

Anti-Diabetic Therapy and Heart Failure: Recent Advances in Clinical Evidence and Molecular Mechanism
 ,
,
Abstract
:1. Introduction
2. Anti-Diabetic Drugs and Heart Failure: Recent Progress from Clinical Trials
2.1. Sodium-Glucose Co-Transporter-2 Inhibitor (SGLT2 Inhibitors)
2.1.1. Prevention of HF in Diabetic Patients
2.1.2. Treatment of Heart Failure in Diabetic Patients
2.2. Glucagon-like Peptide Receptor Agonist (GLP-1 RA)
2.3. Dual Incretin Receptor Agonists
2.4. Bariatric and Metabolic Surgery
2.5. Bromocriptine Mesylate
2.5.1. Cardiovascular Benefits in Diabetic Patients
2.5.2. Therapeutic Efficacy in Peripartum Cardiomyopathy
2.6. Imeglimin
2.7. Metformin
2.8. Insulin
2.9. Sulfonylurea
2.10. Alpha-Glucosidase Inhibitors
2.11. Thiazolidinediones (TZD)
2.12. Dipeptidyl Peptidase-4 (DPP-4) Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Condition | Trial | Reference | Agent | No. | Follow-Up (Years) | Mean Age | Women (%) | DM (%) | HF (%) | CV Death or HHF (HR) | HHF (HR) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SGLT2i | DM | EMPAREG OUTCOME | [5,8] | Empagliflozin | 7020 | 3.1 | 63 | 29 | 100 | 10 | 0.66 * | 0.65 * |
| CANVAS | [6,9] | Canagliflozin | 10,420 | 2.4 | 63 | 36 | 100 | 14 | 0.78 * | 0.67 * | ||
| DECLARE-TIMI 58 | [7,10] | Dapagliflozin | 17,160 | 4.2 | 64 | 37 | 100 | 10 | 0.83 * | 0.73 * | ||
| VERTIS CV | [11,12] | Ertugliflozin | 8246 | 3.5 | 64 | 30 | 100 | 24 | 0.88 | 0.70 * | ||
| CKD | CREDENCE | [15,18] | Canagliflozin | 4041 | 2.6 | 63 | 34 | 100 | 15 | 0.69 * | 0.61 * | |
| SCORED | [16] | Sotagliflozin | 10,508 | 1.3 | 69 | 45 | 100 | 31 | 0.77 * | 0.67 *# | ||
| DAPA-CKD | [17,19] | Dapagliflozin | 4304 | 2.4 | 62 | 33 | 68 | 11 | 0.71 * | 0.51 * | ||
| EMPA-KIDNEY | [20] | Empagliflozin | 6609 | 2 | 64 | 33 | 46 | 10 | 0.84 | 0.8 | ||
| HF | DAPA-HF | [21,22] | Dapagliflozin | 4744 | 1.5 | 66 | 23 | 45 | 100 | 0.75 * | 0.70 * | |
| EMPEROR-Reduced | [23] | Empagliflozin | 3730 | 1.3 | 67 | 24 | 50 | 100 | 0.75 * | 0.69 * | ||
| EMPEROR-Preserved | [24] | Empagliflozin | 5988 | 2.2 | 72 | 45 | 49 | 100 | 0.79 * | 0.71 * | ||
| DELIVER | [25] | Dapagliflozin | 6263 | 2.3 | 72 | 44 | 45 | 100 | 0.80 * | 0.77 * | ||
| SOLOIST-WHF | [26] | Sotagliflozin | 1222 | 0.75 | 70 | 34 | 100 | 100 | 0.71 * | 0.64 *# | ||
| GLP-1 RA | DM | Harmony Outcomes | [28,29] | Albiglutide | 9463 | 1.6 | 64 | 31 | 100 | 20 | 0.85 | 0.71 * |
| AMPLITUDE-O | [30] | Efpeglenatide | 4076 | 1.8 | 65 | 33 | 100 | 18 | - | 0.61 * | ||
| LEADER | [32,33] | Liraglutide | 9340 | 3.8 | 64 | 36 | 100 | 18 | 0.82 * | 0.87 | ||
| EXSCEL | [34] | Exenatide | 14,752 | 3.2 | 62 | 38 | 100 | 16 | - | 0.94 | ||
| SUSTAIN 6 | [35] | Semaglutide | 3297 | 2.1 | 65 | 39 | 100 | 24 | - | 1.11 | ||
| PIONEER 6 | [36] | Semaglutide (oral) | 3183 | 1.3 | 66 | 32 | 100 | 12 | - | 0.86 | ||
| ELIXA | [37] | Lixisenatide | 6068 | 2.1 | 60 | 31 | 100 | 22 | - | 0.96 | ||
| REWIND | [38] | Dulaglutide | 9901 | 5.4 | 66 | 46 | 100 | 9 | - | 0.93 & | ||
| Bromocriptine | DM | [54] | Bromocriptine | 3070 | 1 | 60.1 | 57 | 100 | - | - | 0.77 | |
| Metformin | DM | SAVOR-TIMI 53 (post hoc) | [66] | Metformin | 12,156 | 2.1 | 62.4 | 33.2 | 100 | 13.18 | - | 0.97 |
| Insulin | ORIGIN | [70] | Glargine | 12,537 | 6.2 | 63.5 | 34.8 | 100 | - | - | 0.90 | |
| Insulin or SU | DM + CAD | BARI 2D | [71] | Insulin or SU | 2368 | 5 | 62.4 | 29.6 | 100 | 21.3 | - | 0.86 |
| Insulin or SU | DM | UKPDS 33 | [75] | Insulin or SU | 3867 | 10 | 54 | 38.9 | 100 | - | - | 0.91 |
| α-glucosidase inhibitor | Prediabetes+ CAD | ACE | [80] | Acarbose | 6552 | 5 | 64.3 | 27 | 0 | 0 | 0.89 | |
| TZD | Prediabetes | DREAM | [85] | Rosiglitazone | 5269 | 3 | 54.7 | 59.2 | 0 | 0 | 7.04 * | |
| DM + HF | [86] | Rosiglitazone | 224 | 1 | 62.1 | 18.32 | 100 | 100 | 1.8 @ | |||
| PROactive | [82] | Pioglitazone | 5238 | 2.85 | 61.7 | 33.5 | 100 | 0 | 1.49 * | |||
| DDP-4 inhibitor | DM | TECOS | [99] | Sitagliptin | 14,671 | 3 | 65.5 | 29.3 | 100 | 18 | 1.00 | |
| SAVORP-TIMI53 | [89] | Saxagliptin | 16,492 | 2.1 | 65.1 | 33.1 | 100 | 12.7 | 1.27 * | |||
| EXAMINE | [90] | Alogliptin | 5380 | 1.46 | 61 | 32.1 | 100 | 28.5 | 1.07 * | |||
| CARMELINA | [100] | Linagliptin | 6979 | 2.2 | 65.85 | 35.7 | 100 | 26.8 | 0.90 | |||
| CAROLINA | [101] | Linagliptin | 6033 | 6.3 | 64 | 39.9 | 100 | 4.6 | 1.21 | |||
| Life Style Intervention | DM + Overweight/Obesity | Look AHEAD | [103] | Moderate physical activity and calorie restriction | 5109 | 12.4 | 59.1 | 59.43 | 100 | - | 0.96 |
2.13. Pramlintide
2.14. Lifestyle Intervention
3. Anti-Diabetic Drugs and Heart Failure: Recent Progress of Molecular Mechanism
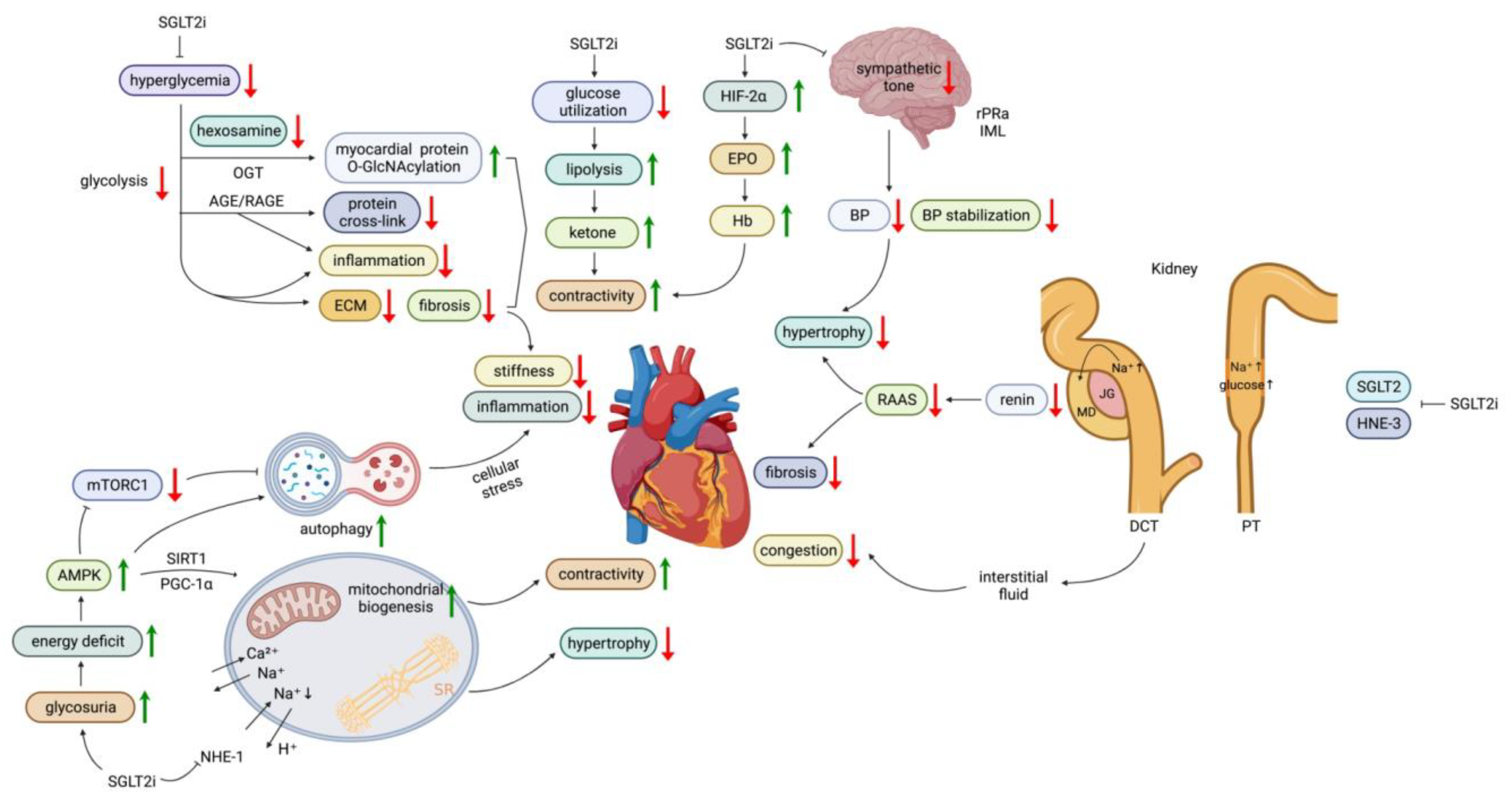
3.1. SGLT2 Inhibitors
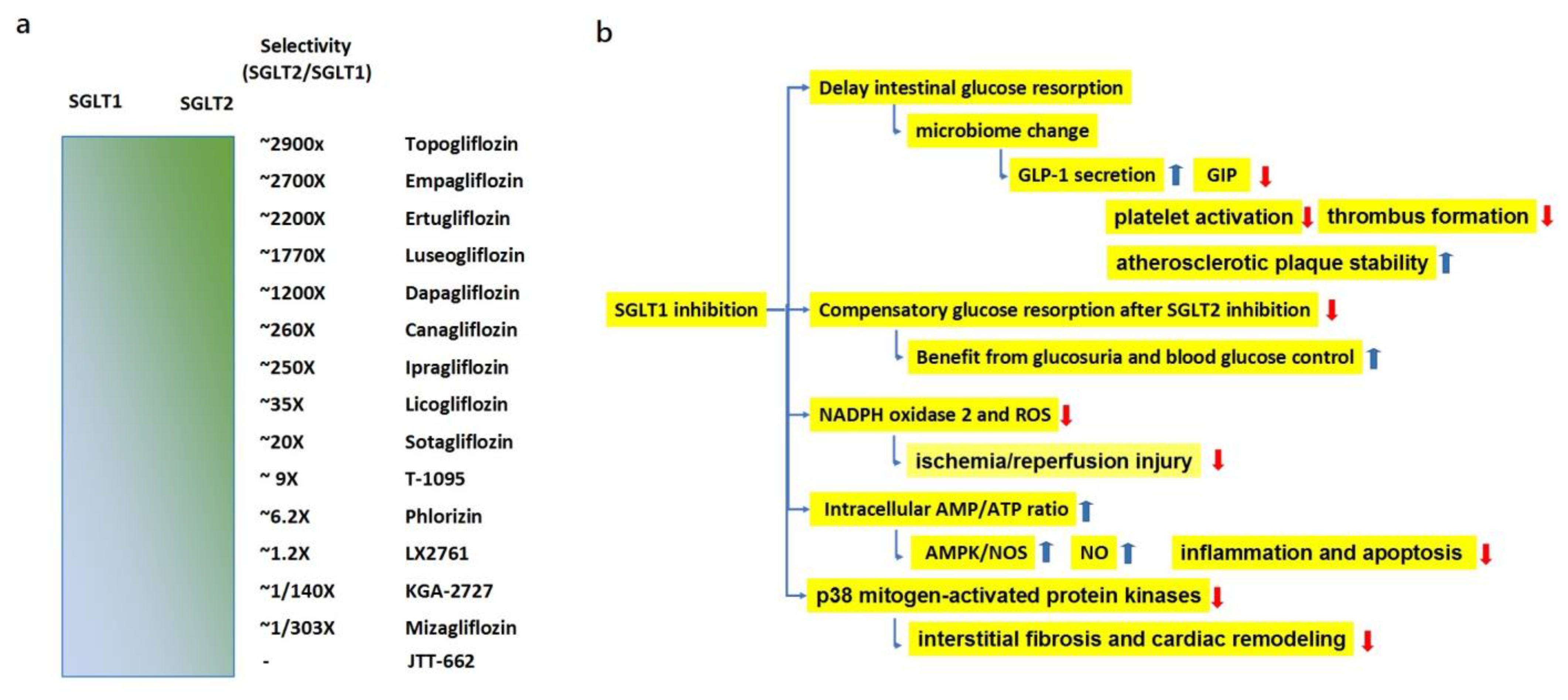
3.1.1. Effects of SGLT1 Inhibition
3.1.2. Effects of SGLT2 Inhibitors on Energy Utilization
3.1.3. Effects of SGLT2 Inhibitors on Renin-Angiotensin-Aldosterone (RAAS) Activation
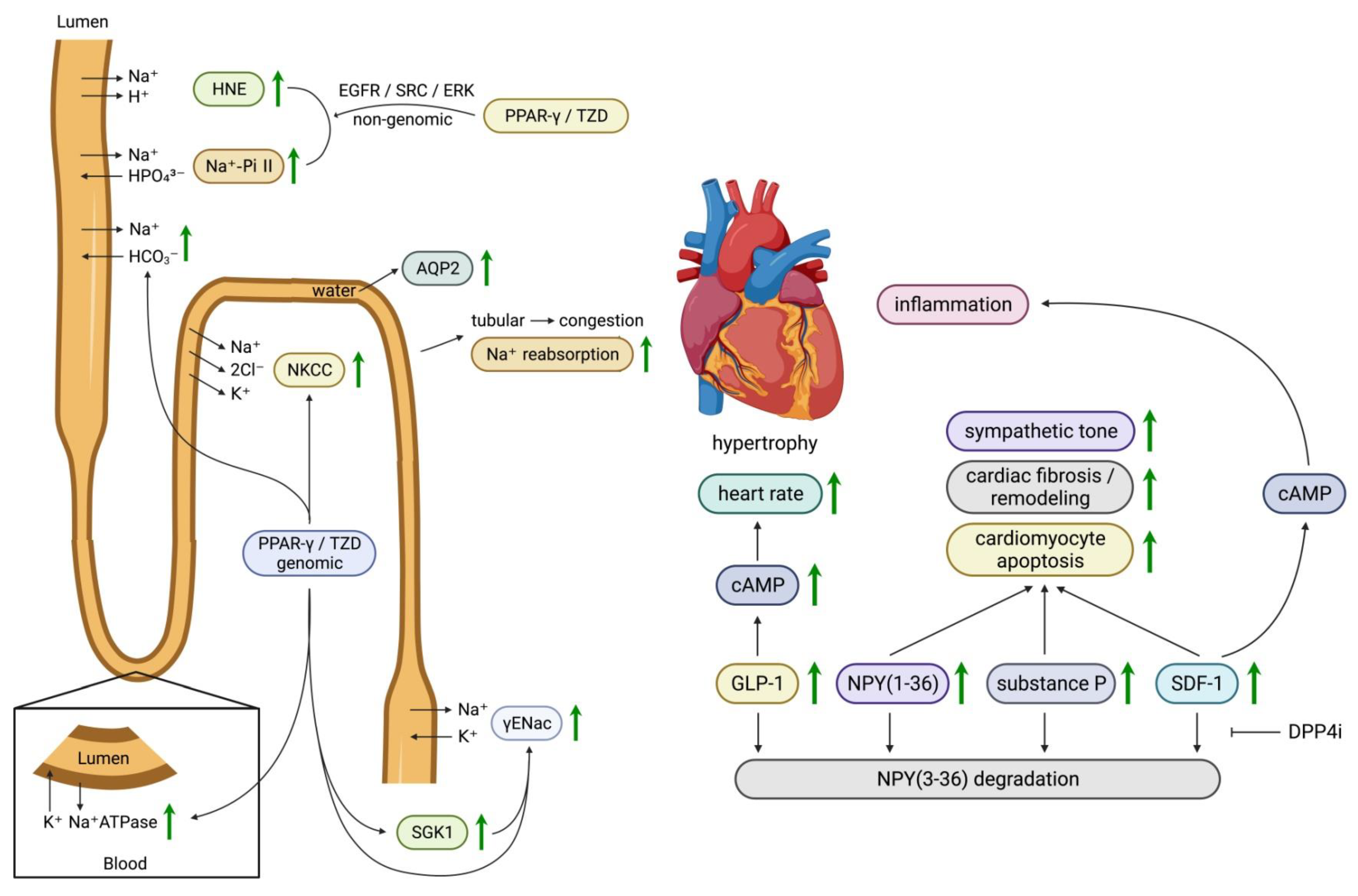
3.1.4. Effects of SGLT2 Inhibitors on Sodium and Fluid Dynamics
3.1.5. Effects of SGLT2 Inhibitors on the Sympathetic Nervous System
3.1.6. Anti-Inflammatory and Anti-Fibrotic Effects of SGLT2 Inhibitors
3.1.7. Effects of SGLT2 Inhibitors on Calcium Homeostasis
3.2. GLP-1 RA
3.3. Bariatric and Metabolic Surgery
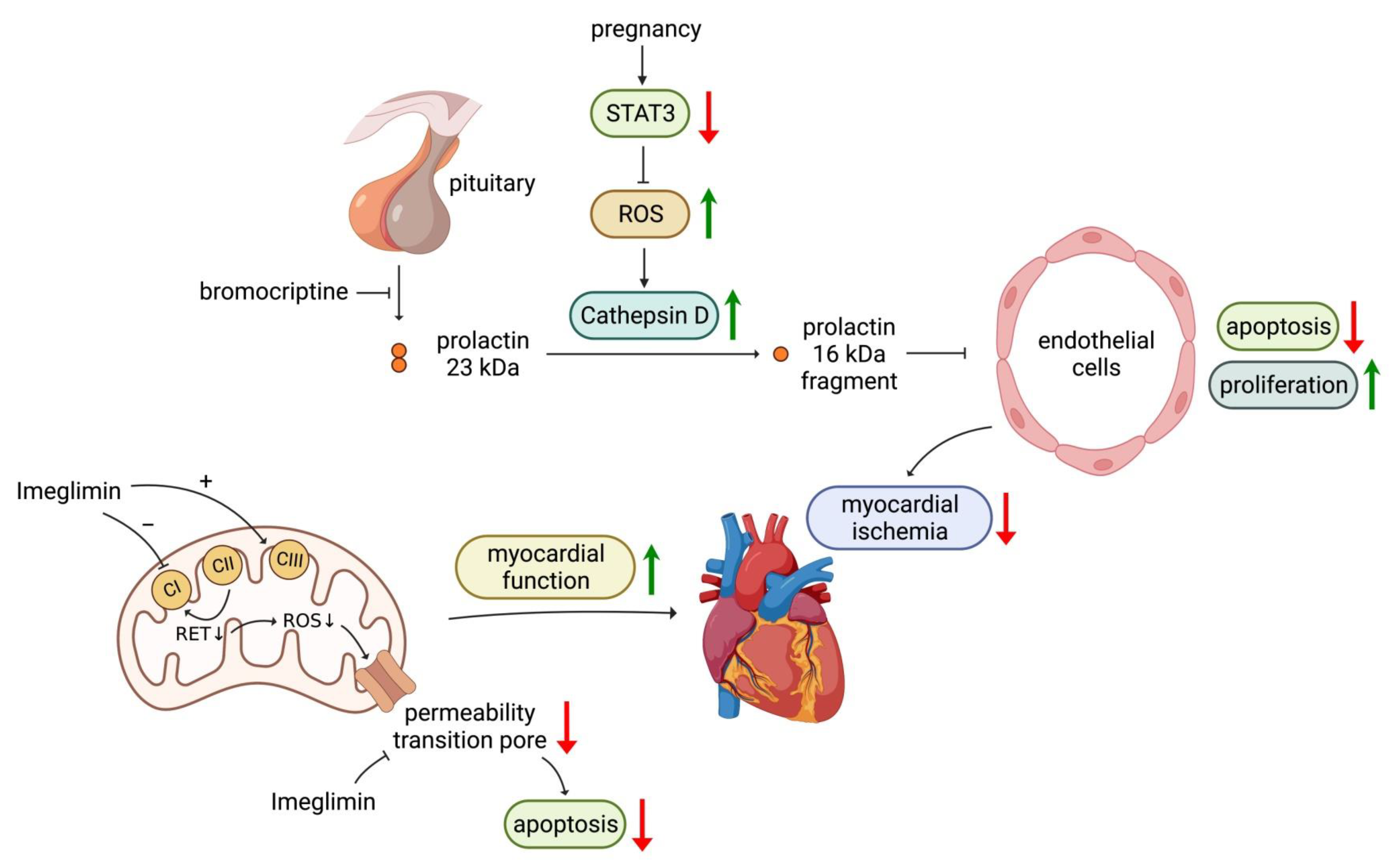
3.4. Bromocriptine
3.5. Imeglimin
3.6. Metformin
3.7. Thiazolidinediones (TZD)
3.8. Dipeptidyl Peptidase-4 (DPP-4) Inhibitors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of Diabetes in Congestive Heart Failure: The Framingham Study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gautham, R.K.; Singh, P.S.; Kumar, G.; Kant, P.; Sharma, H.; Pious, T.; Yadav, S.K. A study of diastolic dysfunction in patients of type-2 diabetes mellitus in rural population of western Uttar Pradesh. J. Evid. Based. Med. Healthc. 2017, 4, 1608–1611. [Google Scholar] [CrossRef] [PubMed]
