
Cardiac Reverse Remodeling in Ischemic Heart Disease with Novel Therapies for Heart Failure with Reduced Ejection Fraction
 , , , , , ,
, , , , , ,
Abstract
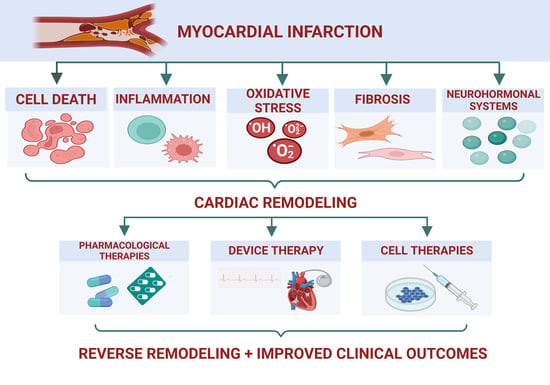
:
{kind=link}
{kind=link}
{kind=link}
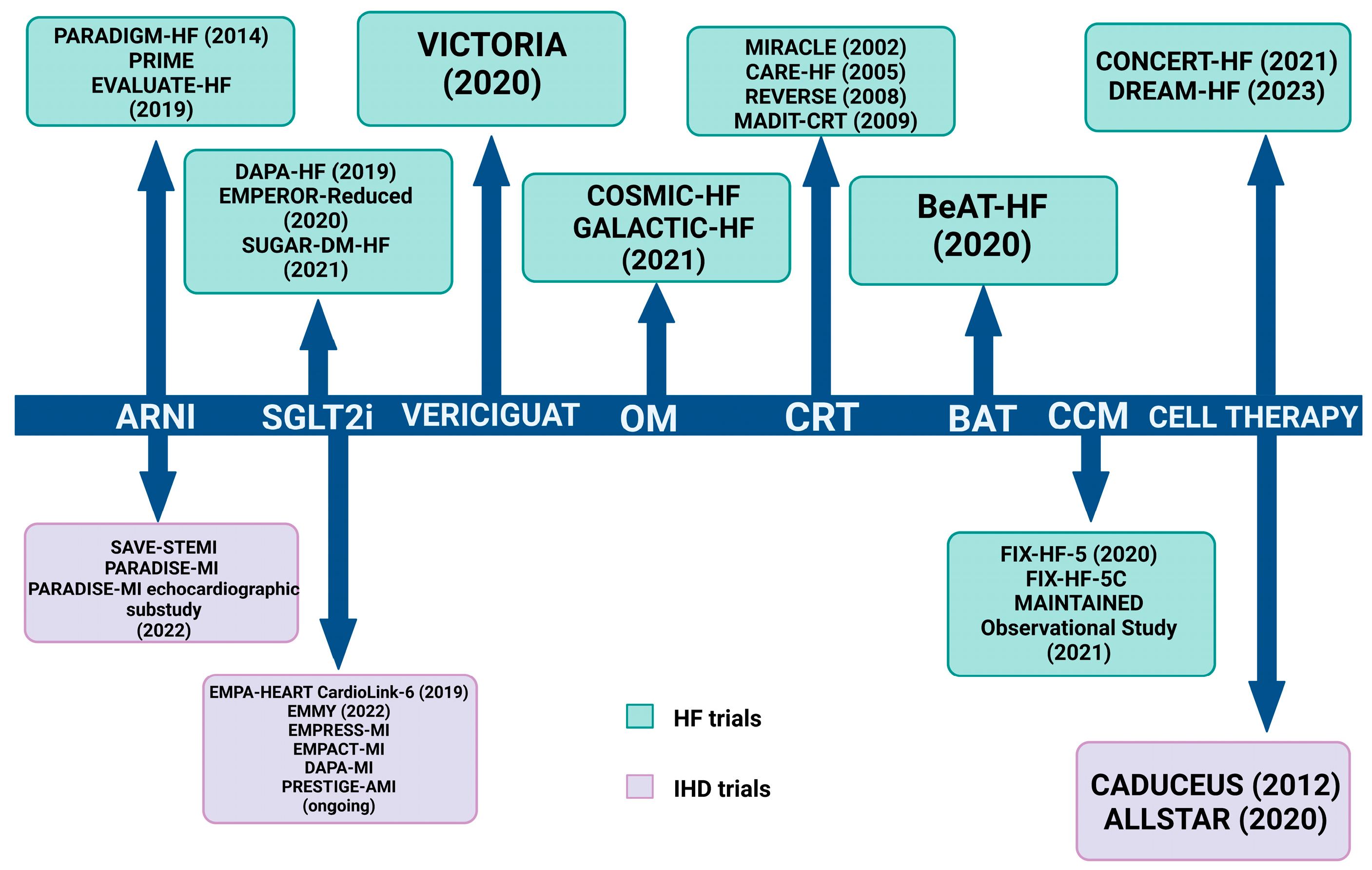{kind=link}
1. Introduction
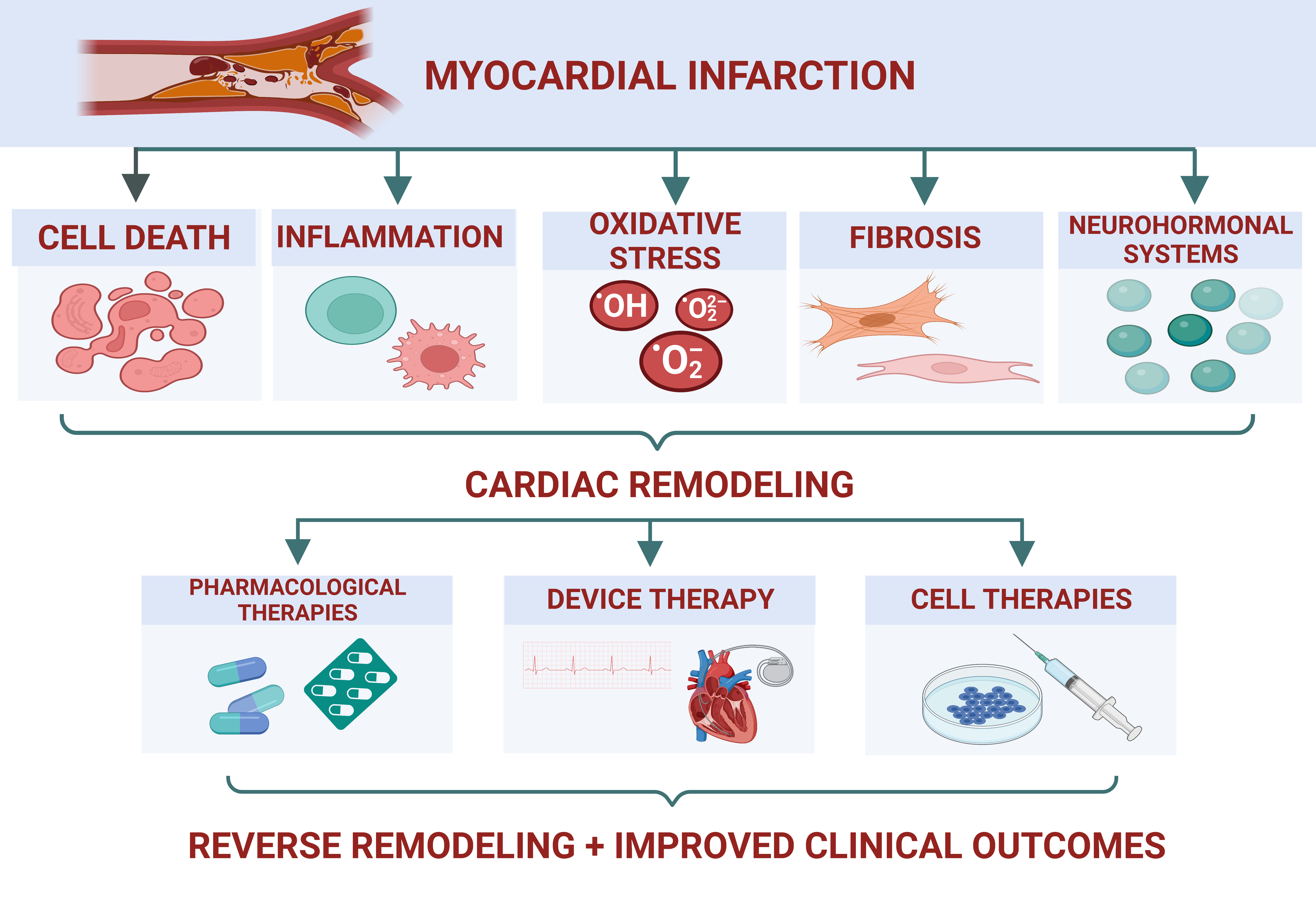
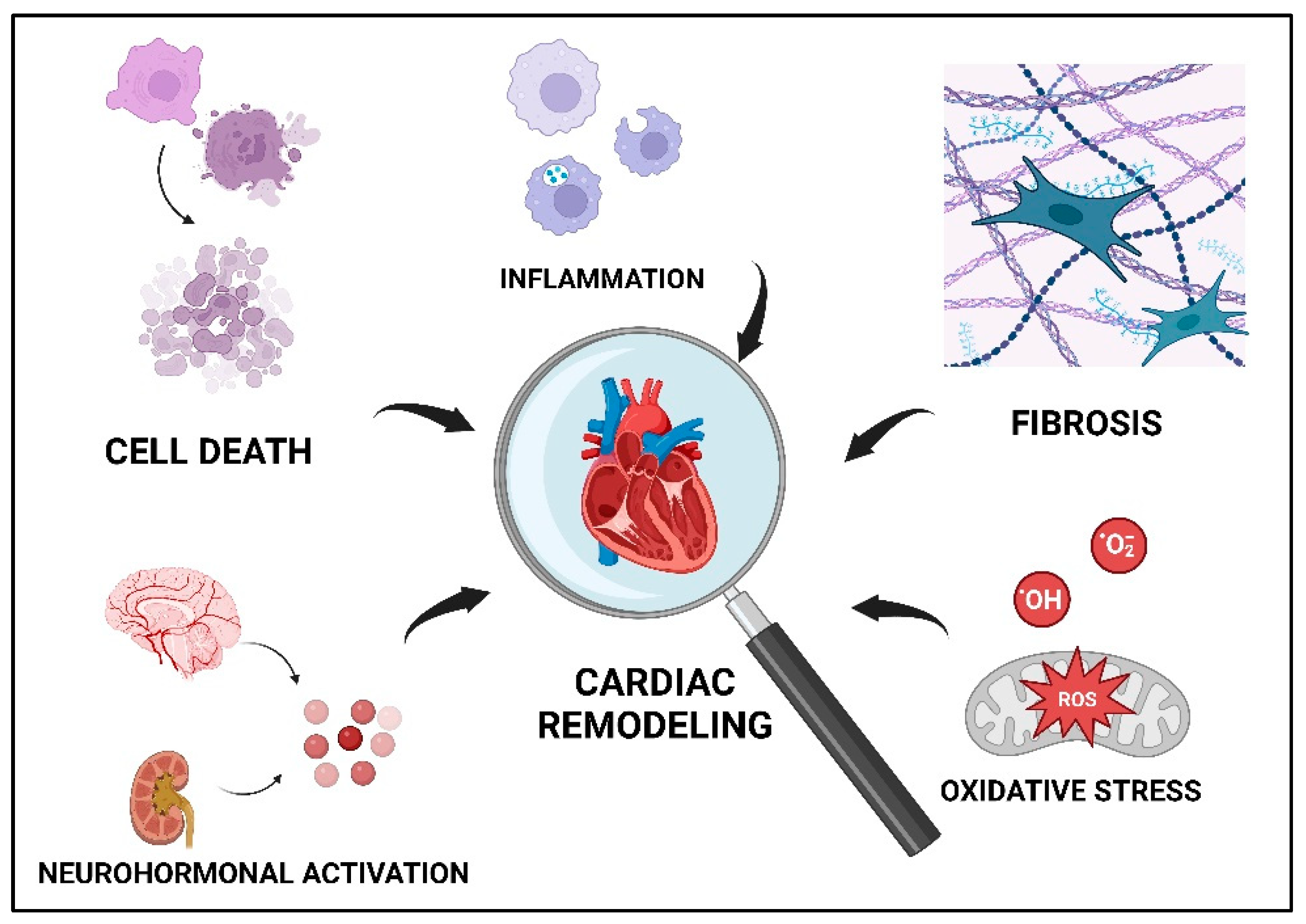
2. Ventricular Remodeling in IHD
2.1. Cell Death
2.2. Inflammation
2.3. Oxidative Stress
2.4. Fibrosis
2.5. Neurohormonal Systems
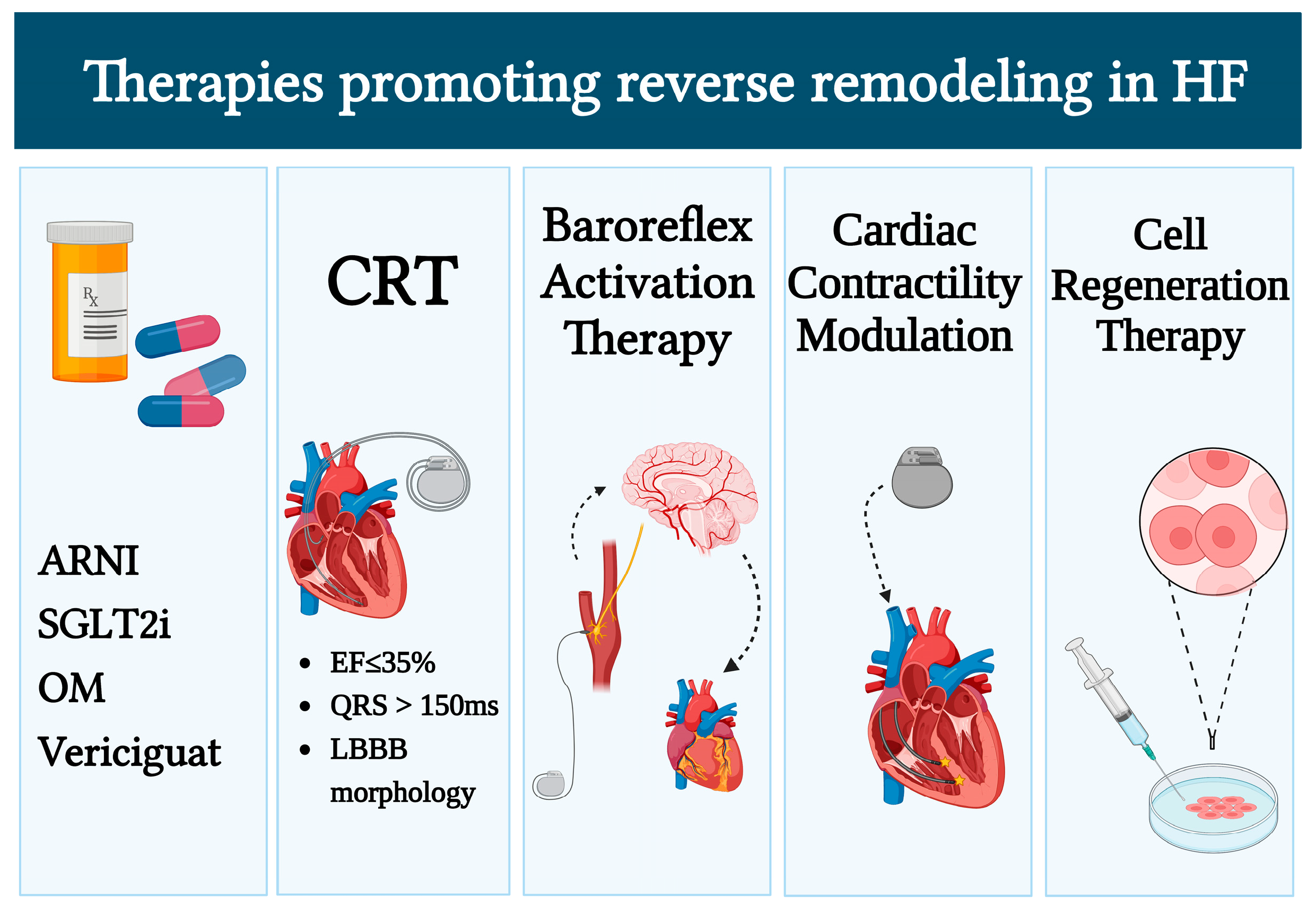
3. Novel Therapeutic Strategies in IHD with Reduced EF
3.1. Pharmacologic Therapies
3.1.1. Angiotensin Receptor Neprilysin Inhibitor
3.1.2. Sodium-Glucose Co-Transporter-2 Inhibitors
3.1.3. Selective Cardiac Myosin Activators
3.1.4. Soluble Guanylate Cyclase Stimulators
3.2. Device Therapy in HF
3.2.1. Cardiac Resynchronization Therapy
3.2.2. Baroreflex Activation Therapy
3.2.3. Cardiac Contractility Modulation (CCM)
3.3. Cell Therapy
3.3.1. Cell Therapy in Acute Myocardial Infarction
3.3.2. Cell Therapy in Ischemic Cardiomyopathy
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxid. Med. Cell Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef] [PubMed]
- Inamdar, A.A.; Inamdar, A.C. Heart Failure: Diagnosis, Management and Utilization. J. Clin. Med. 2016, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Albakri, A. Ischemic Heart Failure: A Review of Clinical Status and Meta-Analysis of Diagnosis and Clinical Management Methods. Clin. Med. Investig. 2018, 3, 1–15. [Google Scholar] [CrossRef]
- Pepine, C.J.; Nichols, W.W. The Pathophysiology of Chronic Ischemic Heart Disease. Clin. Cardiol. 2007, 30, I-4–I-9. [Google Scholar] [CrossRef]
- Filho, J.R.D.A.R.R.; Cardoso, J.N.; Cardoso, C.M.D.R.; Pereira-Barretto, A.C.; Reis, J.R.D.A.R. Reversão Da Remodelação Cardíaca: Um Marcador de Melhor Prognóstico Na Insuficiência Cardíaca. Arq. Bras. Cardiol. 2015, 104, 502–506. [Google Scholar] [CrossRef]
- Kehat, I.; Molkentin, J.D. Molecular Pathways Underlying Cardiac Remodeling during Pathophysiological Stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komajda, M.; Lam, C.S.P. Heart Failure with Preserved Ejection Fraction: A Clinical Dilemma. Eur. Heart J. 2014, 35, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Altay, H.; Pehlivanoglu, S. Heart Failure with Preserved Ejection Fraction. In Cardiomyopathies—Types and Treatments; Intech Open: London, UK, 2017; pp. 39–53. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Luo, H.; Fan, L.; Liu, X.; Gao, C. Heart Failure with Preserved Ejection Fraction: An Update on Pathophysiology, Diagnosis, Treatment, and Prognosis. Braz. J. Med. Biol. Res. 2020, 53, e9646. [Google Scholar] [CrossRef]
- Jenča, D.; Melenovský, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart Failure after Myocardial Infarction: Incidence and Predictors. ESC Heart Fail. 2021, 8, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Wollert, K.C.; Drexler, H. Potential Novel Pharmacological Therapies for Myocardial Remodelling. Cardiovasc. Res. 2009, 81, 519–527. [Google Scholar] [CrossRef]
- Boulet, J.; Mehra, M.R. Left Ventricular Reverse Remodeling in Heart Failure: Remission to Recovery. Struct. Heart 2021, 5, 466–481. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Galuppo, P.; Bauersachs, J. Novel Therapeutic Approaches to Post-Infarction Remodelling. Cardiovasc. Res. 2012, 94, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Cabac-Pogorevici, I.; Muk, B.; Rustamova, Y.; Kalogeropoulos, A.; Tzeis, S.; Vardas, P. Ischaemic Cardiomyopathy. Pathophysiological Insights, Diagnostic Management and the Roles of Revascularisation and Device Treatment. Gaps and Dilemmas in the Era of Advanced Technology. Eur. J. Heart Fail. 2020, 22, 789–799. [Google Scholar] [CrossRef]
- Saraon, T.; Katz, S.D. Reverse Remodeling in Systolic Heart Failure. Cardiol. Rev. 2015, 23, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.S.; Ambrosy, A.P.; Velazquez, E.J. Adverse Remodeling and Reverse Remodeling After Myocardial Infarction. Curr. Cardiol. Rep. 2017, 19, 71. [Google Scholar] [CrossRef]
- Al-Botaty, B.; El-Khoely, A.; Elsayed, E.; Eissa, A. Insight into the Pathophysiology of Myocardial Infarction. J. Adv. Pharm. Res. 2022, 6, 223–237. [Google Scholar] [CrossRef]
- del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Rabinovich-Nikitin, I.; Lieberman, B.; Martino, T.A.; Kirshenbaum, L.A. Circadian-Regulated Cell Death in Cardiovascular Diseases. Circulation 2019, 139, 965–980. [Google Scholar] [CrossRef]
- Mishra, P.K.; Adameova, A.; Hill, J.A.; Baines, C.P.; Kang, P.M.; Downey, J.M.; Narula, J.; Takahashi, M.; Abbate, A.; Piristine, H.C.; et al. REVIEW Guidelines in Cardiovascular Research Guidelines for Evaluating Myocardial Cell Death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, 891–922. [Google Scholar] [CrossRef]
- Briasoulis, A.; Androulakis, E.; Christophides, T.; Tousoulis, D. The Role of Inflammation and Cell Death in the Pathogenesis, Progression and Treatment of Heart Failure. Heart Fail. Rev. 2016, 21, 169–176. [Google Scholar] [CrossRef]
- Webster, K.A. Mitochondrial Membrane Permeabilization and Cell Death during Myocardial Infarction: Roles of Calcium and Reactive Oxygen Species. Future Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef] [Green Version]
