell, T.L.; Abell, C.; Smith, A.G. The Crystal Structure of E. coli Pantothenate Synthetase Confirms It as a Member of the Cytidylyltransferase Superfamily. Structure 2001, 9, 439–450. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Chen, A.F. Immobilized Coenzymes and Derivatives. In The Pyridine Nucleotide Coenzymes; Everse, J., Anderson, B., You, K.-S., Eds.; Academic Press: Cambridge, MA, USA, 1982; pp. 189–224. [Google Scholar] [CrossRef]
- Reitzer, L. Biosynthesis of Glutamate, Aspartate, Asparagine, L-Alanine, and D-Alanine. EcoSal Plus 2004, 1. [Google Scholar] [CrossRef]
- Huber, C.; Wächtershäuser, G. Primordial reductive amination revisited. Tetrahedron Lett. 2003, 44, 1695–1697. [Google Scholar] [CrossRef]
- Barge, L.M.; Flores, E.; Baum, M.M.; VanderVelde, D.G.; Russell, M.J. Redox and pH gradients drive amino acid synthesis in iron oxyhydroxide mineral systems. Proc. Natl. Acad. Sci. USA 2019, 116, 4828–4833. [Google Scholar] [CrossRef]
- Kitadai, N.; Nakamura, R.; Yamamoto, M.; Takai, K.; Yoshida, N.; Oono, Y. Metals likely promoted protometabolism in early ocean alkaline hydrothermal systems. Sci. Adv. 2019, 5, eaav7848. [Google Scholar] [CrossRef]
- Nakada, H.I.; Weinhouse, S. Non-Enzymatic Transamination With Glyoxylic Acid And Various Amino Acids. J. Biol. Chem. 1953, 204, 831–836. [Google Scholar] [CrossRef]
- Cabello, J.; Basilio, C.; Prajoux, V.; Plaza, M. Non-enzymic transamination between ornithine and glyoxylate. Arch. Biochem. Biophys. 1963, 100, 512–515. [Google Scholar] [CrossRef]
- Longenecker, J.B.; Snell, E.E. The Comparative Activities of Metal Ions in Promoting Pyridoxal-catalyzed Reactions of Amino Acids1. J. Am. Chem. Soc. 2002, 79, 142–145. [Google Scholar] [CrossRef]
- Percudani, R.; Peracchi, A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003, 4, 850–854. [Google Scholar] [CrossRef]
- Eliot, A.C.; Kirsch, J.F. Pyridoxal Phosphate Enzymes: Mechanistic, Structural, and Evolutionary Considerations. Annu. Rev. Biochem. 2004, 73, 383–415. [Google Scholar] [CrossRef]
- Meisch, H.U.; Hoffmann, H.; Reinle, W. Vanadium catalysis in the nonenzymatic transamination of δ-aminolevulinic acid. Z. Nat.Sect. C 1978, 33, 623–628. [Google Scholar] [CrossRef]
- Banks, B.E.C.; Diamantis, A.A.; Vernon, C.A. 825. Transamination. Part II. The non-enzymic reactions between pyridoxamine and pyruvic acid and between pyridoxal and alanine. J. Chem. Soc. 1961, 67, 4235–4247. [Google Scholar] [CrossRef]
- Zabinski, R.F.; Toney, M.D. Metal Ion Inhibition of Nonenzymatic Pyridoxal Phosphate Catalyzed Decarboxylation and Transamination. J. Am. Chem. Soc. 2000, 123, 193–198. [Google Scholar] [CrossRef]
- Maltais, T.R.; VanderVelde, D.; LaRowe, D.E.; Goldman, A.D.; Barge, L.M. Reactivity of Metabolic Intermediates and Cofactor Stability under Model Early Earth Conditions. Orig. Life Evol. Biosph. 2020, 50, 35–55. [Google Scholar] [CrossRef]
- Yu, L.; Sivitz, W.I. Oxaloacetate Mediates Mitochondrial Metabolism and Function. Curr. Metab. Syst. Biol. 2020, 7, 11–23. [Google Scholar] [CrossRef]
- Pulletikurti, S.; Yadav, M.; Springsteen, G.; Krishnamurthy, R. Prebiotic synthesis of α-amino acids and orotate from α-ketoacids potentiates transition to extant metabolic pathways. Nat. Chem. 2022, 14, 1142–1150. [Google Scholar] [CrossRef]