- Thrainsdottir, I.S.; Aspelund, T.; Thorgeirsson, G.; Gudnason, V.; Hardarson, T.; Malmberg, K.; Sigurdsson, G.; Ryden, L. The Association between Glucose Abnormalities and Heart Failure in the Population-Based Reykjavik Study. Diabetes Care 2005, 28, 612–616. [Google Scholar] [CrossRef]
- Johansson, I.; Dahlström, U.; Edner, M.; Näsman, P.; Rydén, L.; Norhammar, A. Risk factors, treatment and prognosis in Men and Women with heart failure with and without diabetes. Heart 2015, 10, 1139–1148. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Januzzi, J.; Bruemmer, D.; Butalia, S.; Green, J.; Horton, W.; Knight, C.; Levi, M.; Rasouli, N.; Richardson, C.R. Heart Failure: An Underappreciated Complication of Diabetes. A Consensus Report of the American Diabetes Association. Diabetes Care 2022, 45, 1670–1690. [Google Scholar] [CrossRef]
- Wende, A.R.; Brahma, M.K.; McGinnis, G.R.; Young, M.E. Metabolic origins of heart failure. JACC Basic Transl. Sci. 2017, 2, 297–310. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Dei Cas, A.; Khan, S.S.; Butler, J.; Mentz, R.J.; Bonow, R.O.; Avogaro, A.; Fonarow, G.C. Impact of diabetes on epidemiology, treatment, and outcomes of patients with heart failure. JACC 2015, 3, 136–145. [Google Scholar] [CrossRef]
- Johansson, I.; Edner, M.; Dahlstrom, U.; Nasman, P.; Ryden, L.; Norhammar, A. Is the Prognosis in Patients with Diabetes and Heart Failure a Matter of Unsatisfactory Management? An Observational Study from the Swedish Heart Failure Registry. Eur. J. Heart Fail. 2014, 16, 409–418. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Fitchett, D.; Zinman, B.; Wanner, C.; Lachin, J.; Hantel, S.; Salsali, A.; Johansen, O.; Woerle, H.; Broedl, U.; Inzucchi, S.; et al. Heart Failure Outcomes with Empagliflozin in Patients with Type 2 Diabetes at High Cardiovascular Risk: Results of the Empa-Reg Outcome(R) Trial. Eur. Heart J. 2016, 37, 1526–1534. [Google Scholar] [CrossRef]
- Radholm, K.; Figtree, G.; Perkovic, V.; Solomon, S.D.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Barrett, T.D.; Shaw, W.; Desai, M.; et al. Canagliflozin and Heart Failure in Type 2 Diabetes Mellitus: Results from the Canvas Program. Circulation 2018, 138, 458–468. [Google Scholar] [CrossRef]
- Kato, E.T.; Silverman, M.G.; Mosenzon, O.; Zelniker, T.A.; Cahn, A.; Furtado, R.H.M.; Kuder, J.; Murphy, S.A.; Bhatt, D.L.; Leiter, L.A.; et al. Effect of Dapagliflozin on Heart Failure and Mortality in Type 2 Diabetes Mellitus. Circulation 2019, 139, 2528–2536. [Google Scholar] [CrossRef]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef]
- Cosentino, F.; Cannon, C.P.; Cherney, D.Z.I.; Masiukiewicz, U.; Pratley, R.; Dagogo-Jack, S.; Frederich, R.; Charbonnel, B.; Mancuso, J.; Shih, W.J.; et al. Efficacy of Ertugliflozin on Heart Failure-Related Events in Patients with Type 2 Diabetes Mellitus and Established Atherosclerotic Cardiovascular Disease: Results of the Vertis Cv Trial. Circulation 2020, 142, 2205–2215. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 Aha/Acc/Hfsa Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 79, 1757–1780. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 Esc Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in Patients with Diabetes and Chronic Kidney Disease. N. Engl. J. Med. 2021, 384, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Sarraju, A.; Li, J.; Cannon, C.P.; Chang, T.I.; Agarwal, R.; Bakris, G.; Charytan, D.M.; de Zeeuw, D.; Greene, T.; Heerspink, H.J.L.; et al. Mahaffey Effects of Canagliflozin on Cardiovascular, Renal, and Safety Outcomes in Participants with Type 2 Diabetes and Chronic Kidney Disease According to History of Heart Failure: Results from the Credence Trial. Am. Heart J. 2021, 233, 141–148. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Wheeler, D.C.; Stefansson, B.V.; Jongs, N.; Postmus, D.; Correa-Rotter, R.; Chertow, G.M.; Hou, F.F.; Rossing, P.; Sjostrom, C.D.; et al. Effects of dapagliflozin in patients with kidney disease, with and without heart failure. JACC Heart Fail. 2021, 9, 807–820. [Google Scholar] [CrossRef]
- The Empa-Kidney Collaborative Group; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Docherty, K.F.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Solomon, S.D.; Verma, S.; Belohlavek, J.; et al. Dapagliflozin and the Incidence of Type 2 Diabetes in Patients with Heart Failure and Reduced Ejection Fraction: An Exploratory Analysis from Dapa-Hf. Diabetes Care 2021, 44, 586–594. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Bohm, M.; Rocca, H.P.B.-L.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.; Claggett, B.; de Boer, R.; DeMets, D.; Hernandez, A.; Inzucchi, S.; Kosiborod, M.; Lam, C.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Steg, P.; Cannon, C.; Leiter, L.; McGuire, D.; Lewis, J.; Riddle, M.; Voors, A.; Metra, M.; et al. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar] [CrossRef]
- Voors, A.A.; Angermann, C.; Teerlink, J.; Collins, S.; Kosiborod, M.; Biegus, J.; Ferreira, J.; Nassif, M.; Psotka, M.; Tromp, J.; et al. The SGLT2 Inhibitor Empagliflozin in Patients Hospitalized for Acute Heart Failure: A Multinational Randomized Trial. Nat. Med. 2022, 28, 568–574. [Google Scholar] [CrossRef]
- Spertus, J.A.; Birmingham, M.; Nassif, M.; Damaraju, C.; Abbate, A.; Butler, J.; Lanfear, D.; Lingvay, I.; Kosiborod, M.; Januzzi, J.L. The Sglt2 Inhibitor Canagliflozin in Heart Failure: The Chief-Hf Remote, Patient-Centered Randomized Trial. Nat. Med. 2022, 28, 809–813. [Google Scholar] [CrossRef]