- Teringova, E.; Tousek, P. Apoptosis in Ischemic Heart Disease. J. Transl. Med. 2017, 15, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Hu, S.; Bian, Y.; Yao, J.; Wang, D.; Liu, X.; Guo, Z.; Zhang, S.; Peng, L. Targeting Cell Death: Pyroptosis, Ferroptosis, Apoptosis and Necroptosis in Osteoarthritis. Front. Cell Dev. Biol. 2022, 9, 789948. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Narula, J. Role of Apoptosis in Adverse Ventricular Remodeling. Heart Fail. Clin. 2012, 8, 79–86. [Google Scholar] [CrossRef]
- Moe, G.W.; Marín-García, J. Role of Cell Death in the Progression of Heart Failure. Heart Fail. Rev. 2016, 21, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of Cell Death in Heart Disease. Arter. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Frangogiannis, N.G. Chemokines in Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 14, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Lichý, M.; Szobi, A.; Hrdlička, J.; Neckář, J.; Kolář, F.; Adameová, A. Programmed Cell Death in the Left and Right Ventricle of the Late Phase of Post-Infarction Heart Failure. Int. J. Mol. Sci. 2020, 21, 7782. [Google Scholar] [CrossRef]
- Guo, X.; Chen, Y.; Liu, Q. Necroptosis in Heart Disease: Molecular Mechanisms and Therapeutic Implications. J. Mol. Cell Cardiol. 2022, 169, 74–83. [Google Scholar] [CrossRef]
- Zhe-Wei, S.; Li-Sha, G.; Yue-Chun, L. The Role of Necroptosis in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 721. [Google Scholar] [CrossRef]
- Piamsiri, C.; Maneechote, C.; Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Targeting Necroptosis as Therapeutic Potential in Chronic Myocardial Infarction. J. Biomed. Sci. 2021, 28, 25. [Google Scholar] [CrossRef] [PubMed]
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a Kinase Promoting Necroptotic Cell Death, Mediates Adverse Remodelling Aftermyocardial Infarction. Cardiovasc. Res. 2014, 103, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, L.; Shen, Y.; Ren, C.; Kobayashi, S.; Asahara, T.; Yang, J. Inflammation in Myocardial Infarction: Roles of Mesenchymal Stem Cells and Their Secretome. Cell Death Discov. 2022, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Yee, D.; Shenoy, V.; Nagarajan, N.; Sadoshima, J. The Importance of Autophagy in Cardioprotection. High Blood Press. Cardiovasc. Prev. 2014, 21, 21–28. [Google Scholar] [CrossRef]
- Ibe II, A.C. The Role of Autophagy in Myocardial Ischemia/Reperfusion Injury The Role of Autophagy in Myocardial Ischemia/Reperfusion Injury in Isolated Rat Hearts. Master’s Thesis, Philadelphia College of Osteopathic Medicine, Philadelphia, PA, USA, 2019. [Google Scholar]
- Lin, X.L.; Xiao, W.J.; Xiao, L.; Liu, M.H. Molecular Mechanisms of Autophagy in Cardiac Ischemia/Reperfusion Injury (Review). Mol. Med. Rep. 2018, 18, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riquelme, J.A.; Chavez, M.N.; Mondaca-Ruff, D.; Bustamante, M.; Vicencio, J.M.; Quest, A.F.G.; Lavandero, S. Therapeutic Targeting of Autophagy in Myocardial Infarction and Heart Failure. Expert Rev. Cardiovasc. Ther. 2016, 14, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Ono, K.; Nagao, K.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; et al. Autophagy Limits Acute Myocardial Infarction Induced by Permanent Coronary Artery Occlusion. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, 2261–2271. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.B.; Hernández-Reséndiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation Following Acute Myocardial Infarction: Multiple Players, Dynamic Roles, and Novel Therapeutic Opportunities. Pharmacol. Ther. 2018, 186, 73–87. [Google Scholar] [CrossRef]
- Toldo, S.; Abbate, A. The NLRP3 Inflammasome in Acute Myocardial Infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. NLRP3 Inflammasome in Acute Myocardial Infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef]
- Chua, J.; Crespo-Avilan, G.E.; Ping, Y.E.; Ong, S.-G.; Hernandez-Resendiz, S.; Hausenloy, D.J. Autophagy as a Target for Cardioprotection in Acute Myocardial Infarction and Heart Failure. Cond. Med. 2020, 3, 264–273. [Google Scholar]
- Daskalopoulos, E.P.; Hermans, K.C.M.; van Delft, L.; Altara, R.; Blankesteijn, W.M. The Role of Inflammation in Myocardial Infarction. In Inflammation in Heart Failure; Blankesteijn, W.M., Altara, R., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 39–65. ISBN 978-0-12-800039-7. [Google Scholar]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Rosa, C.C.D.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Huang, S.; Frangogiannis, N.G. Anti-Inflammatory Therapies in Myocardial Infarction: Failures, Hopes and Challenges. Br. J. Pharmacol. 2018, 175, 1377–1400. [Google Scholar] [CrossRef] [Green Version]
- Westman, P.C.; Lipinski, M.J.; Luger, D.; Waksman, R.; Bonow, R.O.; Wu, E.; Epstein, S.E. Inflammation as a Driver of Adverse Left Ventricular Remodeling After Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2016, 67, 2050–2260. [Google Scholar] [CrossRef]
- Duncan, S.E.; Gao, S.; Sarhene, M.; Coffie, J.W.; Linhua, D.; Bao, X.; Jing, Z.; Li, S.; Guo, R.; Su, J.; et al. Macrophage Activities in Myocardial Infarction and Heart Failure. Cardiol. Res. Pract. 2020, 2020, 4375127. [Google Scholar] [CrossRef]
- Oliveira, J.B.; Soares, A.A.S.M.; Sposito, A.C. Inflammatory Response During Myocardial Infarction. Adv. Clin. Chem. 2018, 84, 39–79. [Google Scholar] [CrossRef]
- Yin, C.; Heit, B. Cellular Responses to the Efferocytosis of Apoptotic Cells. Front. Immunol. 2021, 12, 631714. [Google Scholar] [CrossRef]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac Monocytes and Macrophages after Myocardial Infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Li, J. Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clin. Med. Insights Cardiol. 2016, 10, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An Introduction to Immunology and Immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef] [Green Version]