- Mayer, R.J.; Moran, J. Quantifying Reductive Amination in Nonenzymatic Amino Acid Synthesis. Angew. Chem. Int. Ed. 2022, 61, e202212237. [Google Scholar] [CrossRef]
- Sogin, E.M.; Puskás, E.; Dubilier, N.; Liebeke, M. Marine Metabolomics: A Method for Nontargeted Measurement of Metabolites in Seawater by Gas Chromatography–Mass Spectrometry. Msystems 2019, 4, e00638-19. [Google Scholar] [CrossRef]
- Jámbor, A.; Molnár-Perl, I. Amino acid analysis by high-performance liquid chromatography after derivatization with 9-fluorenylmethyloxycarbonyl chloride: Literature overview and further study. J. Chromatogr. A 2009, 1216, 3064–3077. [Google Scholar] [CrossRef]
- Wang, J.; Huang, Z.-H.; Gage, D.A.; Watson, J. Analysis of amino acids by gas chromatography—Flame ionization detection and gas chromatography—Mass spectrometry: Simultaneous derivatization of functional groups by an aqueous-phase chloroformate-mediated reaction. J. Chromatogr. A 1994, 663, 71–78. [Google Scholar] [CrossRef]
- Yin, J.; Wei, Y.; Liu, D.; Hu, Y.; Lu, Q.; Ang, E.L.; Zhao, H.; Zhang, Y. An extended bacterial reductive pyrimidine degradation pathway that enables nitrogen release from β-alanine. J. Biol. Chem. 2019, 294, 15662–15671. [Google Scholar] [CrossRef]
- Martin, W.F.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Philos. Trans. R. Soc. B Biol. Sci. 2006, 362, 1887–1926. [Google Scholar] [CrossRef]
- Sojo, V.; Herschy, B.; Whicher, A.; Camprubí, E.; Lane, N. The Origin of Life in Alkaline Hydrothermal Vents. Astrobiology 2016, 16, 181–197. [Google Scholar] [CrossRef]
- Herschy, B.; Whicher, A.; Camprubi, E.; Watson, C.; Dartnell, L.; Ward, J.; Evans, J.R.G.; Lane, N. An Origin-of-Life Reactor to Simulate Alkaline Hydrothermal Vents. J. Mol. Evol. 2014, 79, 213–227. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J. The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J. Geol. Soc. 1997, 154, 377–402. [Google Scholar] [CrossRef]
- Asano, T.; Le Noble, W.J. Activation and Reaction Volumes in Solution. Chem. Rev. 1978, 78, 407–489. [Google Scholar] [CrossRef]
- Krebs, H.A. The effect of inorganic salts on the ketone decomposition of oxaloacetic acid. Biochem. J. 1942, 36, 303–305. [Google Scholar] [CrossRef]
- Ito, H.; Kobayashi, H.; Nomiya, K. Metal-ion catalyzed decarboxylation of oxaloacetic acid. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1973, 69, 113–121. [Google Scholar] [CrossRef]
- Holland, H.D. The Chemical Evolution of the Atmosphere and Oceans; Princeton University Press: Princeton, NJ, USA, 2020. [Google Scholar] [CrossRef]
- Jordan, S.F.; Nee, E.; Lane, N. Isoprenoids enhance the stability of fatty acid membranes at the emergence of life potentially leading to an early lipid divide. Interface Focus 2019, 9, 20190067. [Google Scholar] [CrossRef]
- Walton, C.R.; Rimmer, P.; Shorttle, O. Can prebiotic systems survive in the wild? An interference chemistry approach. Front. Earth Sci. 2022, 10, 1011717. [Google Scholar] [CrossRef]
- Charlou, J.; Donval, J.; Fouquet, Y.; Jean-Baptiste, P.; Holm, N. Geochemistry of high H2 and CH4 vent fluids issuing from ultramafic rocks at the Rainbow hydrothermal field (36°14′N, MAR). Chem. Geol. 2002, 191, 345–359. [Google Scholar] [CrossRef]
- EniG. Periodic Table of the Elements. Solubility Product Constants 2022. Available online: https://www.periodni.com/solubility_product_constants.html (accessed on 27 February 2022).