- Anker, S.D.; Usman, M.; Butler, J. SGLT2 Inhibitors: From Antihyperglycemic Agents to All-Around Heart Failure Therapy. Circulation 2022, 146, 299–302. [Google Scholar] [CrossRef]
- Hernandez, A.F.; Green, J.; Janmohamed, S.; D’Agostino, R.B.; Granger, C.; Jones, N.; Leiter, L.; Rosenberg, A.; Sigmon, K.; Somerville, M.; et al. Albiglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes and Cardiovascular Disease (Harmony Outcomes): A Double-Blind, Randomised Placebo-Controlled Trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Sharma, A.; Vasques-Novoa, F.; Angelico-Goncalves, A.; Leite, A.; Borges-Canha, M.; Carvalho, D.; Packer, M.; Zannad, F.; Leite-Moreira, A.; et al. Albiglutide in Patients with Type 2 Diabetes and Heart Failure: A Post-Hoc Analysis from Harmony Outcomes. Eur. J. Heart Fail. 2022, 24, 1792–1801. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Sattar, N.; Rosenstock, J.; Ramasundarahettige, C.; Pratley, R.; Lopes, R.; Lam, C.; Khurmi, N.; Heenan, L.; Del Prato, S.; et al. Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 896–907. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Ramasundarahettige, C.; Branch, K.; Sattar, N.; Rosenstock, J.; Pratley, R.; Del Prato, S.; Lopes, R.; Niemoeller, E.; Khurmi, N.; et al. Efpeglenatide and Clinical Outcomes with and without Concomitant Sodium-Glucose Cotransporter-2 Inhibition Use in Type 2 Diabetes: Exploratory Analysis of the Amplitude-O Trial. Circulation 2022, 145, 565–574. [Google Scholar] [CrossRef]
- Marso, S.P.; Baeres, F.; Bain, S.; Goldman, B.; Husain, M.; Nauck, M.; Poulter, N.; Pratley, R.; Thomsen, A.; Buse, J.; et al. Effects of Liraglutide on Cardiovascular Outcomes in Patients with Diabetes with or without Heart Failure. J. Am. Coll. Cardiol. 2020, 75, 1128–1141. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.; Nauck, M.; Nissen, S.; Pocock, S.; Poulter, N.; Ravn, L.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.R.; Bethel, M.; Mentz, R.; Thompson, V.; Lokhnygina, Y.; Buse, J.; Chan, J.; Choi, J.; Gustavson, S.; Iqbal, N.; et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Bain, S.; Consoli, A.; Eliaschewitz, F.; Jodar, E.; Leiter, L.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Birkenfeld, A.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.; Franco, D.; Jeppesen, O.; Lingvay, I.; Mosenzon, O.; Pedersen, S.; et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Claggett, B.; Diaz, R.; Dickstein, K.; Gerstein, H.; Kober, L.; Lawson, F.; Ping, L.; Wei, X.; Lewis, E.; et al. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. N. Engl. J. Med. 2015, 373, 2247–2257. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Colhoun, H.; Dagenais, G.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.; Riddle, M.; Ryden, L.; et al. Dulaglutide and Cardiovascular Outcomes in Type 2 Diabetes (Rewind): A Double-Blind, Randomised Placebo-Controlled Trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; Lee, M.; Kristensen, S.; Branch, K.; Del Prato, S.; Khurmi, N.; Lam, C.; Lopes, R.; McMurray, J.; Pratley, R.; et al. Cardiovascular, Mortality, and Kidney Outcomes with Glp-1 Receptor Agonists in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis of Randomised Trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef]
- Jorsal, A.; Kistorp, C.; Holmager, P.; Tougaard, R.; Nielsen, R.; Hanselmann, A.; Nilsson, B.; Moller, J.; Hjort, J.; Rasmussen, J.; et al. Effect of Liraglutide, a Glucagon-Like Peptide-1 Analogue, on Left Ventricular Function in Stable Chronic Heart Failure Patients with and without Diabetes (Live)-a Multicentre, Double-Blind, Randomised, Placebo-Controlled Trial. Eur. J. Heart Fail. 2017, 19, 69–77. [Google Scholar] [CrossRef]
- Margulies, K.B.; Hernandez, A.; Redfield, M.; Givertz, M.; Oliveira, G.; Cole, R.; Mann, D.; Whellan, D.; Kiernan, M.; Felker, G.; et al. Effects of Liraglutide on Clinical Stability among Patients with Advanced Heart Failure and Reduced Ejection Fraction: A Randomized Clinical Trial. JAMA 2016, 316, 500–508. [Google Scholar] [CrossRef]
- Lepore, J.J.; Olson, E.; Demopoulos, L.; Haws, T.; Fang, Z.; Barbour, A.; Fossler, M.; Davila-Roman, V.; Russell, S.; Gropler, R.J. Effects of the Novel Long-Acting Glp-1 Agonist, Albiglutide, on Cardiac Function, Cardiac Metabolism, and Exercise Capacity in Patients with Chronic Heart Failure and Reduced Ejection Fraction. JACC Heart Fail. 2016, 4, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Jastreboff, A.M.; Aronne, L.; Ahmad, N.; Wharton, S.; Connery, L.; Alves, B.; Kiyosue, A.; Zhang, S.; Liu, B.; Bunck, M.; et al. Tirzepatide Once Weekly for the Treatment of Obesity. N. Engl. J. Med. 2022, 387, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Frias, J.P.; Davies, M.; Rosenstock, J.; Manghi, F.P.; Lando, L.F.; Bergman, B.; Liu, B.; Cui, X.; Brown, K.; SURPASS-2 Investigators. Tirzepatide Versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; McGuire, D.; Pavo, I.; Weerakkody, G.; Nishiyama, H.; Wiese, R.; Zoungas, S. Tirzepatide Cardiovascular Event Risk Assessment: A Pre-Specified Meta-Analysis. Nat. Med. 2022, 28, 591–598. [Google Scholar] [CrossRef] [PubMed]
- A Study of Tirzepatide (Ly3298176) Compared with Dulaglutide on Major Cardiovascular Events in Participants with Type 2 Diabetes (Surpass-Cvot). Available online: https://clinicaltrials.gov/ct2/show/NCT04255433 (accessed on 1 March 2023).
- A Study of Tirzepatide (Ly3298176) in Participants with Heart Failure with Preserved Ejection Fraction and Obesity (Summit). Available online: https://clinicaltrials.gov/ct2/show/NCT04847557 (accessed on 1 March 2023).