- Blanton, R.M.; Carrillo-Salinas, F.J.; Alcaide, P. T-Cell Recruitment to the Heart: Friendly Guests or Unwelcome Visitors? Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H124–H140. [Google Scholar] [CrossRef] [PubMed]
- Boag, S.E.; Andreano, E.; Spyridopoulos, I. Lymphocyte Communication in Myocardial Ischemia/Reperfusion Injury. Antioxid. Redox. Signal. 2017, 26, 660–675. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Prabhu, S.D.; Bansal, S.S. CD4+ T-Lymphocytes Exhibit Biphasic Kinetics Post-Myocardial Infarction. Front. Cardiovasc. Med. 2022, 9, 992653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, W.; Liu, H. The Role of Immune Cells in Cardiac Remodeling After Myocardial Infarction. J. Cardiovasc. Pharmacol. 2020, 76, 407–413. [Google Scholar] [CrossRef]
- Santos-Zas, I.; Lemarié, J.; Tedgui, A.; Ait-Oufella, H. Adaptive Immune Responses Contribute to Post-Ischemic Cardiac Remodeling. Front. Cardiovasc. Med. 2019, 5, 198. [Google Scholar] [CrossRef]
- Lu, Y.; Xia, N.; Cheng, X. Regulatory T Cells in Chronic Heart Failure. Front. Immunol. 2021, 12, 732794. [Google Scholar] [CrossRef]
- Weiß, E.; Ramos, G.C.; Delgobo, M. Myocardial-Treg Crosstalk: How to Tame a Wolf. Front. Immunol. 2022, 13, 914033. [Google Scholar] [CrossRef]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Zhou, G.; Rokosh, G.; Hamid, T.; Prabhu, S.D. Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy. Circulation 2019, 139, 206–221. [Google Scholar] [CrossRef]
- Ilatovskaya, D.V.; Pitts, C.; Clayton, J.; Domondon, M.; Troncoso, M.; Pippin, S.; Kristine, X.; Deleon-Pennell, Y. CD8 T-Cells Negatively Regulate Inflammation Post-Myocardial Infarction. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, 581–596. [Google Scholar] [CrossRef]
- Kino, T.; Khan, M.; Mohsin, S. The Regulatory Role of T Cell Responses in Cardiac Remodeling Following Myocardial Infarction. Int. J. Mol. Sci. 2020, 21, 5013. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating Oxidative Stress in Heart Failure: Past, Present and Future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef] [Green Version]
- Kamiński, K.; Bonda, T.; Wojtkowska, I.; Dobrzycki, S.; Kralisz, P.; Nowak, K.; Prokopczuk, P.; Skrzydlewska, E.; Kozuch, M.; Musial, W.J. Oxidative Stress and Antioxidative Defense Parameters Early after Reperfusion Therapy for Acute Myocardial Infarction. Acute. Cardiac. Care 2008, 10, 121–126. [Google Scholar] [CrossRef]
- Neri, M.; Fineschi, V.; di Paolo, M.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Cerretani, D. Cardiac Oxidative Stress and Inflammatory Cytokines Response after Myocardial Infarction. Curr. Vasc. Pharmacol. 2015, 13, 26–36. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial Dysfunction and Oxidative Stress in Heart Disease. Exp. Mol. Med. 2019, 51, 162. [Google Scholar] [CrossRef]
- Matin, E.; Ghaffari, S.; Garjani, A.; Roshanravan, N.; Matin, S.; Mesri Alamdari, N.; Safaie, N. Oxidative Stress and Its Association with ST Resolution and Clinical Outcome Measures in Patients with ST-Segment Elevation Myocardial Infarction (STEMI) Undergoing Primary Percutaneous Coronary Intervention. BMC Res. Notes 2020, 13, 525. [Google Scholar] [CrossRef]
- van Nieuwenhoven, F.A.; Turner, N.A. The Role of Cardiac Fibroblasts in the Transition from Inflammation to Fibrosis Following Myocardial Infarction. Vascul. Pharmacol. 2013, 58, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Varzideh, F.; Kansakar, U.; Donkor, K.; Wilson, S.; Jankauskas, S.S.; Mone, P.; Wang, X.; Lombardi, A.; Santulli, G. Cardiac Remodeling After Myocardial Infarction: Functional Contribution of MicroRNAs to Inflammation and Fibrosis. Front. Cardiovasc. Med. 2022, 9, 863238. [Google Scholar] [CrossRef]
- Nagaraju, C.K.; Dries, E.; Popovic, N.; Singh, A.A.; Haemers, P.; Roderick, H.L.; Claus, P.; Sipido, K.R.; Driesen, R.B. Global Fibroblast Activation throughout the Left Ventricle but Localized Fibrosis after Myocardial Infarction. Sci. Rep. 2017, 7, 10801. [Google Scholar] [CrossRef] [Green Version]
- Xue, K.; Zhang, J.; Li, C.; Li, J.; Wang, C.; Zhang, Q.; Chen, X.; Yu, X.; Sun, L.; Yu, X. The Role and Mechanism of Transforming Growth Factor Beta 3 in Human Myocardial Infarction-Induced Myocardial Fibrosis. J. Cell Mol. Med. 2019, 23, 4229–4243. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Shi, H.; Chang, S.; Gao, Y.; Li, X.; Lv, C.; Yang, H.; Xiang, H.; Yang, J.; Xu, L.; et al. The Selective NLRP3-Inflammasome Inhibitor MCC950 Reduces Myocardial Fibrosis and Improves Cardiac Remodeling in a Mouse Model of Myocardial Infarction. Int. Immunopharmacol. 2019, 74, 105575. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.G.; Balding, L.C.; Candido, R.; Burns, W.C.; Cao, Z.; Twigg, S.M.; Burrell, L.M. Connective Tissue Growth Factor and Cardiac Fibrosis after Myocardial Infarction. J. Histochem. Cytochem. 2005, 53, 1245–1256. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- D’elia, N.; D’hooge, J.; Marwick, T.H. Association Between Myocardial Mechanics and Ischemic LV Remodeling. JACC Cardiovasc. Imaging 2015, 8, 1430–1443. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair after Myocardial Infarction. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac Fibrosis: Cell Biological Mechanisms, Molecular Pathways and Therapeutic Opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, J.; Qin, Z.; Lu, Y.; Xu, Y.; Guo, J.; Cui, X.; Zhang, J.; Tang, J. The Present Clinical Treatment and Future Emerging Interdisciplinary for Heart Failure: Where We Are and What We Can Do. Intensive Care Res. 2023, 3, 3–11. [Google Scholar] [CrossRef]