- Fuchs, G. Alternative Pathways of Carbon Dioxide Fixation: Insights into the Early Evolution of Life? Annu. Rev. Microbiol. 2011, 65, 631–658. [Google Scholar] [CrossRef]
- Hudson, R.; de Graaf, R.; Rodin, M.S.; Ohno, A.; Lane, N.; McGlynn, S.E.; Yamada, Y.M.A.; Nakamura, R.; Barge, L.M.; Braun, D.; et al. CO 2 reduction driven by a pH gradient. Proc. Natl. Acad. Sci. USA 2020, 117, 22873–22879. [Google Scholar] [CrossRef]
- Raschle, T.; Arigoni, D.; Brunisholz, R.; Rechsteiner, H.; Amrhein, N.; Fitzpatrick, T.B. Reaction Mechanism of Pyridoxal 5′-Phosphate Synthase. Detection of an enzyme-bound chromophoric intermediate. J. Biol. Chem. 2007, 282, 6098–6105. [Google Scholar] [CrossRef]
- Camprubi, E.; Harrison, S.; Jordan, S.; Bonnel, J.; Pinna, S.; Lane, N. Do Soluble Phosphates Direct the Formose Reaction towards Pentose Sugars? Astrobiology 2022, 22, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Kopetzki, D.; Antonietti, M. Hydrothermal formose reaction. New J. Chem. 2011, 35, 1787–1794. [Google Scholar] [CrossRef]
- Omran, A. Plausibility of the Formose Reaction in Alkaline Hydrothermal Vent Environments. Orig. Life Evol. Biosph. 2020, 1–13. [Google Scholar] [CrossRef]
- Austin, S.M.; Waddell, T.G. Prebiotic Synthesis of Vitamin B6-type Compounds. Orig. Life Evol. Biosph. 1999, 29, 287–296. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (mins) | Mobile Phase A (%) | Mobile Phase B (%) |
|---|---|---|
| 0.00 | 72 | 28 |
| 2.38 | 72 | 28 |
| 21.42 | 55 | 45 |
| 25.39 | 5 | 95 |
| 29.36 | 5 | 95 |
| 30.95 | 72 | 28 |
| 50.00 | 72 | 28 |
| Amino Acid (Retention Time) | Primary Ion, m/z | Qualifier Ions (Relative Intensity %) |
|---|---|---|
| Alanine (15.49) | 144.0 | 88.0 (20.2), 116.0 (11.3) |
| Glycine (15.69) | 133.0 | 102.0 (32.4), 120.0 (18.0), 176.0 (17.6) |
| β-Alanine (16.66) | 116.0 | 88.0 (38.1), 98.0 (37.6), 143 (45.4) |
| Aspartate (20.85) | 244.0 | 88.0 (43.5), 144 (23.8), 160.0 (12.4) |
| Serine (21.52) | 204.0 | 86.0 (90.9), 148.0 (64.2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison, S.A.; Webb, W.L.; Rammu, H.; Lane, N. Prebiotic Synthesis of Aspartate Using Life’s Metabolism as a Guide. Life 2023, 13, 1177. https://doi.org/10.3390/life13051177
Harrison SA, Webb WL, Rammu H, Lane N. Prebiotic Synthesis of Aspartate Using Life’s Metabolism as a Guide. Life. 2023; 13(5):1177. https://doi.org/10.3390/life13051177
Chicago/Turabian StyleHarrison, Stuart A., William L. Webb, Hanadi Rammu, and Nick Lane. 2023. "Prebiotic Synthesis of Aspartate Using Life’s Metabolism as a Guide" Life 13, no. 5: 1177. https://doi.org/10.3390/life13051177