- Aminian, A.; Zajichek, A.; Arterburn, D.; Wolski, K.; Brethauer, S.; Schauer, P.; Kattan, M.; Nissen, S.E. Association of Metabolic Surgery with Major Adverse Cardiovascular Outcomes in Patients with Type 2 Diabetes and Obesity. JAMA 2019, 322, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Hoskuldsdottir, G.; Sattar, N.; Miftaraj, M.; Naslund, I.; Ottosson, J.; Franzen, S.; Svensson, A.; Eliasson, B. Potential Effects of Bariatric Surgery on the Incidence of Heart Failure and Atrial Fibrillation in Patients with Type 2 Diabetes Mellitus and Obesity and on Mortality in Patients with Preexisting Heart Failure: A Nationwide, Matched, Observational Cohort Study. J. Am. Heart Assoc. 2021, 10, e019323. [Google Scholar]
- Aminian, A.; Wilson, R.; Zajichek, A.; Tu, C.; Wolski, K.; Schauer, P.; Kattan, M.; Nissen, S.; Brethauer, S.A. Cardiovascular Outcomes in Patients with Type 2 Diabetes and Obesity: Comparison of Gastric Bypass, Sleeve Gastrectomy, and Usual Care. Diabetes Care 2021, 44, 2552–2563. [Google Scholar] [CrossRef]
- Chamarthi, B.; Ezrokhi, M.; Rutty, D.; Cincotta, A.H. Impact of Bromocriptine-Qr Therapy on Cardiovascular Outcomes in Type 2 Diabetes Mellitus Subjects on Metformin. Postgrad. Med. 2016, 128, 761–769. [Google Scholar] [CrossRef]
- Chamarthi, B.; Gaziano, J.; Blonde, L.; Vinik, A.; Scranton, R.; Ezrokhi, M.; Rutty, D.; Cincotta, A.H. Timed Bromocriptine-Qr Therapy Reduces Progression of Cardiovascular Disease and Dysglycemia in Subjects with Well-Controlled Type 2 Diabetes Mellitus. J. Diabetes Res. 2015, 2015, 157698. [Google Scholar] [CrossRef]
- Chamarthi, B.; Cincotta, A.H. Effect of Bromocriptine-Qr Therapy on Glycemic Control in Subjects with Type 2 Diabetes Mellitus Whose Dysglycemia Is Inadequately Controlled on Insulin. Postgrad. Med. 2017, 129, 446–455. [Google Scholar] [CrossRef]
- Gaziano, J.M.; Cincotta, A.; Vinik, A.; Blonde, L.; Bohannon, N.; Scranton, R. Effect of Bromocriptine-Qr (a Quick-Release Formulation of Bromocriptine Mesylate) on Major Adverse Cardiovascular Events in Type 2 Diabetes Subjects. J. Am. Heart Assoc. 2012, 1, e002279. [Google Scholar] [CrossRef]
- Trongtorsak, A.; Kittipibul, V.; Mahabir, S.; Ibrahim, M.; Croix, G.S.; Hernandez, G.; Chaparro, S. Effects of Bromocriptine in Peripartum Cardiomyopathy: A Systematic Review and Meta-Analysis. Heart Fail. Rev. 2022, 27, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Badianyama, M.; Das, P.; Gaddameedi, S.; Saukhla, S.; Nagammagari, T.; Bandari, V.; Mohammed, L. A Systematic Review of the Utility of Bromocriptine in Acute Peripartum Cardiomyopathy. Cureus 2021, 13, e18248. [Google Scholar] [PubMed]
- Konkwo, C.; Perry, R.J. Imeglimin: Current Development and Future Potential in Type 2 Diabetes. Drugs 2021, 81, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Kitakata, H.; Endo, J.; Hashimoto, S.; Mizuno, E.; Moriyama, H.; Shirakawa, K.; Goto, S.; Katsumata, Y.; Fukuda, K.; Sano, M. Imeglimin Prevents Heart Failure with Preserved Ejection Fraction by Recovering the Impaired Unfolded Protein Response in Mice Subjected to Cardiometabolic Stress. Biochem. Biophys. Res. Commun. 2021, 572, 185–190. [Google Scholar] [CrossRef]
- Lachaux, M.; Soulie, M.; Hamzaoui, M.; Bailly, A.; Nicol, L.; Remy-Jouet, I.; Renet, S.; Vendeville, C.; Gluais-Dagorn, P.; Hallakou-Bozec, S.; et al. Short-and Long-Term Administration of Imeglimin Counters Cardiorenal Dysfunction in a Rat Model of Metabolic Syndrome. Endocrinol. Diabetes Metab. 2020, 3, e00128. [Google Scholar] [CrossRef]
- Detaille, D.; Vial, G.; Borel, A.; Cottet-Rouselle, C.; Hallakou-Bozec, S.; Bolze, S.; Fouqueray, P.; Fontaine, E. Imeglimin Prevents Human Endothelial Cell Death by Inhibiting Mitochondrial Permeability Transition without Inhibiting Mitochondrial Respiration. Cell Death Discov. 2016, 2, 15072. [Google Scholar] [CrossRef]
- Crowley, M.J.; Diamantidis, C.; McDuffie, J.; Cameron, C.; Stanifer, J.; Mock, C.; Wang, X.; Tang, S.; Nagi, A.; Kosinski, A.; et al. Clinical Outcomes of Metformin Use in Populations with Chronic Kidney Disease, Congestive Heart Failure, or Chronic Liver Disease: A Systematic Review. Ann. Intern. Med. 2017, 166, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, H.; Wu, C.; Zheng, Y.; Jiang, J. Effect of Metformin on Adverse Outcomes in T2dm Patients: Systemic Review and Meta-Analysis of Observational Studies. Front. Cardiovasc. Med. 2022, 9, 944902. [Google Scholar] [CrossRef]
- Benes, J.; Kotrc, M.; Kroupova, K.; Wohlfahrt, P.; Kovar, J.; Franekova, J.; Hegarova, M.; Hoskova, L.; Hoskova, E.; Pelikanova, T.; et al. Metformin Treatment Is Associated with Improved Outcome in Patients with Diabetes and Advanced Heart Failure (Hfref). Sci. Rep. 2022, 12, 13038. [Google Scholar] [CrossRef]
- Monami, M.; Candido, R.; Pintaudi, B.; Targher, G.; Mannucci, E.; Type, S.-A.P.I.G.T. Effect of Metformin on All-Cause Mortality and Major Adverse Cardiovascular Events: An Updated Meta-Analysis of Randomized Controlled Trials. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 699–704. [Google Scholar] [CrossRef]
- Griffin, S.J.; Leaver, J.; Irving, G.J. Impact of Metformin on Cardiovascular Disease: A Meta-Analysis of Randomised Trials among People with Type 2 Diabetes. Diabetologia 2017, 60, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Bergmark, B.A.; Bhatt, D.; McGuire, D.; Scirica, B.M. Response by Bergmark Et Al to Letter Regarding Article, Metformin Use and Clinical Outcomes among Patients with Diabetes Mellitus with or without Heart Failure or Kidney Dysfunction: Observations from the Savor-Timi 53 Trial. Circulation 2020, 141, e57–e58. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.H.; Jessen, N.; Norrelund, H.; Tolbod, L.; Harms, H.; Feddersen, S.; Nielsen, F.; Brosen, K.; Hansson, N.; Frokiaer, J.; et al. A Randomised, Double-Blind, Placebo-Controlled Trial of Metformin on Myocardial Efficiency in Insulin-Resistant Chronic Heart Failure Patients without Diabetes. Eur. J. Heart Fail. 2020, 22, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Wiggers, H.; Kober, L.; Gislason, G.; Schou, M.; Poulsen, M.; Vraa, S.; Nielsen, O.; Bruun, N.; Norrelund, H.; Hollingdal, M.; et al. The Danish Randomized, Double-Blind, Placebo Controlled Trial in Patients with Chronic Heart Failure (Danheart): A 2 X 2 Factorial Trial of Hydralazine-Isosorbide Dinitrate in Patients with Chronic Heart Failure (H-Heft) and Metformin in Patients with Chronic Heart Failure and Diabetes or Prediabetes (Met-Heft). Am. Heart J. 2021, 231, 137–146. [Google Scholar]
- Investigation of Metformin in Pre-Diabetes on Atherosclerotic Cardiovascular Outcomes (Va-Impact). Available online: https://clinicaltrials.gov/ct2/show/NCT02915198 (accessed on 1 March 2023).
- Origin Trial Investigators; Gerstein, H.; Bosch, J.; Dagenais, G.; Diaz, R.; Jung, H.; Maggioni, A.; Pogue, J.; Probstfield, J.; Ramachandran, A.; et al. Basal Insulin and Cardiovascular and Other Outcomes in Dysglycemia. N. Engl. J. Med. 2012, 367, 319–328. [Google Scholar]
- The BARI 2D Study Group; Frye, R.; August, P.; Brooks, M.; Hardison, R.; Kelsey, S.; MacGregor, J.; Orchard, T.; Chaitman, B.; Genuth, S.; et al. A Randomized Trial of Therapies for Type 2 Diabetes and Coronary Artery Disease. N. Engl. J. Med. 2009, 360, 2503–2515. [Google Scholar]
- Shen, L.; Rorth, R.; Cosmi, D.; Kristensen, S.; Petrie, M.; Cosmi, F.; Latini, R.; Kober, L.; Anand, I.; Carson, P.; et al. Insulin Treatment and Clinical Outcomes in Patients with Diabetes and Heart Failure with Preserved Ejection Fraction. Eur. J. Heart Fail. 2019, 21, 974–984. [Google Scholar] [CrossRef]
- Cosmi, F.; Shen, L.; Magnoli, M.; Abraham, W.; Anand, I.; Cleland, J.; Cohn, J.; Cosmi, D.; De Berardis, G.; Dickstein, K.; et al. Treatment with Insulin Is Associated with Worse Outcome in Patients with Chronic Heart Failure and Diabetes. Eur. J. Heart Fail. 2018, 20, 888–895. [Google Scholar] [CrossRef]
- Riehle, C.; Abel, E.D. Insulin Signaling and Heart Failure. Circ. Res. 2016, 118, 1151–1169. [Google Scholar] [CrossRef]
- Uk Prospective Diabetes Study (Ukpds) Group. Intensive Blood-Glucose Control with Sulphonylureas or Insulin Compared with Conventional Treatment and Risk of Complications in Patients with Type 2 Diabetes (Ukpds 33). Lancet 1998, 352, 837–853. [Google Scholar]
- Hougen, I.; Whitlock, R.; Komenda, P.; Rigatto, C.; Clemens, K.; Tangri, N. Safety of Add-on Sulfonylurea Therapy in Patients with Type 2 Diabetes Using Metformin: A Population-Based Real-World Study. BMJ Open Diabetes Res. Care 2021, 9, e002352. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Solomon, N.; DeVore, A.; Sharma, A.; Felker, G.; Hernandez, A.; Heidenreich, P.; Matsouaka, R.; Green, J.; Butler, J.; et al. Clinical Outcomes with Metformin and Sulfonylurea Therapies among Patients with Heart Failure and Diabetes. JACC Heart Fail. 2022, 10, 198–210. [Google Scholar] [CrossRef]
- Richardson, T.L., Jr.; Hackstadt, A.; Hung, A.; Greevy, R.; Grijalva, C.; Griffin, M.; Elasy, T.; Roumie, C.L. Hospitalization for Heart Failure among Patients with Diabetes Mellitus and Reduced Kidney Function Treated with Metformin Versus Sulfonylureas: A Retrospective Cohort Study. J. Am. Heart Assoc. 2021, 10, e019211. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, M.; Cagatay, M.; Petrowitsch, T.; Neuser, D.; Petzinna, D.; Rupp, M. Acarbose Reduces the Risk for Myocardial Infarction in Type 2 Diabetic Patients: Meta-Analysis of Seven Long-Term Studies. Eur. Heart J. 2004, 25, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.R.; Coleman, R.; Chan, J.; Chiasson, J.; Feng, H.; Ge, J.; Gerstein, H.; Gray, R.; Huo, Y.; Lang, Z.; et al. Effects of Acarbose on Cardiovascular and Diabetes Outcomes in Patients with Coronary Heart Disease and Impaired Glucose Tolerance (Ace): A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Diabetes Endocrinol. 2017, 5, 877–886. [Google Scholar] [CrossRef]