- Jorbenadze, A.; Fudim, M.; Mahfoud, F.; Adamson, P.B.; Bekfani, T.; Wachter, R.; Sievert, H.; Ponikowski, P.P.; Cleland, J.G.F.; Anker, S.D. Extra-Cardiac Targets in the Management of Cardiometabolic Disease: Device-Based Therapies. ESC Heart Fail. 2021, 8, 3327–3338. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Xu, X.; Li, Y.; Shi, S.; Lv, J.; Wang, Y.; Zheng, H.; Mao, X.; Wu, H.; Zhang, B.; Song, Q. The Application of Angiotensin Receptor Neprilysin Inhibitor in Cardiovascular Diseases: A Bibliometric Review From 2000 to 2022. Front. Cardiovasc. Med. 2022, 9, 899235. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, I.Y.; Mahtta, D.; Pepine, C.J. Medical Therapy for Heart Failure Caused by Ischemic Heart Disease. Circ. Res. 2019, 124, 1520–1535. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Claggett, B.; McMurray, J.J.V.; Hernandez, A.F.; Fonarow, G.C. Combined Neprilysin and Renin–Angiotensin System Inhibition in Heart Failure with Reduced Ejection Fraction: A Meta-Analysis. Eur. J. Heart Fail. 2016, 18, 1238–1243. [Google Scholar] [CrossRef]
- Abdin, A.; Schulz, M.; Riemer, U.; Hadëri, B.; Wachter, R.; Laufs, U.; Bauersachs, J.; Kindermann, I.; Vukadinović, D.; Böhm, M. Sacubitril/Valsartan in Heart Failure: Efficacy and Safety in and Outside Clinical Trials. ESC Heart Fail. 2022, 9, 3737–3750. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, P.; Beliën, H.; Dupont, M.; Vandervoort, P.; Mullens, W. The Reverse Remodeling Response to Sacubitril/Valsartan Therapy in Heart Failure with Reduced Ejection Fraction. Cardiovasc. Ther. 2018, 36, e12435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almufleh, A.; Marbach, J.; Chih, S.; Stadnick, E.; Davies, R.; Liu, P.; Mielniczuk, L. Ejection fraction improvement and reverse remodeling achieved with Sacubitril/Valsartan in heart failure with reduced ejection fraction patients. Am. J. Cardiovasc. Dis. 2017, 7, 108–113. [Google Scholar]
- Kang, D.H.; Park, S.J.; Shin, S.H.; Hong, G.R.; Lee, S.; Kim, M.S.; Yun, S.C.; Song, J.M.; Park, S.W.; Kim, J.J. Angiotensin Receptor Neprilysin Inhibitor for Functional Mitral Regurgitation. Circulation 2019, 139, 1354–1365. [Google Scholar] [CrossRef]
- Desai, A.S.; Solomon, S.D.; Shah, A.M.; Claggett, B.L.; Fang, J.C.; Izzo, J.; McCague, K.; Abbas, C.A.; Rocha, R.; Mitchell, G.F. Effect of Sacubitril-Valsartan vs Enalapril on Aortic Stiffness in Patients with Heart Failure and Reduced Ejection Fraction: A Randomized Clinical Trial. JAMA 2019, 322, 1077–1084. [Google Scholar] [CrossRef]
- Rezq, A.; Saad, M.; el Nozahi, M. Comparison of the Efficacy and Safety of Sacubitril/Valsartan versus Ramipril in Patients with ST-Segment Elevation Myocardial Infarction. Am. J. Cardiol. 2021, 143, 7–13. [Google Scholar] [CrossRef]
- Pathak, P. Angiotensin Receptor-Neprilysin Inhibitor in Acute Myocardial Infarction. J. Assoc. Physicians India. 2022, 70, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.M.; Claggett, B.; Prasad, N.; Li, G.; Volquez, M.; Jering, K.; Cikes, M.; Kovacs, A.; Mullens, W.; Nicolau, J.C.; et al. Impact of Sacubitril/Valsartan Compared with Ramipril on Cardiac Structure and Function After Acute Myocardial Infarction: The PARADISE-MI Echocardiographic Substudy. Circulation 2022, 146, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhu, H.; Zheng, Y.; Tan, X.; Tong, X. A Systematic Review and Meta-Analysis of Sacubitril-Valsartan in the Treatment of Ventricular Remodeling in Patients with Heart Failure after Acute Myocardial Infarction. Front. Cardiovasc. Med. 2022, 9, 953948. [Google Scholar] [CrossRef]
- Vargas Delgado, A.P.; Requena Ibañez, J.A.; Santos-Gallego, C.G.; Badimon, J.J. Are the Antidiabetic SGLT2 Inhibitors a Cardiovascular Treatment? Clin. E Investig. En Arterioscler. 2021, 33, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Singh, T.; Newby, D.E.; Singh, J. Sodium-Glucose Co-Transporter 2 Inhibitor Therapy: Mechanisms of Action in Heart Failure. Heart 2021, 107, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Osasan, S.; Sethia, S.; Batool, T.; Bambhroliya, Z.; Sandrugu, J.; Lowe, M.; Okunlola, O.; Hamid, P. A Systematic Review of Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors and Sympathetic Nervous System Inhibition: An Underrated Mechanism of Cardiorenal Protection. Cureus 2022, 14, e26313. [Google Scholar] [CrossRef] [PubMed]
- Aimo, A.; Gaggin, H.K.; Barison, A.; Emdin, M.; Januzzi, J.L. Imaging, Biomarker, and Clinical Predictors of Cardiac Remodeling in Heart Failure with Reduced Ejection Fraction. JACC Heart Fail. 2019, 7, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Kurczyński, D.; Hudzik, B.; Jagosz, M.; Zabierowski, J.; Nowak, J.; Tomasik, A.; Badziński, A.; Rozentryt, P.; Gąsior, M. Sodium-Glucose Cotransporter-2 Inhibitors-from the Treatment of Diabetes to Therapy of Chronic Heart Failure. J. Cardiovasc. Dev. Dis. 2022, 9, 225. [Google Scholar] [CrossRef]