- Young, L.H.; Viscoli, C.; Schwartz, G.; Inzucchi, S.; Curtis, J.; Gorman, M.; Furie, K.; Conwit, R.; Spatz, E.; Lovejoy, A.; et al. Heart Failure after Ischemic Stroke or Transient Ischemic Attack in Insulin-Resistant Patients without Diabetes Mellitus Treated with Pioglitazone. Circulation 2018, 138, 1210–1220. [Google Scholar] [CrossRef]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.; Skene, A.; Tan, M.; Lefebvre, P.; Murray, G.; et al. Secondary Prevention of Macrovascular Events in Patients with Type 2 Diabetes in the Proactive Study (Prospective Pioglitazone Clinical Trial in Macrovascular Events): A Randomised Controlled Trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Nissen, S.E.; Nicholls, S.; Wolski, K.; Nesto, R.; Kupfer, S.; Perez, A.; Jure, H.; De Larochelliere, R.; Staniloae, C.; Mavromatis, K.; et al. Comparison of Pioglitazone Vs Glimepiride on Progression of Coronary Atherosclerosis in Patients with Type 2 Diabetes: The Periscope Randomized Controlled Trial. JAMA 2008, 299, 1561–1573. [Google Scholar] [CrossRef]
- Guan, Y.; Hao, C.; Cha, D.; Rao, R.; Lu, W.; Kohan, D.; Magnuson, M.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones Expand Body Fluid Volume through Ppargamma Stimulation of Enac-Mediated Renal Salt Absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef]
- Dream Trial Investigators; Dagenais, G.; Gerstein, H.; Holman, R.; Budaj, A.; Escalante, A.; Hedner, T.; Keltai, M.; Lonn, E.; McFarlane, S.; et al. Effects of Ramipril and Rosiglitazone on Cardiovascular and Renal Outcomes in People with Impaired Glucose Tolerance or Impaired Fasting Glucose: Results of the Diabetes Reduction Assessment with Ramipril and Rosiglitazone Medication (Dream) Trial. Diabetes Care 2008, 31, 1007–1014. [Google Scholar]
- Dargie, H.J.; Hildebrandt, P.; Riegger, G.; McMurray, J.; McMorn, S.; Roberts, J.; Zambanini, A.; Wilding, J.P. A Randomized, Placebo-Controlled Trial Assessing the Effects of Rosiglitazone on Echocardiographic Function and Cardiac Status in Type 2 Diabetic Patients with New York Heart Association Functional Class I or Ii Heart Failure. J. Am. Coll. Cardiol. 2007, 49, 1696–1704. [Google Scholar] [CrossRef]
- Lago, R.M.; Singh, P.; Nesto, R.W. Congestive Heart Failure and Cardiovascular Death in Patients with Prediabetes and Type 2 Diabetes Given Thiazolidinediones: A Meta-Analysis of Randomised Clinical Trials. Lancet 2007, 370, 1129–1136. [Google Scholar] [CrossRef]
- Nesto, R.W.; Bell, D.; Bonow, R.; Fonseca, V.; Grundy, S.; Horton, E.; Le Winter, M.; Porte, D.; Semenkovich, C.; Smith, S.; et al. Thiazolidinedione Use, Fluid Retention, and Congestive Heart Failure: A Consensus Statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004, 27, 256–263. [Google Scholar] [CrossRef]
- Scirica, B.M.; Braunwald, E.; Raz, I.; Cavender, M.; Morrow, D.; Jarolim, P.; Udell, J.; Mosenzon, O.; Im, K.; Umez-Eronini, A.; et al. Heart Failure, Saxagliptin, and Diabetes Mellitus: Observations from the Savor-Timi 53 Randomized Trial. Circulation 2015, 132, e198. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Cannon, C.; Cushman, W.; Bakris, G.; Menon, V.; Perez, A.; Fleck, P.; Mehta, C.; Kupfer, S.; Wilson, C.; et al. Heart Failure and Mortality Outcomes in Patients with Type 2 Diabetes Taking Alogliptin Versus Placebo in Examine: A Multicentre, Randomised, Double-Blind Trial. Lancet 2015, 385, 2067–2076. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Ponikowski, P.; Bolli, G.; Lukashevich, V.; Kozlovski, P.; Kothny, W.; Lewsey, J.; Krum, H.; VIVIDD Trial Committees and Investigators. Effects of Vildagliptin on Ventricular Function in Patients with Type 2 Diabetes Mellitus and Heart Failure: A Randomized Placebo-Controlled Trial. JACC Heart Fail. 2018, 6, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Lovshin, J.A.; Rajasekeran, H.; Lytvyn, Y.; Lovblom, L.; Khan, S.; Alemu, R.; Locke, A.; Lai, V.; He, H.; Hittle, L.; et al. Dipeptidyl Peptidase 4 Inhibition Stimulates Distal Tubular Natriuresis and Increases in Circulating Sdf-1alpha(1-67) in Patients with Type 2 Diabetes. Diabetes Care 2017, 40, 1073–1081. [Google Scholar] [CrossRef]
- Aoyama, M.; Kawase, H.; Bando, Y.; Monji, A.; Murohara, T. Dipeptidyl Peptidase 4 Inhibition Alleviates Shortage of Circulating Glucagon-Like Peptide-1 in Heart Failure and Mitigates Myocardial Remodeling and Apoptosis Via the Exchange Protein Directly Activated by Cyclic Amp 1/Ras-Related Protein 1 Axis. Circ. Heart Fail. 2016, 9, e002081. [Google Scholar] [CrossRef]
- Dehlin, H.M.; Manteufel, E.; Monroe, A.; Reimer, M., Jr.; Levick, S.P. Substance P Acting Via the Neurokinin-1 Receptor Regulates Adverse Myocardial Remodeling in a Rat Model of Hypertension. Int. J. Cardiol. 2013, 168, 4643–4651. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Gillespie, D.; Jackson, E.K. Npy1-36 and Pyy1-36 Activate Cardiac Fibroblasts: An Effect Enhanced by Genetic Hypertension and Inhibition of Dipeptidyl Peptidase 4. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1528–H1542. [Google Scholar] [CrossRef]
- Devin, J.K.; Pretorius, M.; Nian, H.; Yu, C.; Billings, F.; Brown, N.J. Substance P Increases Sympathetic Activity During Combined Angiotensin-Converting Enzyme and Dipeptidyl Peptidase-4 Inhibition. Hypertension 2014, 63, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.G.; Zhang, Z.; Yu, Y.; Felder, R.B. Central Sdf-1/Cxcl12 Expression and Its Cardiovascular and Sympathetic Effects: The Role of Angiotensin Ii, Tnf-Alpha, and Map Kinase Signaling. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1643–H1654. [Google Scholar] [CrossRef]
- Jarrah, A.A.; Schwarskopf, M.; Wang, E.; LaRocca, T.; Dhume, A.; Zhang, S.; Hadri, L.; Hajjar, R.; Schecter, A.; Tarzami, S.T. Sdf-1 Induces Tnf-Mediated Apoptosis in Cardiac Myocytes. Apoptosis 2018, 23, 79–91. [Google Scholar] [CrossRef]
- Green, J.B.; Bethel, M.; Armstrong, P.; Buse, J.; Engel, S.; Garg, J.; Josse, R.; Kaufman, K.; Koglin, J.; Korn, S.; et al. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 232–242. [Google Scholar] [CrossRef]
- Rosenstock, J.; Perkovic, V.; Johansen, O.; Cooper, M.; Kahn, S.; Marx, N.; Alexander, J.; Pencina, M.; Toto, R.; Wanner, C.; et al. Effect of Linagliptin Vs Placebo on Major Cardiovascular Events in Adults with Type 2 Diabetes and High Cardiovascular and Renal Risk: The Carmelina Randomized Clinical Trial. JAMA 2019, 321, 69–79. [Google Scholar] [CrossRef]
- Rosenstock, J.; Kahn, S.; Johansen, O.; Zinman, B.; Espeland, M.; Woerle, H.; Pfarr, E.; Keller, A.; Mattheus, M.; Baanstra, D.; et al. Effect of Linagliptin Vs Glimepiride on Major Adverse Cardiovascular Outcomes in Patients with Type 2 Diabetes: The Carolina Randomized Clinical Trial. JAMA 2019, 322, 1155–1166. [Google Scholar] [CrossRef]
- Herrmann, K.; Zhou, M.; Wang, A.; de Bruin, T.W.A. Cardiovascular Safety Assessment of Pramlintide in Type 2 Diabetes: Results from a Pooled Analysis of Five Clinical Trials. Clin. Diabetes Endocrinol. 2016, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Gregg, E.W.; Chen, H.; Wagenknecht, L.; Clark, J.; Delahanty, L.; Bantle, J.; Pownall, H.; Johnson, K.; Safford, M.; Kitabchi, A.; et al. Association of an Intensive Lifestyle Intervention with Remission of Type 2 Diabetes. JAMA 2012, 308, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.G.; Savarese, G.; Haynes, R.; Marx, N.; Mellbin, L.; Lund, L.H.; Dendale, P.; Seferovic, P.; Rosano, G.; Staplin, N.; et al. Cardiac, renal, and metabolic effects of sodium-glucose co-transporter 2 inhibitors: A position paper from the European Society of Cardiology ad-hoc task force on sodium-glucose co-transporter 2 inhibitors. Eur. J. Heart Fail. 2021, 23, 1260–1275. [Google Scholar] [CrossRef]
- Cinti, F.; Moffa, S.; Impronta, F.; Cefalo, C.M.; Sun, V.A.; Sorice, G.P.; Giaccari, A. Spotlight on ertugliflozin and its potential in the treatment of type 2 diabetes: Evidence to date. Drug Des. Dev. Ther. 2017, 11, 2905–2919. [Google Scholar] [CrossRef]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol. Ren. Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef]
- Cefalo, C.M.A.; Cinti, F.; Moffa, S.; Impronta, F.; Sorice, G.P.; Mezza, T.; Pontecorvi, A.; Giaccari, A. Sotagliflozin, the first dual SGLT inhibitor: Current outlook and perspectives. Cardiovasc. Diabetol. 2019, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, R.L.; Greenway, F.L.; Chen, L.; Liu, Y.; Breed, S.L.; Andrews, S.M.; Wald, J.A.; Walker, A.C.; Smith, C.D. Selective sodium-dependent glucose transporter 1 inhibitors block glucose absorption and impair glucose-dependent insulinotropic peptide release. Am. J. Physiol. Gastrointest. Liver. Physiol. 2015, 308, G946–G954. [Google Scholar] [CrossRef]
- Zambrowicz, B.; Freiman, J.; Brown, P.M.; Frazier, K.S.; Turnage, A.; Bronner, J.; Ruff, D.; Shadoan, M.; Banks, P.; Mseeh, F.; et al. LX4211, a dual SGLT1/SGLT2 inhibitor, improved glycemic control in patients with type 2 diabetes in a randomized, placebo-controlled trial. Clin. Pharmacol. Ther. 2012, 92, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Täger, T.; Frankenstein, L.; Atar, D.; Agewall, S.; Frey, N.; Grundtvig, M.; Clark, A.L.; Cleland, J.G.F.; Fröhlich HFröhlich, H. Influence of receptor selectivity on benefits from SGLT2 inhibitors in patients with heart failure: A systematic review and head-to-head comparative efficacy network meta-analysis. Clin. Res. Cardiol. 2022, 111, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Cryan, E.V.; D’Andrea, M.R.; Belkowski, S.; Conway, B.R.; Demarest, K.T. Human cardiomyocytes express high level of Na+/glucose cotransporter 1 (SGLT1). J. Cell. Biochem. 2003, 90, 339–346. [Google Scholar] [CrossRef]
- von Lewinski, D.; Gasser, R.; Rainer, P.P.; Huber, M.S.; Wilhelm, B.; Roessl, U.; Haas, T.; Wasler, A.; Grimm, M.; Bisping, E.; et al. Functional effects of glucose transporters in human ventricular myocardium. Eur. J. Heart Fail. 2010, 12, 106–113. [Google Scholar] [CrossRef]
- Sayour, A.A.; Oláh, A.; Ruppert, M.; Barta, B.A.; Horváth, E.M.; Benke, K.; Pólos, M.; Hartyánszky, I.; Merkely, B.; Radovits, T. Characterization of left ventricular myocardial sodium-glucose cotransporter 1 expression in patients with end-stage heart failure. Cardiovasc. Diabetol. 2020, 19, 159. [Google Scholar] [CrossRef]
- Li, Z.; Agrawal, V.; Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Sincoular, A.; Jakubiak, M.; Music, M.L.; Kutschke, W.J.; Huang, X.N.; et al. Cardiac sodium-dependent glucose cotransporter 1 is a novel mediator of ischaemia/reperfusion injury. Cardiovasc. Res. 2019, 115, 1646–1658. [Google Scholar] [CrossRef]
- Kondo, H.; Akoumianakis, I.; Badi, I.; Akawi, N.; Kotanidis, C.P.; Polkinghorne, M.; Stadiotti, I.; Sommariva, E.; Antonopoulos, A.S.; Carena, M.C.; et al. Effects of canagliflozin on human myocardial redox signaling: Clinical implications. Eur. Heart J. 2021, 42, 4947–4960. [Google Scholar] [CrossRef]
- Lin, H.; Guan, L.; Meng, L.; Uzui, H.; Guo, H. SGLT1 knockdown attenuates cardiac fibroblast activation in diabetic cardiac fibrosis. Front. Pharmacol. 2021, 12, 700366. [Google Scholar] [CrossRef]
- Seidelmann, S.B.; Feofanova, E.; Yu, B.; Franceschini, N.; Claggett, B.; Kuokkanen, M.; Puolijoki, H.; Ebeling, T.; Perola, M.; Salomaa, V.; et al. Genetic variants in SGLT1, glucose tolerance, and cardiometabolic risk. J. Am. Coll. Cardiol. 2018, 72, 1763–1773. [Google Scholar] [CrossRef]
- Lin, N.; Lin, H.; Yang, Q.; Lu, W.; Sun, Z.; Sun, S.; Meng, L.; Chi, J.; Guo, H. SGLT1 inhibition attenuates apoptosis in diabetic cardiomyopathy via the JNK and p38 pathway. Front. Pharmacol. 2020, 11, 598353. [Google Scholar] [CrossRef] [PubMed]
- Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Lee, S.J.; Wang, D.; Huang, X.Y.; Ahmad, F. Transgenic knockdown of cardiac sodium/glucose cotransporter 1 (SGLT1) attenuates PRKAG2 cardiomyopathy, whereas transgenic overexpression of cardiac SGLT1 causes pathologic hypertrophy and dysfunction in mice. J. Am. Heart Assoc. 2014, 3, e000899. [Google Scholar] [CrossRef]
- Connelly, K.A.; Zhang, Y.; Desjardins, J.-F.; Thai, K.; Gilbert, R.E. Dual inhibition of sodium-glucose linked cotransporters 1 and 2 exacerbates cardiac dysfunction following experimental myocardial infarction. Cardiovasc. Diabetol. 2018, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Sawa, Y.; Saito, M.; Ishida, N.; Ibi, M.; Matsushita, N.; Morino, Y.; Taira, E.; Hirose, M. Pretreatment with KGA-2727, a selective SGLT1 inhibitor, is protective against myocardial infarction-induced ventricular remodeling and heart failure in mice. J. Pharmacol. Sci. 2020, 142, 16–25. [Google Scholar] [CrossRef]
- Powell, D.R.; Smith, M.G.; Doree, D.D.; Harris, A.L.; Greer, J.; Dacosta, C.M.; Thompson, A.; Jeter-Jones, S.; Xiong, W.; Carson, K.G.; et al. LX2761, a Sodium/Glucose Cotransporter 1 Inhibitor Restricted to the Intestine, Improves Glycemic Control in Mice. J. Pharmacol. Exp. Ther. 2017, 362, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Kelly, D.; Margulies, K.B. Implications of Altered Ketone Metabolism and Therapeutic Ketosis in Heart Failure. Circulation 2020, 141, 1800–1812. [Google Scholar] [CrossRef]
- Schugar, R.C.; Moll, A.; Andre d’Avignon, D.; Weinheimer, C.; Kovacs, A.; Crawford, P.A. Cardiomyocyte-Specific Deficiency of Ketone Body Metabolism Promotes Accelerated Pathological Remodeling. Mol. Metab. 2014, 3, 754–769. [Google Scholar] [CrossRef]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the Empa-Reg Outcome Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef]
- Yuan, T.; Rafizadeh, S.; Gorrepati, K.; Lupse, B.; Oberholzer, J.; Maedler, K.; Ardestani, A. Reciprocal Regulation of Mtor Complexes in Pancreatic Islets from Humans with Type 2 Diabetes. Diabetologia 2017, 60, 668–678. [Google Scholar] [CrossRef]
- Jiang, P.; Ren, L.; Zhi, L.; Yu, Z.; Lv, F.; Xu, F.; Peng, W.; Bai, X.; Cheng, K.; Quan, L.; et al. Negative Regulation of Ampk Signaling by High Glucose Via E3 Ubiquitin Ligase Mg53. Mol. Cell 2021, 81, 629–637.e5. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. Ampk: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Paramesha, B.; Anwar, M.; Meghwani, H.; Maulik, S.; Arava, S.; Banerjee, S.K. Sirt1 and Sirt3 Activation Improved Cardiac Function of Diabetic Rats Via Modulation of Mitochondrial Function. Antioxidants 2021, 10, 338. [Google Scholar] [CrossRef]
- Oshima, H.; Miki, T.; Kuno, A.; Mizuno, M.; Sato, T.; Tanno, M.; Yano, T.; Nakata, K.; Kimura, Y.; Abe, K.; et al. Empagliflozin, an Sglt2 Inhibitor, Reduced the Mortality Rate after Acute Myocardial Infarction with Modification of Cardiac Metabolomes and Antioxidants in Diabetic Rats. J. Pharmacol. Exp. Ther. 2019, 368, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a Sodium Glucose Co-Transporter-2 Inhibitor, Alleviates Atrial Remodeling and Improves Mitochondrial Function in High-Fat Diet/Streptozotocin-Induced Diabetic Rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin Rescues Diabetic Myocardial Microvascular Injury Via Ampk-Mediated Inhibition of Mitochondrial Fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef]
- Aragon-Herrera, A.; Feijoo-Bandin, S.; Santiago, M.O.; Barral, L.; Campos-Toimil, M.; Gil-Longo, J.; Pereira, T.C.; Garcia-Caballero, T.; Rodriguez-Segade, S.; Rodriguez, J.; et al. Empagliflozin Reduces the Levels of Cd36 and Cardiotoxic Lipids While Improving Autophagy in the Hearts of Zucker Diabetic Fatty Rats. Biochem. Pharmacol. 2019, 170, 113677. [Google Scholar] [CrossRef]
- Li, X.; Flynn, E.; do Carmo, J.M.; Wang, Z.; da Silva, A.; Mouton, A.; Omoto, A.; Hall, M.; Hall, J.E. Direct Cardiac Actions of Sodium-Glucose Cotransporter 2 Inhibition Improve Mitochondrial Function and Attenuate Oxidative Stress in Pressure Overload-Induced Heart Failure. Front. Cardiovasc. Med. 2022, 9, 859253. [Google Scholar] [CrossRef]
- Packer, M. Mechanisms Leading to Differential Hypoxia-Inducible Factor Signaling in the Diabetic Kidney: Modulation by Sglt2 Inhibitors and Hypoxia Mimetics. Am. J. Kidney Dis. 2021, 77, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, T.; Xian, J.; Chen, J.; Huang, Y.; Zhang, Q.; Lin, X.; Lu, H.; Lin, Y. Sglt2 Inhibitor Dapagliflozin Attenuates Cardiac Fibrosis and Inflammation by Reverting the Hif-2alpha Signaling Pathway in Arrhythmogenic Cardiomyopathy. FASEB J. 2022, 36, e22410. [Google Scholar] [CrossRef] [PubMed]
- Curthoys, N.P.; Moe, O.W. Proximal Tubule Function and Response to Acidosis. Clin. J. Am. Soc. Nephrol. 2014, 9, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Bjornstad, P.; Nelson, R.; Pavkov, M.E. Do Sodium-Glucose Cotransporter-2 Inhibitors Affect Renal Hemodynamics by Different Mechanisms in Type 1 and Type 2 Diabetes? Kidney Int. 2020, 97, 31–33. [Google Scholar] [CrossRef] [PubMed]
- van Raalte, D.H.; Bjornstad, P. Role of Sodium-Glucose Cotransporter 2 Inhibition to Mitigate Diabetic Kidney Disease Risk in Type 1 Diabetes. Nephrol. Dial. Transp. 2020, 35 (Suppl. S1), i24–i32. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, T.D.; Campos, L.; Carraro-Lacroix, L.; Girardi, A.; Malnic, G. Functional Role of Glucose Metabolism, Osmotic Stress, and Sodium-Glucose Cotransporter Isoform-Mediated Transport on Na+/H+ Exchanger Isoform 3 Activity in the Renal Proximal Tubule. J. Am. Soc. Nephrol. 2014, 25, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.D.; Polidoro, J.; Borges-Junior, F.; Girardi, A.C.C. Cardioprotection Conferred by Sodium-Glucose Cotransporter 2 Inhibitors: A Renal Proximal Tubule Perspective. Am. J. Physiol. Cell Physiol. 2020, 318, C328–C336. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Kim, S.; Son, M.; Kim, M.; Koh, E.; Shin, S.; Ko, S.; Kim, H.S. Empagliflozin Contributes to Polyuria Via Regulation of Sodium Transporters and Water Channels in Diabetic Rat Kidneys. Front. Physiol. 2019, 10, 271. [Google Scholar] [CrossRef]
- Lambers Heerspink, H.J.; de Zeeuw, D.; Wie, L.; Leslie, B.; List, J. Dapagliflozin a Glucose-Regulating Drug with Diuretic Properties in Subjects with Type 2 Diabetes. Diabetes Obes. Metab. 2013, 15, 853–862. [Google Scholar] [CrossRef]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.; McMurray, J.; Boulton, D.W. Why Do Sglt2 Inhibitors Reduce Heart Failure Hospitalization? A Differential Volume Regulation Hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487. [Google Scholar] [CrossRef]
- Maruyama, T.; Takashima, H.; Oguma, H.; Nakamura, Y.; Ohno, M.; Utsunomiya, K.; Furukawa, T.; Tei, R.; Abe, M. Canagliflozin Improves Erythropoiesis in Diabetes Patients with Anemia of Chronic Kidney Disease. Diabetes Technol. Ther. 2019, 21, 713–720. [Google Scholar] [CrossRef]
- Jordan, J.; Tank, J.; Heusser, K.; Heise, T.; Wanner, C.; Heer, M.; Macha, S.; Mattheus, M.; Lund, S.; Woerle, H.; et al. The Effect of Empagliflozin on Muscle Sympathetic Nerve Activity in Patients with Type Ii Diabetes Mellitus. J. Am. Soc. Hypertens. 2017, 11, 604–612. [Google Scholar] [CrossRef]
- Reed, J.W. Impact of Sodium-Glucose Cotransporter 2 Inhibitors on Blood Pressure. Vasc. Health Risk Manag. 2016, 12, 393–405. [Google Scholar] [CrossRef]
- Basalay, M.; Davidson, S.; Yellon, D. Bs16 the Role of Parasympathetic Nervous System in the Infarct-Limiting Effect of Sglt2 Inhibitors. Heart 2021, 107, A164. [Google Scholar]