- Verma, S.; Mcmurray, J.J.V. The Serendipitous Story of SGLT2 Inhibitors in Heart Failure: New Insights From DECLARE-TIMI 58. Circulation 2019, 139, 2537–2541. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Verma, S.; Mazer, C.D.; Yan, A.T.; Mason, T.; Garg, V.; Teoh, H.; Zuo, F.; Quan, A.; Farkouh, M.E.; Fitchett, D.H.; et al. Effect of Empagliflozin on Left Ventricular Mass in Patients with Type 2 Diabetes Mellitus and Coronary Artery Disease: The EMPA-HEART CardioLink-6 Randomized Clinical Trial. Circulation 2019, 140, 1693–1702. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Gitto, M.; Vrachatis, D.A.; Condorelli, G.; Papathanasiou, K.; Reimers, B.; Deftereos, S.; Stefanini, G.G. Potential Therapeutic Benefits of Sodium-Glucose Cotransporter 2 Inhibitors in the Context of Ischemic Heart Failure: A State-of-the-Art Review. Cardiovasc. Hematol. Agents Med. Chem. 2022, 20, 90–102. [Google Scholar] [CrossRef]
- Lee, M.M.Y.; Brooksbank, K.J.M.; Wetherall, K.; Mangion, K.; Roditi, G.; Campbell, R.T.; Berry, C.; Chong, V.; Coyle, L.; Docherty, K.F.; et al. Effect of Empagliflozin on Left Ventricular Volumes in Patients with Type 2 Diabetes, or Prediabetes, and Heart Failure with Reduced Ejection Fraction (SUGAR-DM-HF). Circulation 2021, 143, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Tentolouris, A.; Vlachakis, P.; Tzeravini, E.; Eleftheriadou, I.; Tentolouris, N. SGLT2 Inhibitors: A Review of Their Antidiabetic and Cardioprotective Effects. Int. J. Environ. Res. Public Health 2019, 16, 2965. [Google Scholar] [CrossRef] [Green Version]
- von Lewinski, D.; Kolesnik, E.; Tripolt, N.J.; Pferschy, P.N.; Benedikt, M.; Wallner, M.; Alber, H.; Berger, R.; Lichtenauer, M.; Saely, C.H.; et al. Empagliflozin in Acute Myocardial Infarction: The EMMY Trial. Eur. Heart. J. 2022, 43, 4421–4432. [Google Scholar] [CrossRef]
- Udell, J.A.; Jones, W.S.; Petrie, M.C.; Harrington, J.; Anker, S.D.; Bhatt, D.L.; Hernandez, A.F.; Butler, J. Sodium Glucose Cotransporter-2 Inhibition for Acute Myocardial Infarction: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2022, 79, 2058–2068. [Google Scholar] [CrossRef]
- Harrington, J.; Udell, J.A.; Jones, W.S.; Anker, S.D.; Bhatt, D.L.; Petrie, M.C.; Vedin, O.; Sumin, M.; Zwiener, I.; Hernandez, A.F.; et al. Empagliflozin in Patients Post Myocardial Infarction Rationale and Design of the EMPACT-MI Trial. Am. Heart J. 2022, 253, 86–98. [Google Scholar] [CrossRef]
- Nicholson, M.K.; Ghazal Asswad, R.; Wilding, J.P.H. Dapagliflozin for the Treatment of Type 2 Diabetes Mellitus–an Update. Expert Opin. Pharm. 2021, 22, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Kaplinsky, E.; Mallarkey, G. Cardiac Myosin Activators for Heart Failure Therapy: Focus on Omecamtiv Mecarbil. Drugs Context 2018, 7, 212518. [Google Scholar] [CrossRef] [PubMed]
- Tarone, G.; Balligand, J.L.; Bauersachs, J.; Clerk, A.; de Windt, L.; Heymans, S.; Hilfiker-Kleiner, D.; Hirsch, E.; Iaccarino, G.; Knöll, R.; et al. Targeting Myocardial Remodelling to Develop Novel Therapies for Heart Failure: A Position Paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur. J. Heart Fail. 2014, 16, 494–508. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, B.H.; Chou, W.; Saikali, K.G.; Escandón, R.; Lee, J.H.; Chen, M.M.; Treshkur, T.; Megreladze, I.; Wasserman, S.M.; Eisenberg, P.; et al. Safety and Tolerability of Omecamtiv Mecarbil During Exercise in Patients with Ischemic Cardiomyopathy and Angina. JACC Heart Fail. 2015, 3, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.H.; Nguyen, M.; Rodriguez, R.; Surani, S.; Udeani, G. Omecamtiv Mecarbil: A Novel Mechanistic and Therapeutic Approach to Chronic Heart Failure Management. Cureus 2021, 13, e12419. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.J.; Solomon, S.D.; Adams, K.F.; Cleland, J.G.; Ezekowitz, J.A.; Goudev, A.; Macdonald, P.; Metra, M.; et al. Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF): A phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet 2016, 388, 2895–2903. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.P. Omecamtiv Mecarbil: A Personalized Treatment for Patients with Severely Impaired Ejection Fraction. J. Am. Coll. Cardiol. 2021, 78, 109–111. [Google Scholar] [CrossRef]
- Felker, G.M.; Solomon, S.D.; McMurray, J.J.V.; Cleland, J.G.F.; Abbasi, S.A.; Malik, F.I.; Zhang, H.; Globe, G.; Teerlink, J.R. Effects of Omecamtiv Mecarbil on Symptoms and Health-Related Quality of Life in Patients with Chronic Heart Failure: Results From the COSMIC-HF Study. Circ. Heart Fail. 2020, 13, E007814. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sørensen, T.; et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. N. Engl. J. Med. 2021, 384, 105–116. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Biering-Sørensen, T.; Böhm, M.; Bonderman, D.; Fang, J.C.; et al. Effect of Ejection Fraction on Clinical Outcomes in Patients Treated with Omecamtiv Mecarbil in GALACTIC-HF. J. Am. Coll. Cardiol. 2021, 78, 97–108. [Google Scholar] [CrossRef]
- Lewis, G.D.; Voors, A.A.; Cohen-Solal, A.; Metra, M.; Whellan, D.J.; Ezekowitz, J.A.; Böhm, M.; Teerlink, J.R.; Docherty, K.F.; Lopes, R.D.; et al. Effect of Omecamtiv Mecarbil on Exercise Capacity in Chronic Heart Failure with Reduced Ejection Fraction: The METEORIC-HF Randomized Clinical Trial. JAMA 2022, 328, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Stroethoff, M.; Behmenburg, F.; Meierkord, S.; Bunte, S.; Mayer, F.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R. Cardioprotective Properties of Omecamtiv Mecarbil against Ischemia and Reperfusion Injury. J. Clin. Med. 2019, 8, 375. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, P.W.; Roessig, L.; Patel, M.J.; Anstrom, K.J.; Butler, J.; Voors, A.A.; Lam, C.S.P.; Ponikowski, P.; Temple, T.; Pieske, B.; et al. A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial of the Efficacy and Safety of the Oral Soluble Guanylate Cyclase Stimulator: The VICTORIA Trial. JACC Heart Fail. 2018, 6, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Vannuccini, F.; Campora, A.; Barilli, M.; Palazzuoli, A. Vericiguat in Heart Failure: Characteristics, Scientific Evidence and Potential Clinical Applications. Biomedicines 2022, 10, 2471. [Google Scholar] [CrossRef]