- Yoshikawa, T.; Kishi, T.; Shinohara, K.; Takesue, K.; Shibata, R.; Sonoda, N.; Inoguchi, T.; Sunagawa, K.; Tsutsui, H.; Hirooka, Y. Arterial Pressure Lability Is Improved by Sodium-Glucose Cotransporter 2 Inhibitor in Streptozotocin-Induced Diabetic Rats. Hypertens. Res. 2017, 40, 646–651. [Google Scholar] [CrossRef]
- Chiba, Y.; Yamada, T.; Tsukita, S.; Takahashi, K.; Munakata, Y.; Shirai, Y.; Kodama, S.; Asai, Y.; Sugisawa, T.; Uno, K.; et al. Dapagliflozin, a Sodium-Glucose Co-Transporter 2 Inhibitor, Acutely Reduces Energy Expenditure in Bat Via Neural Signals in Mice. PLoS ONE 2016, 11, e0150756. [Google Scholar] [CrossRef]
- Nguyen, T.; Wen, S.; Gong, M.; Yuan, X.; Xu, D.; Wang, C.; Jin, J.; Zhou, L. Dapagliflozin Activates Neurons in the Central Nervous System and Regulates Cardiovascular Activity by Inhibiting Sglt-2 in Mice. Diabetes Metab. Syndr. Obes. 2020, 13, 2781–2799. [Google Scholar] [CrossRef]
- Sardu, C.; Massetti, M.M.; Rambaldi, P.; Gatta, G.; Cappabianca, S.; Sasso, F.; Santamaria, M.; Volpicelli, M.; Ducceschi, V.; Signoriello, G.; et al. Sglt2-Inhibitors Reduce the Cardiac Autonomic Neuropathy Dysfunction and Vaso-Vagal Syncope Recurrence in Patients with Type 2 Diabetes Mellitus: The Scan Study. Metabolism 2022, 137, 155243. [Google Scholar] [CrossRef]
- Aronson, D. Cross-Linking of Glycated Collagen in the Pathogenesis of Arterial and Myocardial Stiffening of Aging and Diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef]
- Oelze, M.; Kroller-Schon, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; Mader, M.; Zinssius, E.; Agdauletova, S.; et al. The Sodium-Glucose Co-Transporter 2 Inhibitor Empagliflozin Improves Diabetes-Induced Vascular Dysfunction in the Streptozotocin Diabetes Rat Model by Interfering with Oxidative Stress and Glucotoxicity. PLoS ONE 2014, 9, e112394. [Google Scholar] [CrossRef]
- Ducheix, S.; Magre, J.; Cariou, B.; Prieur, X. Chronic O-Glcnacylation and Diabetic Cardiomyopathy: The Bitterness of Glucose. Front. Endocrinol. 2018, 9, 642. [Google Scholar] [CrossRef]
- Joubert, M.; Jagu, B.; Montaigne, D.; Marechal, X.; Tesse, A.; Ayer, A.; Dollet, L.; Le May, C.; Toumaniantz, G.; Manrique, A.; et al. The Sodium-Glucose Cotransporter 2 Inhibitor Dapagliflozin Prevents Cardiomyopathy in a Diabetic Lipodystrophic Mouse Model. Diabetes 2017, 66, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Koya, D.; Jirousek, M.; Lin, Y.; Ishii, H.; Kuboki, K.; King, G.L. Characterization of Protein Kinase C Beta Isoform Activation on the Gene Expression of Transforming Growth Factor-Beta, Extracellular Matrix Components, and Prostanoids in the Glomeruli of Diabetic Rats. J. Clin. Investig. 1997, 100, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Wang, J.; Lin, S.B. Antisense Oligonucleotides Targeting Protein Kinase C-Alpha, -Beta I, or -Delta but Not -Eta Inhibit Lipopolysaccharide-Induced Nitric Oxide Synthase Expression in Raw 264.7 Macrophages: Involvement of a Nuclear Factor Kappa B-Dependent Mechanism. J. Immunol. 1998, 161, 6206–6214. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ford, R.; Smith, B.; Gowans, G.; Mancini, S.; Pitt, R.; Day, E.; Salt, I.; Steinberg, G.; Hardie, D.G. The Naþ/glucose Co-Transporter Inhibitor Canagliflozin Activates AMP-Activated Protein Kinase by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Schiattarella, G.; Jiang, N.; Gillette, T.; Hill, J.A. Impaired Ampk Signaling in Hfpef-Associated Atrial Fibrillation. J. Am. Coll. Cardiol. 2020, 75 (Suppl. S1), 347. [Google Scholar] [CrossRef]
- Zhang, P.; Hu, X.; Xu, X.; Fassett, J.; Zhu, G.; Viollet, B.; Xu, W.; Wiczer, B.; Bernlohr, D.; Bache, R.; et al. AMP Activated Protein Kinase-Alpha2 Deficiency Exacerbates Pressure-Overload-Induced Left Ventricular Hypertrophy and Dysfunction in Mice. Hypertension 2008, 52, 918–924. [Google Scholar] [CrossRef]
- Liao, Y.; Takashima, S.; Maeda, N.; Ouchi, N.; Komamura, K.; Shimomura, I.; Hori, M.; Matsuzawa, Y.; Funahashi, T.; Kitakaze, M. Exacerbation of Heart Failure in Adiponectin-Deficient Mice Due to Impaired Regulation of AMPK and Glucose Metabolism. Cardiovasc. Res. 2005, 67, 705–713. [Google Scholar] [CrossRef]
- Sung, M.M.; Zordoky, B.; Bujak, A.; Lally, J.; Fung, D.; Young, M.; Horman, S.; Miller, E.; Light, P.; Kemp, B.; et al. AMPK Deficiency in Cardiac Muscle Results in Dilated Cardiomyopathy in the Absence of Changes in Energy Metabolism. Cardiovasc. Res. 2015, 107, 235–245. [Google Scholar] [CrossRef]
- Elrakaybi, A.; Laubner, K.; Zhou, Q.; Hug, M.J.; Seufert, J. Cardiovascular protection by SGLT2 inhibitors—Do anti-inflammatory mechanisms play a role? Mol. Metab. 2022, 64, 101549. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin Alleviates Cardiac Fibrosis through Suppressing EndMT and Fibroblast Activation via AMPKα/TGF-β/SmadSignalling in Type 2 Diabetic Rats. J. Cell. Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.; Perez-Polo, J.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Fan, X.; Wang, J.; Hou, J.; Lin, C.; Bensoussan, A.; Chang, D.; Liu, J.; Wang, B. Berberine Alleviates Ox-LDL Induced Inflammatory Factors by Up-Regulation of Autophagy via AMPK/mTOR Signaling Pathway. J. Transl. Med. 2015, 13, 92. [Google Scholar] [CrossRef]
- Ansari, M.Y.; Ahmad, N.; Haqqi, T.M. Butein Activates Autophagy through AMPK/TSC2/ULK1/mTOR Pathway to Inhibit IL-6 Expression in IL-1β Stimulated Human Chondrocytes. Cell. Physiol. Biochem. 2018, 49, 932–946. [Google Scholar] [CrossRef]
- Ren, C.; Sun, K.; Zhang, Y.; Hu, Y.; Hu, B.; Zhao, J.; He, Z.; Ding, R.; Wang, W.; Liang, C. Sodium-Glucose CoTransporter-2 Inhibitor Empagliflozin Ameliorates Sunitinib-Induced Cardiac Dysfunction via Regulation of AMPK-mTOR Signaling Pathway-Mediated Autophagy. Front. Pharmacol. 2021, 12, 664181. [Google Scholar] [CrossRef]
- Karmazyn, M.; Gan, X.; Humphreys, R.; Yoshida, H.; Kusumoto, K. The Myocardial Na(+)-H(+) Exchange: Structure, Regulation, and Its Role in Heart Disease. Circ. Res. 1999, 85, 777–786. [Google Scholar] [CrossRef]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.; Fiolet, J.; Koeman, A.; Jancev, M.; Hollmann, M.; Weber, N.; Coronel, R.; et al. Class Effects of Sglt2 Inhibitors in Mouse Cardiomyocytes and Hearts: Inhibition of Na+/H+ Exchanger, Lowering of Cytosolic Na+ and Vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.; Kim, S.; Kim, J.; Choi, E.; Cho, W.; Rim, J.; Hwang, I.; Lee, C.; Lee, M.; et al. Sglt2 Inhibition Modulates Nlrp3 Inflammasome Activity Via Ketones and Insulin in Diabetes with Cardiovascular Disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Smits, M.M.; Muskiet, M.; Tonneijck, L.; Hoekstra, T.; Kramer, M.; Diamant, M.; van Raalte, D.H. Exenatide Acutely Increases Heart Rate in Parallel with Augmented Sympathetic Nervous System Activation in Healthy Overweight Males. Br. J. Clin. Pharmacol. 2016, 81, 613–620. [Google Scholar] [CrossRef]
- Baggio, L.L.; Ussher, J.; McLean, B.; Cao, X.; Kabir, M.; Mulvihill, E.; Mighiu, A.; Zhang, H.; Ludwig, A.; Seeley, R.; et al. The Autonomic Nervous System and Cardiac Glp-1 Receptors Control Heart Rate in Mice. Mol. Metab. 2017, 6, 1339–1349. [Google Scholar] [CrossRef]
- Ibrahim, N.E.; Gaggin, H.; Turchin, A.; Patel, H.; Song, Y.; Trebnick, A.; Doros, G.; Maya, J.; Cannon, C.; Januzzi, J.L. Heart Rate, Beta-Blocker Use, and Outcomes of Heart Failure with Reduced Ejection Fraction. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 3–11. [Google Scholar] [CrossRef]
- Wallner, M.; Kolesnik, E.; Ablasser, K.; Khafaga, M.; Wakula, P.; Ljubojevic, S.; Thon-Gutschi, E.; Sourij, H.; Kapl, M.; Edmunds, N.; et al. Exenatide Exerts a Pka-Dependent Positive Inotropic Effect in Human Atrial Myocardium: Glp-1r Mediated Effects in Human Myocardium. J. Mol. Cell Cardiol. 2015, 89 Pt B, 365–375. [Google Scholar] [CrossRef]
- Packer, M.; Carver, J.; Rodeheffer, R.; Ivanhoe, R.; DiBianco, R.; Zeldis, S.; Hendrix, G.; Bommer, W.; Elkayam, U.; Kukin, M.; et al. Effect of Oral Milrinone on Mortality in Severe Chronic Heart Failure. The Promise Study Research Group. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef]
- Shan, Y.; Gao, L.; Wang, X.; Wang, X.; Wang, F. The Glp-1 Analog Liraglutide Protects against Angiotensin Ii and Pressure Overload-Induced Cardiac Hypertrophy Via Pi3k/Akt1 and Ampka Signaling. Front. Pharmacol. 2019, 10, 537. [Google Scholar]
- Nguyen, T.D.; Shingu, Y.; Amorim, P.; Schenkl, C.; Schwarzer, M.; Doenst, T. Glp-1 Improves Diastolic Function and Survival in Heart Failure with Preserved Ejection Fraction. J. Cardiovasc. Transl. Res. 2018, 11, 259–267. [Google Scholar] [CrossRef]
- Liu, Q.; Anderson, C.; Broyde, A.; Polizzi, C.; Fernandez, R.; Baron, A.; Parkes, D.G. Glucagon-Like Peptide-1 and the Exenatide Analogue Ac3174 Improve Cardiac Function, Cardiac Remodeling, and Survival in Rats with Chronic Heart Failure. Cardiovasc. Diabetol. 2010, 9, 76. [Google Scholar] [CrossRef]
- Heneghan, H.M.; Meron-Eldar, S.; Brethauer, S.; Schauer, P.; Young, J.B. Effect of Bariatric Surgery on Cardiovascular Risk Profile. Am. J. Cardiol. 2011, 108, 1499–1507. [Google Scholar] [CrossRef]
- Benotti, P.N.; Wood, G.; Carey, D.; Mehra, V.; Mirshahi, T.; Lent, M.; Petrick, A.; Still, C.; Gerhard, G.; Hirsch, A.G. Gastric Bypass Surgery Produces a Durable Reduction in Cardiovascular Disease Risk Factors and Reduces the Long-Term Risks of Congestive Heart Failure. J. Am. Heart Assoc. 2017, 6, e005126. [Google Scholar] [CrossRef]
- Cuspidi, C.; Rescaldani, M.; Tadic, M.; Sala, C.; Grassi, G. Effects of Bariatric Surgery on Cardiac Structure and Function: A Systematic Review and Meta-Analysis. Am. J. Hypertens. 2014, 27, 146–156. [Google Scholar] [CrossRef]
- Shin, S.H.; Lee, Y.; Heo, Y.; Park, S.; Kwon, S.; Woo, S.; Kim, D.; Park, K.; Kwan, J. Beneficial Effects of Bariatric Surgery on Cardiac Structure and Function in Obesity. Obes. Surg. 2017, 27, 620–625. [Google Scholar] [CrossRef]