- González-Juanatey, J.R.; Anguita-Sánchez, M.; Bayes-Genís, A.; Comín-Colet, J.; García-Quintana, A.; Recio-Mayoral, A.; Zamorano-Gómez, J.L.; Cepeda-Rodrigo, J.M.; Manzano, L. Vericiguat in Heart Failure: From Scientific Evidence to Clinical Practice. Rev. Clínica Española 2022, 222, 359–369. [Google Scholar] [CrossRef]
- Kassis-George, H.; Verlinden, N.J.; Fu, S.; Kanwar, M. Vericiguat in Heart Failure with a Reduced Ejection Fraction: Patient Selection and Special Considerations. Ther. Clin. Risk Manag. 2022, 18, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Saldarriaga, C.; Atar, D.; Stebbins, A.; Lewis, B.S.; Abidin, I.Z.; Blaustein, R.O.; Butler, J.; Ezekowitz, J.A.; Hernandez, A.F.; Lam, C.S.P.; et al. Vericiguat in patients with coronary artery disease and heart failure with reduced ejection fraction. Eur. J. Heart Fail. 2022, 24, 782–790. [Google Scholar] [CrossRef]
- Mann, D.L.; Felker, G.M. Mechanisms and Models in Heart Failure: A Translational Approach. Circ. Res. 2021, 128, 1435–1450. [Google Scholar] [CrossRef]
- Methner, C.; Buonincontri, G.; Hu, C.I.; Vujic, A.; Kretschmer, A.; Sawiak, S.; Carpenter, A.; Stasch, J.P.; Krieg, T. Riociguat Reduces Infarct Size and Post-Infarct Heart Failure in Mouse Hearts: Insights from MRI/PET Imaging. PLoS ONE 2013, 8, e83910. [Google Scholar] [CrossRef]
- Sandner, P. From Molecules to Patients: Exploring the Therapeutic Role of Soluble Guanylate Cyclase Stimulators. Biol. Chem. 2018, 399, 679–690. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Galuppo, P.; Motschenbacher, S.; Ruetten, H.; Schäfer, A.; Bauersachs, J. Soluble Guanylyl Cyclase Activation Improves Progressive Cardiac Remodeling and Failure after Myocardial Infarction. Cardioprotection over ACE Inhibition. Basic Res. Cardiol. 2014, 109, 421. [Google Scholar] [CrossRef] [PubMed]
- Breitenstein, S.; Roessig, L.; Sandner, P.; Lewis, K.S. Novel SGC Stimulators and SGC Activators for the Treatment of Heart Failure. In Handbook of Experimental Pharmacology; Bauersachs, J., Butler, J., Sandner, P., Eds.; Springer: Cham, Switzerland, 2017; Volume 243, pp. 225–247. ISBN 978-3-319-59659-4. [Google Scholar]
- Tomassoni, G.; Baker, J.; Corbisiero, R.; Love, C.; Martin, D.; Sheppard, R.; Worley, S.J.; Lee, K.; Niazi, I. Rationale and design of a randomized trial to assess the safety and efficacy of MultiPoint Pacing (MPP) in cardiac resynchronization therapy: The MPP Trial. Ann. Noninvasive Electrocardiol. 2017, 22, e12448. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, C.; Bleeker, G.B.; Linde, C.; Donal, E.; Bax, J.J.; Schalij, M.J.; Daubert, C. Cardiac Resynchronization Therapy: Clinical Results and Evolution of Candidate Selection. Eur. Heart J. 2007, 9, I94–I106. [Google Scholar] [CrossRef] [Green Version]
- Sieniewicz, B.J.; Gould, J.; Porter, B.; Sidhu, B.S.; Teall, T.; Webb, J.; Carr-White, G.; Rinaldi, C.A. Understanding Non-Response to Cardiac Resynchronisation Therapy: Common Problems and Potential Solutions. Heart Fail. Rev. 2019, 24, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoshiki, H.; Mitsuyama, H.; Watanabe, M.; Mitsuhashi, T.; Shimizu, A. Cardiac Resynchronization Therapy in Ischemic and Non-Ischemic Cardiomyopathy. J. Arrhythm. 2017, 33, 410–416. [Google Scholar] [CrossRef]
- Cleland, J.G.; Daubert, J.C.; Erdmann, E.; Freemantle, N.; Gras, D.; Kappenberger, L.; Tavazzi, L. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N. Engl. J. Med. 2005, 352, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Abraham, W.T.; Kuck, K.H.; Goldsmith, R.L.; Lindenfeld, J.A.; Reddy, V.Y.; Carson, P.E.; Mann, D.L.; Saville, B.; Parise, H.; Chan, R.; et al. A Randomized Controlled Trial to Evaluate the Safety and Efficacy of Cardiac Contractility Modulation. JACC Heart Fail. 2018, 6, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Hall, W.J.; Cannom, D.S.; Klein, H.; Brown, M.W.; Daubert, J.P.; Estes, N.A.M.; Foster, E.; Greenberg, H.; Higgins, S.L.; et al. Cardiac-Resynchronization Therapy for the Prevention of Heart-Failure Events. N. Engl. J. Med. 2009, 361, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Barsheshet, A.; Goldenberg, I.; Moss, A.J.; Eldar, M.; Huang, D.T.; McNitt, S.; Klein, H.U.; Hall, W.J.; Brown, M.W.; Goldberger, J.J.; et al. Response to Preventive Cardiac Resynchronization Therapy in Patients with Ischaemic and Nonischaemic Cardiomyopathy in MADIT-CRT. Eur. Heart J. 2011, 32, 1622–1630. [Google Scholar] [CrossRef]
- Linde, C.; Abraham, W.T.; Gold, M.R.; Sutton, M.S.J.; Ghio, S.; Daubert, C. Randomized Trial of Cardiac Resynchronization in Mildly Symptomatic Heart Failure Patients and in Asymptomatic Patients with Left Ventricular Dysfunction and Previous Heart Failure Symptoms. J. Am. Coll. Cardiol. 2008, 52, 1834–1843. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, I.; Moss, A.J.; Jackson Hall, W.; Foster, E.; Goldberger, J.J.; Santucci, P.; Shinn, T.; Solomon, S.; Steinberg, J.S.; Wilber, D.; et al. Heart Failure Predictors of Response to Cardiac Resynchronization Therapy in the Multicenter Automatic Defibrillator Implantation Trial with Cardiac Resynchronization Therapy (MADIT-CRT). Circulation 2011, 124, 1527–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, S.; Marek, J.; Schwartzman, D.; Jain, S.; Adelstein, E.; White, P.; Oyenuga, O.A.; Onishi, T.; Soman, P.; Gorcsan, J. Echocardiography-Guided Left Ventricular Lead Placement for Cardiac Resynchronization Therapy Results of the Speckle Tracking Assisted Resynchronization Therapy for Electrode Region Trial. Circ. Heart Fail. 2013, 6, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Marsan, N.A.; Bleeker, G.B.; van Bommel, R.J.; Borleffs, J.W.; Bertini, M.; Holman, E.R.; van der Wall, E.E.; Schalij, M.J.; Bax, J.J. Cardiac Resynchronization Therapy in Patients with Ischemic versus Non-Ischemic Heart Failure: Differential Effect of Optimizing Interventricular Pacing Interval. Am. Heart J. 2009, 158, 769–776. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, E895–E1032. [Google Scholar] [CrossRef] [PubMed]
- Coats, A.J.S.; Abraham, W.T.; Zile, M.R.; Lindenfeld, J.A.; Weaver, F.A.; Fudim, M.; Bauersachs, J.; Duval, S.; Galle, E.; Zannad, F. Baroreflex Activation Therapy with the BarostimTM Device in Patients with Heart Failure with Reduced Ejection Fraction: A Patient Level Meta-Analysis of Randomized Controlled Trials. Eur. J. Heart Fail. 2022, 24, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Halbach, M.; Abraham, W.T.; Butter, C.; Ducharme, A.; Klug, D.; Little, W.C.; Reuter, H.; Schafer, J.E.; Senni, M.; Swarup, V.; et al. Baroreflex Activation Therapy for the Treatment of Heart Failure with Reduced Ejection Fraction in Patients with and without Coronary Artery Disease. Int. J. Cardiol. 2018, 266, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Zile, M.R.; Lindenfeld, J.A.; Weaver, F.A.; Zannad, F.; Galle, E.; Rogers, T.; Abraham, W.T. Baroreflex Activation Therapy in Patients with Heart Failure with Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2020, 76, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rao, I.V.; Burkhoff, D. Cardiac Contractility Modulation for the Treatment of Moderate to Severe HF. Expert Rev. Med. Devices 2021, 18, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Mando, R.; Goel, A.; Habash, F.; Saad, M.; Ayoub, K.; Vallurupalli, S.; Maskoun, W. Outcomes of Cardiac Contractility Modulation: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Cardiovasc. Ther. 2019, 2019, 9769724. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.A.; Nadarajah, R.; Ali, N.; Gierula, J.; Witte, K.K. Cardiac Contractility Modulation for the Treatment of Heart Failure with Reduced Ejection Fraction. Heart Fail. Rev. 2021, 26, 217–226. [Google Scholar] [CrossRef]
- Kadish, A.; Nademanee, K.; Volosin, K.; Krueger, S.; Neelagaru, S.; Raval, N.; Obel, O.; Weiner, S.; Wish, M.; Carson, P.; et al. A Randomized Controlled Trial Evaluating the Safety and Efficacy of Cardiac Contractility Modulation in Advanced Heart Failure. Am. Heart J. 2011, 161, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Fastner, C.; Yuecel, G.; Rudic, B.; Schmiel, G.; Toepel, M.; Burkhoff, D.; Liebe, V.; Kruska, M.; Hetjens, S.; Borggrefe, M.; et al. Cardiac Contractility Modulation in Patients with Ischemic versus Non-Ischemic Cardiomyopathy: Results from the MAINTAINED Observational Study. Int. J. Cardiol. 2021, 342, 49–55. [Google Scholar] [CrossRef]
- Zhang, Q.; Chan, Y.S.; Liang, Y.J.; Fang, F.; Lam, Y.Y.; Chan, C.P.; Lee, A.P.W.; Chan, K.C.Y.; Wu, E.B.; Yu, C.M. Comparison of Left Ventricular Reverse Remodeling Induced by Cardiac Contractility Modulation and Cardiac Resynchronization Therapy in Heart Failure Patients with Different QRS Durations. Int. J. Cardiol. 2013, 167, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.K.; Rhee, J.W.; Wu, J.C. Adult Stem Cell Therapy and Heart Failure, 2000 to 2016: A Systematic Review. JAMA Cardiol. 2016, 1, 831–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, K.U.; Guo, Y.; Li, Q.-H.; Cao, P.; Al-Maqtari, T.; Vajravelu, B.N.; Du, J.; Book, M.J.; Zhu, X.; Nong, Y.; et al. C-Kit+ Cardiac Stem Cells Alleviate Post-Myocardial Infarction Left Ventricular Dysfunction Despite Poor Engraftment and Negligible Retention in the Recipient Heart. PLoS ONE 2014, 9, e96725. [Google Scholar] [CrossRef] [Green Version]
- Bolli, R.; Mitrani, R.D.; Hare, J.M.; Pepine, C.J.; Perin, E.C.; Willerson, J.T.; Traverse, J.H.; Henry, T.D.; Yang, P.C.; Murphy, M.P.; et al. A Phase II Study of Autologous Mesenchymal Stromal Cells and C-kit Positive Cardiac Cells, Alone or in Combination, in Patients with Ischaemic Heart Failure: The CCTRN CONCERT-HF Trial. Eur. J. Heart Fail. 2021, 23, 661–674. [Google Scholar] [CrossRef]
- Cambria, E.; Pasqualini, F.S.; Wolint, P.; Günter, J.; Steiger, J.; Bopp, A.; Hoerstrup, S.P.; Emmert, M.Y. Translational Cardiac Stem Cell Therapy: Advancing from First-Generation to next-Generation Cell Types. NPJ Regen. Med. 2017, 2, 17. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Mobashery, S.; Chang, M. Roles of Matrix Metalloproteinases in Cutaneous Wound Healing. In Wound Healing—New Insights into Ancient Challenges; Alexandrescu, V., Ed.; Intechopen: London, UK, 2016. [Google Scholar] [CrossRef] [Green Version]