- Ashrafian, H.; Athanasiou, T.; le Roux, C.W. Heart Remodelling and Obesity: The Complexities and Variation of Cardiac Geometry. Heart 2011, 97, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Batandier, C.; Guigas, B.; Detaille, D.; El-Mir, M.; Fontaine, E.; Rigoulet, M.; Leverve, X.M. The Ros Production Induced by a Reverse-Electron Flux at Respiratory-Chain Complex 1 Is Hampered by Metformin. J. Bioenerg. Biomembr. 2006, 38, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Kaminski, K.; Podewski, E.; Bonda, T.; Schaefer, A.; Sliwa, K.; Forster, O.; Quint, A.; Landmesser, U.; Doerries, C.; et al. A Cathepsin D-Cleaved 16 Kda Form of Prolactin Mediates Postpartum Cardiomyopathy. Cell 2007, 128, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, K.; Negoro, S.; Tone, E.; Funamoto, M.; Osugi, T.; Yamada, S.; Okabe, M.; Kishimoto, T.; Yamauchi-Takihara, K. Signal Transducer and Activator of Transcription 3 in the Heart Transduces Not Only a Hypertrophic Signal but a Protective Signal against Doxorubicin-Induced Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2000, 97, 315–319. [Google Scholar] [CrossRef]
- Hallakou-Bozec, S.; Vial, G.; Kergoat, M.; Fouqueray, P.; Bolze, S.; Borel, A.; Fontaine, E.; Moller, D.E. Mechanism of Action of Imeglimin: A Novel Therapeutic Agent for Type 2 Diabetes. Diabetes Obes. Metab. 2021, 23, 664–673. [Google Scholar] [CrossRef]
- Markowicz-Piasecka, M.; Sadkowska, A.; Huttunen, K.; Podsiedlik, M.; Mikiciuk-Olasik, E.; Sikora, J. An Investigation into the Pleiotropic Activity of Metformin. A Glimpse of Haemostasis. Eur. J. Pharmacol. 2020, 872, 172984. [Google Scholar] [CrossRef]
- Russell, R.R., 3rd; Li, J.; Coven, D.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.; Mu, J.; Birnbaum, M.; Young, L.H. Amp-Activated Protein Kinase Mediates Ischemic Glucose Uptake and Prevents Postischemic Cardiac Dysfunction, Apoptosis, and Injury. J. Clin. Investig. 2004, 114, 495–503. [Google Scholar] [CrossRef]
- Gao, C.; Fang, L.; Zhang, H.; Zhang, W.; Li, X.; Du, S.Y. Metformin Induces Autophagy Via the Ampk-Mtor Signaling Pathway in Human Hepatocellular Carcinoma Cells. Cancer Manag. Res. 2020, 12, 5803–5811. [Google Scholar] [CrossRef]
- Szrejder, M.; Rachubik, P.; Rogacka, D.; Audzeyenka, I.; Rychlowski, M.; Kreft, E.; Angielski, S.; Piwkowska, A. Metformin Reduces Trpc6 Expression through Ampk Activation and Modulates Cytoskeleton Dynamics in Podocytes under Diabetic Conditions. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165610. [Google Scholar] [CrossRef]
- Yu, J.W.; Deng, Y.; Han, X.; Ren, G.; Cai, J.; Jiang, G.J. Metformin Improves the Angiogenic Functions of Endothelial Progenitor Cells Via Activating Ampk/Enos Pathway in Diabetic Mice. Cardiovasc. Diabetol. 2016, 15, 88. [Google Scholar] [CrossRef]
- Kim, S.A.; Choi, H.C. Metformin Inhibits Inflammatory Response Via Ampk-Pten Pathway in Vascular Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 2012, 425, 866–872. [Google Scholar] [CrossRef] [PubMed]
- de Maranon, A.M.; Diaz-Pozo, P.; Canet, F.; Diaz-Morales, N.; Abad-Jimenez, Z.; Lopez-Domenech, S.; Vezza, T.; Apostolova, N.; Morillas, C.; Rocha, M.; et al. Metformin Modulates Mitochondrial Function and Mitophagy in Peripheral Blood Mononuclear Cells from Type 2 Diabetic Patients. Redox Biol. 2022, 53, 102342. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Ma, X.; Feng, W.; Fu, Y.; Lu, Z.; Xu, M.; Shen, Q.; Zhu, Y.; Zhang, Y. Metformin Attenuates Cardiac Fibrosis by Inhibiting the Tgfbeta1-Smad3 Signalling Pathway. Cardiovasc. Res. 2010, 87, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Pan, S.; Meng, R.; Peng, C.; Xiong, Z.; Chen, B.; Chen, G.; Yao, F.; Chen, Y.; Ma, Y.; et al. Metformin Attenuates Ventricular Hypertrophy by Activating the Amp-Activated Protein Kinase-Endothelial Nitric Oxide Synthase Pathway in Rats. Clin. Exp. Pharmacol. Physiol. 2011, 38, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Cheng, P.; Tu, S.; Hsu, S.; Cheng, Y.; Liu, Y.H. Effect of Metformin Monotherapy on Serum Lipid Profile in Statin-Naive Individuals with Newly Diagnosed Type 2 Diabetes Mellitus: A Cohort Study. PeerJ 2018, 6, e4578. [Google Scholar] [CrossRef]
- Titus, A.S.; Ushakumary, M.G.; Venugopal, H.; Wang, M.; Lakatta, E.G.; Kailasam, S. Metformin Attenuates Hyperglycaemia-Stimulated Pro-Fibrotic Gene Expression in Adventitial Fibroblasts via Inhibition of Discoidin Domain Receptor 2. Int. J. Mol. Sci. 2022, 24, 585. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, A.; Kohan, D.; Nelson, R.; Gonzalez, F.; Yang, T. Collecting Duct-Specific Deletion of Peroxisome Proliferator-Activated Receptor Gamma Blocks Thiazolidinedione-Induced Fluid Retention. Proc. Natl. Acad. Sci. USA 2005, 102, 9406–9411. [Google Scholar] [CrossRef]
- Hong, G.; Lockhart, A.; Davis, B.; Rahmoune, H.; Baker, S.; Ye, L.; Thompson, P.; Shou, Y.; O’Shaughnessy, K.; Ronco, P.; et al. Ppargamma Activation Enhances Cell Surface Enacalpha Via up-Regulation of Sgk1 in Human Collecting Duct Cells. FASEB J. 2003, 17, 1966–1968. [Google Scholar] [CrossRef] [PubMed]
- Nofziger, C.; Chen, L.; Shane, M.; Smith, C.; Brown, K.; Blazer-Yost, B.L. Ppargamma Agonists Do Not Directly Enhance Basal or Insulin-Stimulated Na+ Transport Via the Epithelial Na+ Channel. Pflugers Arch. 2005, 451, 445–453. [Google Scholar] [CrossRef]
- Song, J.; Knepper, M.; Hu, X.; Verbalis, J.; Ecelbarger, C.A. Rosiglitazone Activates Renal Sodium- and Water-Reabsorptive Pathways and Lowers Blood Pressure in Normal Rats. J. Pharmacol. Exp. Ther. 2004, 308, 426–433. [Google Scholar] [CrossRef]
- Vallon, V.; Hummler, E.; Rieg, T.; Pochynyuk, O.; Bugaj, V.; Schroth, J.; Dechenes, G.; Rossier, B.; Cunard, R.; Stockand, J. Thiazolidinedione-Induced Fluid Retention Is Independent of Collecting Duct Alphaenac Activity. J. Am. Soc. Nephrol. 2009, 20, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, B.; McNulty, J.; Clifton, L.; Binz, J.; Grimes, A.; Strum, J.; Harrington, W.; Chen, Z.; Balon, T.; et al. Gi262570, a Peroxisome Proliferator-Activated Receptor Gamma Agonist, Changes Electrolytes and Water Reabsorption from the Distal Nephron in Rats. J. Pharmacol. Exp. Ther. 2005, 312, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Suzuki, M.; Yamada, H.; Horita, S.; Kunimi, M.; Yamazaki, O.; Shirai, A.; Nakamura, M.; Iso, O.; Li, Y.; et al. Thiazolidinediones Enhance Sodium-Coupled Bicarbonate Absorption from Renal Proximal Tubules Via Ppargamma-Dependent Nongenomic Signaling. Cell. Metab. 2011, 13, 550–561. [Google Scholar] [CrossRef]
- Chu, P.Y.; Zatta, A.; Kiriazis, H.; Chin-Dusting, J.; Du, X.; Marshall, T.; Kaye, D.M. Cxcr4 Antagonism Attenuates the Cardiorenal Consequences of Mineralocorticoid Excess. Circ. Heart Fail. 2011, 4, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.Y.; Walder, K.; Horlock, D.; Williams, D.; Nelson, E.; Byrne, M.; Jandeleit-Dahm, K.; Zimmet, P.; Kaye, D.M. Cxcr4 Antagonism Attenuates the Development of Diabetic Cardiac Fibrosis. PLoS ONE 2015, 10, e0133616. [Google Scholar] [CrossRef]
- Wei, S.G.; Zhang, Z.; Yu, Y.; Weiss, R.; Felder, R.B. Central Actions of the Chemokine Stromal Cell-Derived Factor 1 Contribute to Neurohumoral Excitation in Heart Failure Rats. Hypertension 2012, 59, 991–998. [Google Scholar] [CrossRef]
- Arif, E.; Wang, C.; Swiderska-Syn, M.; Solanki, A.; Rahman, B.; Manka, P.; Coombes, J.; Canbay, A.; Papa, S.; Nihalani, D.; et al. Targeting Myosin 1c Inhibits Murine Hepatic Fibrogenesis. Am. J. Physiol. Gastrointest. Liver. Physiol. 2021, 320, G1044–G1053. [Google Scholar] [CrossRef]
- Dzurik, M.V.; Diedrich, A.; Black, B.; Paranjape, S.; Raj, S.; Byrne, D.; Robertson, D. Endogenous Substance P Modulates Human Cardiovascular Regulation at Rest and During Orthostatic Load. J. Appl. Physiol. 2007, 102, 2092–2097. [Google Scholar] [CrossRef]
- Shanks, J.; Herring, N. Peripheral Cardiac Sympathetic Hyperactivity in Cardiovascular Disease: Role of Neuropeptides. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R1411–R1420. [Google Scholar] [CrossRef]
- Luo, G.; Xu, X.; Guo, W.; Luo, C.; Wang, H.; Meng, X.; Zhu, S.; Wei, Y. Neuropeptide Y Damages the Integrity of Mitochondrial Structure and Disrupts Energy Metabolism in Cultured Neonatal Rat Cardiomyocytes. Peptides 2015, 71, 162–169. [Google Scholar] [CrossRef]
- Robinson, P.; Kasembeli, M.; Bharadwaj, U.; Engineer, N.; Eckols, K.; Tweardy, D.J. Substance P Receptor Signaling Mediates Doxorubicin-Induced Cardiomyocyte Apoptosis and Triple-Negative Breast Cancer Chemoresistance. BioMed Res. Int. 2016, 2016, 1959270. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, E.E.; Varin, E.; Ussher, J.; Campbell, J.; Bang, K.; Abdullah, T.; Baggio, L.; Drucker, D.J. Inhibition of Dipeptidyl Peptidase-4 Impairs Ventricular Function and Promotes Cardiac Fibrosis in High Fat-Fed Diabetic Mice. Diabetes 2016, 65, 742–754. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-N.; Hsuan, C.-F.; Liao, D.; Chang, J.K.-J.; Chang, A.J.-W.; Hee, S.-W.; Lee, H.-L.; Teng, S.I.F. Anti-Diabetic Therapy and Heart Failure: Recent Advances in Clinical Evidence and Molecular Mechanism. Life 2023, 13, 1024. https://doi.org/10.3390/life13041024
Hsu C-N, Hsuan C-F, Liao D, Chang JK-J, Chang AJ-W, Hee S-W, Lee H-L, Teng SIF. Anti-Diabetic Therapy and Heart Failure: Recent Advances in Clinical Evidence and Molecular Mechanism. Life. 2023; 13(4):1024. https://doi.org/10.3390/life13041024
Chicago/Turabian StyleHsu, Chih-Neng, Chin-Feng Hsuan, Daniel Liao, Jack Keng-Jui Chang, Allen Jiun-Wei Chang, Siow-Wey Hee, Hsiao-Lin Lee, and Sean I. F. Teng. 2023. "Anti-Diabetic Therapy and Heart Failure: Recent Advances in Clinical Evidence and Molecular Mechanism" Life 13, no. 4: 1024. https://doi.org/10.3390/life13041024