- Wollert, K.C.; Meyer, G.P.; Lotz, J.; Ringes Lichtenberg, S.; Lippolt, P.; Breidenbach, C.; Fichtner, S.; Korte, T.; Hornig, B.; Messinger, D.; et al. Intracoronary Autologous Bone-Marrow Cell Transfer after Myocardial Infarction: The BOOST Randomised Controlled Clinical Trial. Lancet 2004, 364, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.P.; Wollert, K.C.; Lotz, J.; Steffens, J.; Lippolt, P.; Fichtner, S.; Hecker, H.; Schaefer, A.; Arseniev, L.; Hertenstein, B.; et al. Intracoronary Bone Marrow Cell Transfer After Myocardial Infarction. Circulation 2006, 113, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Schächinger, V.; Erbs, S.; Elsässer, A.; Haberbosch, W.; Hambrecht, R.; Hölschermann, H.; Yu, J.; Corti, R.; Mathey, D.G.; Hamm, C.W.; et al. Intracoronary Bone Marrow–Derived Progenitor Cells in Acute Myocardial Infarction. N. Engl. J. Med. 2006, 355, 1210–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudry, F.; Hamshere, S.; Saunders, N.; Veerapen, J.; Bavnbek, K.; Knight, C.; Pellerin, D.; Locca, D.; Westwood, M.; Rakhit, R.; et al. A Randomized Double-Blind Control Study of Early Intra-Coronary Autologous Bone Marrow Cell Infusion in Acute Myocardial Infarction: The REGENERATE-AMI Clinical Trial. Eur. Heart J. 2016, 37, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Nicolau, J.C.; Furtado, R.H.M.; Silva, S.A.; Rochitte, C.E.; Rassi, A.; Moraes, J.B.M.C.; Quintella, E.; Costantini, C.R.; Korman, A.P.M.; Mattos, M.A.; et al. Stem-Cell Therapy in ST-Segment Elevation Myocardial Infarction with Reduced Ejection Fraction: A Multicenter, Double-Blind Randomized Trial. Clin. Cardiol. 2018, 41, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tendera, M.; Wojakowski, W.; Rużyłło, W.; Chojnowska, L.; Kępka, C.; Tracz, W.; Musiałek, P.; Piwowarska, W.; Nessler, J.; Buszman, P.; et al. Intracoronary Infusion of Bone Marrow-Derived Selected CD34+CXCR4+ Cells and Non-Selected Mononuclear Cells in Patients with Acute STEMI and Reduced Left Ventricular Ejection Fraction: Results of Randomized, Multicentre Myocardial Regeneration by Intracoronary Infusion of Selected Population of Stem Cells in Acute Myocardial Infarction (REGENT) Trial. Eur. Heart J. 2009, 30, 1313–1321. [Google Scholar] [CrossRef] [Green Version]
- Wollert, K.C.; Meyer, G.P.; Müller-Ehmsen, J.; Tschöpe, C.; Bonarjee, V.; Larsen, A.I.; May, A.E.; Empen, K.; Chorianopoulos, E.; Tebbe, U.; et al. Intracoronary Autologous Bone Marrow Cell Transfer after Myocardial Infarction: The BOOST-2 Randomised Placebo-Controlled Clinical Trial. Eur. Heart J. 2017, 38, 2936–2943. [Google Scholar] [CrossRef] [Green Version]
- Mathur, A.; Sim, D.S.; Choudry, F.; Veerapen, J.; Colicchia, M.; Turlejski, T.; Hussain, M.; Hamshere, S.; Locca, D.; Rakhit, R.; et al. Five-year Follow-up of Intracoronary Autologous Cell Therapy in Acute Myocardial Infarction: The REGENERATE-AMI Trial. ESC Heart Fail. 2022, 9, 1152–1159. [Google Scholar] [CrossRef]
- Fisher, S.A.; Zhang, H.; Doree, C.; Mathur, A.; Martin-Rendon, E. Stem Cell Treatment for Acute Myocardial Infarction. Cochrane Database Syst. Rev. 2015, 2015, CD006536. [Google Scholar] [CrossRef]
- Hosseinpour, A.; Kheshti, F.; Kazemi, A.; Attar, A. Comparing the Effect of Bone Marrow Mono-Nuclear Cells with Mesenchymal Stem Cells after Acute Myocardial Infarction on Improvement of Left Ventricular Function: A Meta-Analysis of Clinical Trials. Stem. Cell Res. Ther. 2022, 13, 203. [Google Scholar] [CrossRef]
- Attar, A.; Bahmanzadegan Jahromi, F.; Kavousi, S.; Monabati, A.; Kazemi, A. Mesenchymal Stem Cell Transplantation after Acute Myocardial Infarction: A Meta-Analysis of Clinical Trials. Stem. Cell Res. Ther. 2021, 12, 600. [Google Scholar] [CrossRef]
- Makkar, R.R.; Smith, R.R.; Cheng, K.; Malliaras, K.; Thomson, L.E.; Berman, D.; Czer, L.S.; Marbán, L.; Mendizabal, A.; Johnston, P.V.; et al. Intracoronary Cardiosphere-Derived Cells for Heart Regeneration after Myocardial Infarction (CADUCEUS): A Prospective, Randomised Phase 1 Trial. Lancet 2012, 379, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Makkar, R.R.; Kereiakes, D.J.; Aguirre, F.; Kowalchuk, G.; Chakravarty, T.; Malliaras, K.; Francis, G.S.; Povsic, T.J.; Schatz, R.; Traverse, J.H.; et al. Intracoronary ALLogeneic Heart STem Cells to Achieve Myocardial Regeneration (ALLSTAR): A Randomized, Placebo-Controlled, Double-Blinded Trial. Eur. Heart J. 2020, 41, 3451–3458. [Google Scholar] [CrossRef]
- Bolli, R.; Solankhi, M.; Tang, X.-L.; Kahlon, A. Cell Therapy in Patients with Heart Failure: A Comprehensive Review and Emerging Concepts. Cardiovasc. Res. 2022, 118, 951–976. [Google Scholar] [CrossRef]
- Perin, E.C.; Borow, K.M.; Henry, T.D.; Mendelsohn, F.O.; Miller, L.W.; Swiggum, E.; Adler, E.D.; Chang, D.H.; Fish, R.D.; Bouchard, A.; et al. Randomized Trial of Targeted Transendocardial Mesenchymal Precursor Cell Therapy in Patients with Heart Failure. J. Am. Coll. Cardiol. 2023, 81, 849–863. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leancă, S.A.; Afrăsânie, I.; Crișu, D.; Matei, I.T.; Duca, Ș.T.; Costache, A.D.; Onofrei, V.; Tudorancea, I.; Mitu, O.; Bădescu, M.C.; et al. Cardiac Reverse Remodeling in Ischemic Heart Disease with Novel Therapies for Heart Failure with Reduced Ejection Fraction. Life 2023, 13, 1000. https://doi.org/10.3390/life13041000
Leancă SA, Afrăsânie I, Crișu D, Matei IT, Duca ȘT, Costache AD, Onofrei V, Tudorancea I, Mitu O, Bădescu MC, et al. Cardiac Reverse Remodeling in Ischemic Heart Disease with Novel Therapies for Heart Failure with Reduced Ejection Fraction. Life. 2023; 13(4):1000. https://doi.org/10.3390/life13041000
Chicago/Turabian StyleLeancă, Sabina Andreea, Irina Afrăsânie, Daniela Crișu, Iulian Theodor Matei, Ștefania Teodora Duca, Alexandru Dan Costache, Viviana Onofrei, Ionuţ Tudorancea, Ovidiu Mitu, Minerva Codruța Bădescu, and et al. 2023. "Cardiac Reverse Remodeling in Ischemic Heart Disease with Novel Therapies for Heart Failure with Reduced Ejection Fraction" Life 13, no. 4: 1000. https://doi.org/10.3390/life13041000