
Perplexing Polyphenolics: The Isolations, Syntheses, Reappraisals, and Bioactivities of Flavonoids, Isoflavonoids, and Neoflavonoids from 2016 to 2022

Abstract
:1. Introduction
2. Isolation of Novel Flavonoids, Isoflavonoids, and Neoflavonoids
2.1. Source Analysis
2.2. Isolation Methods
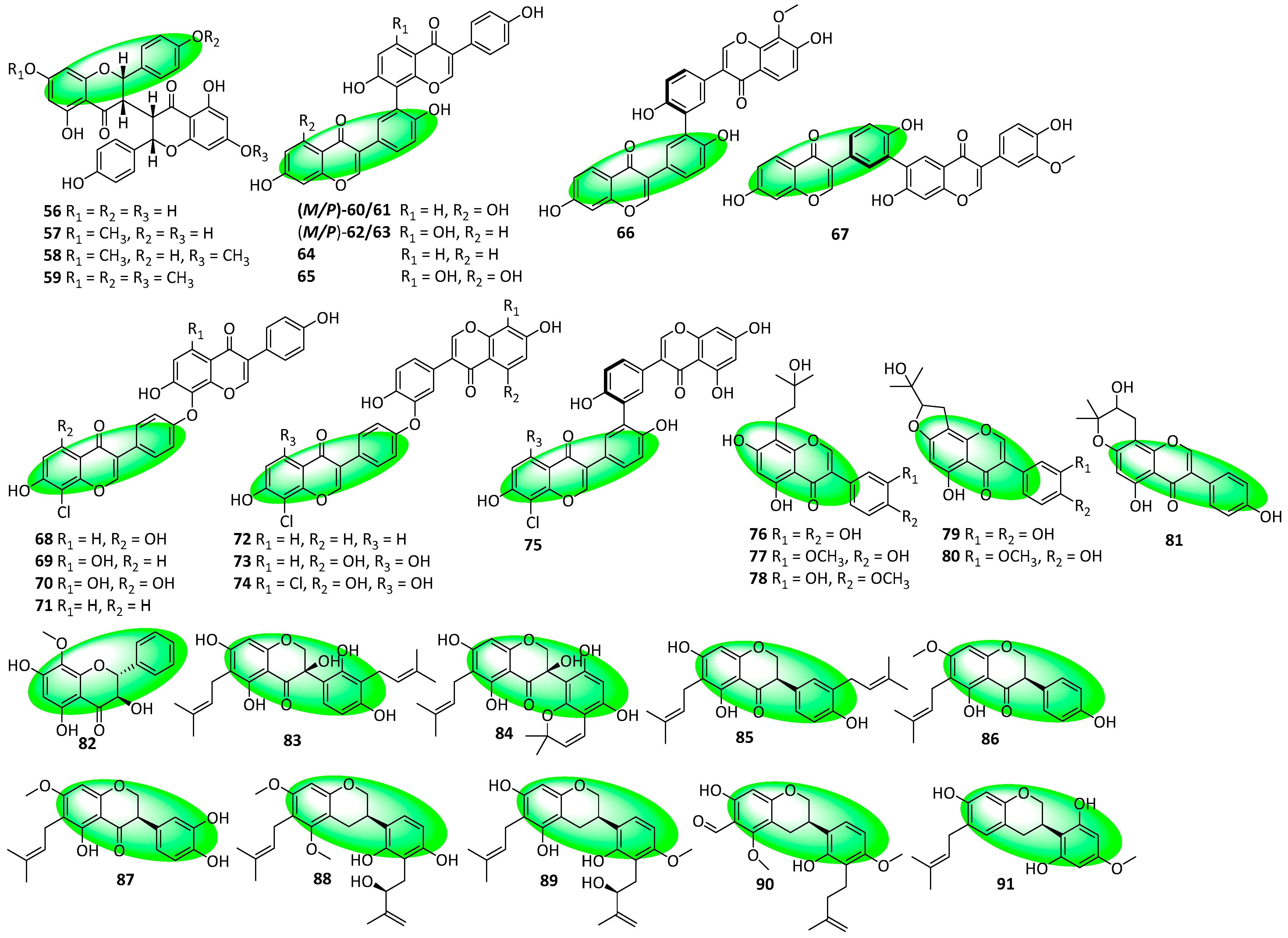
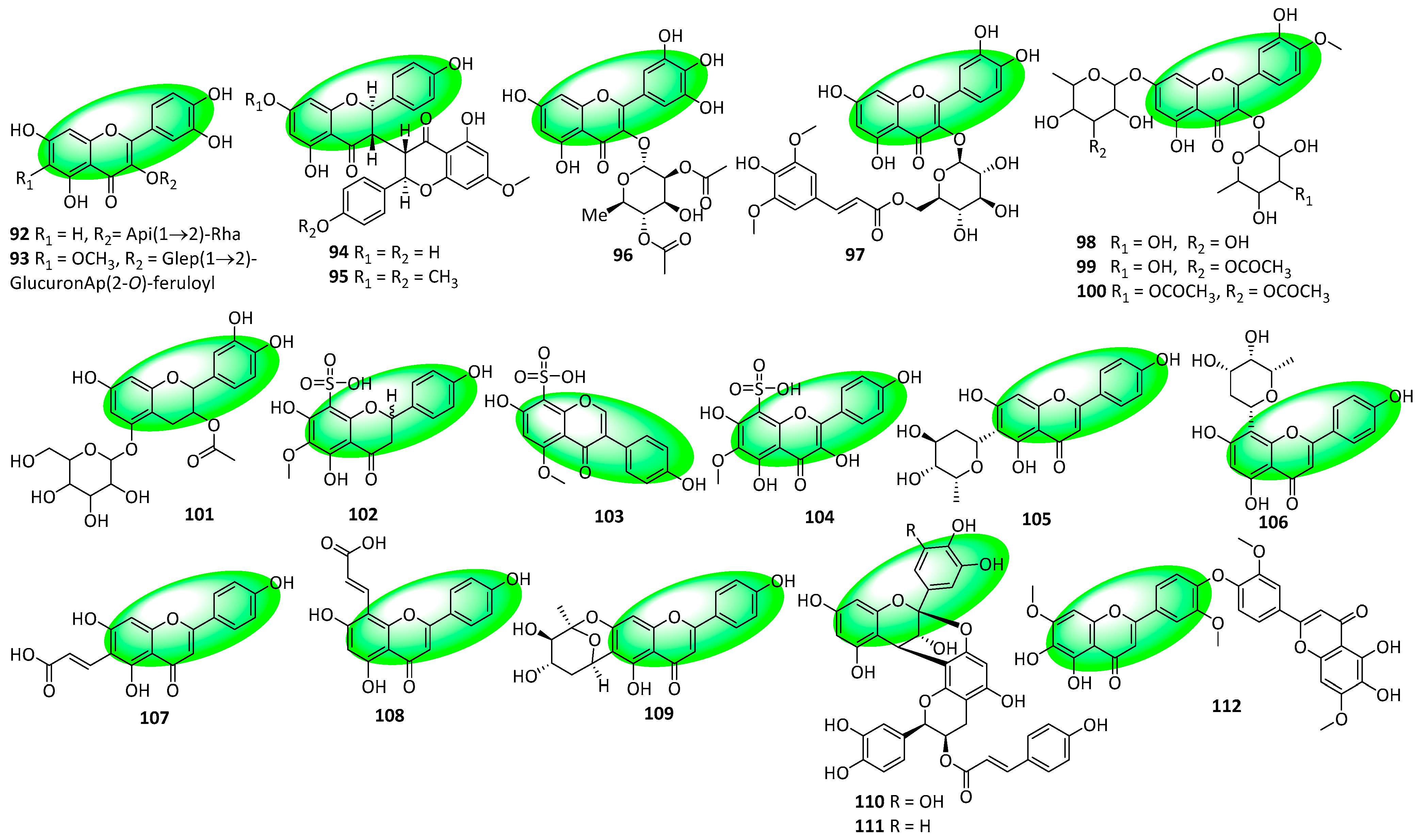
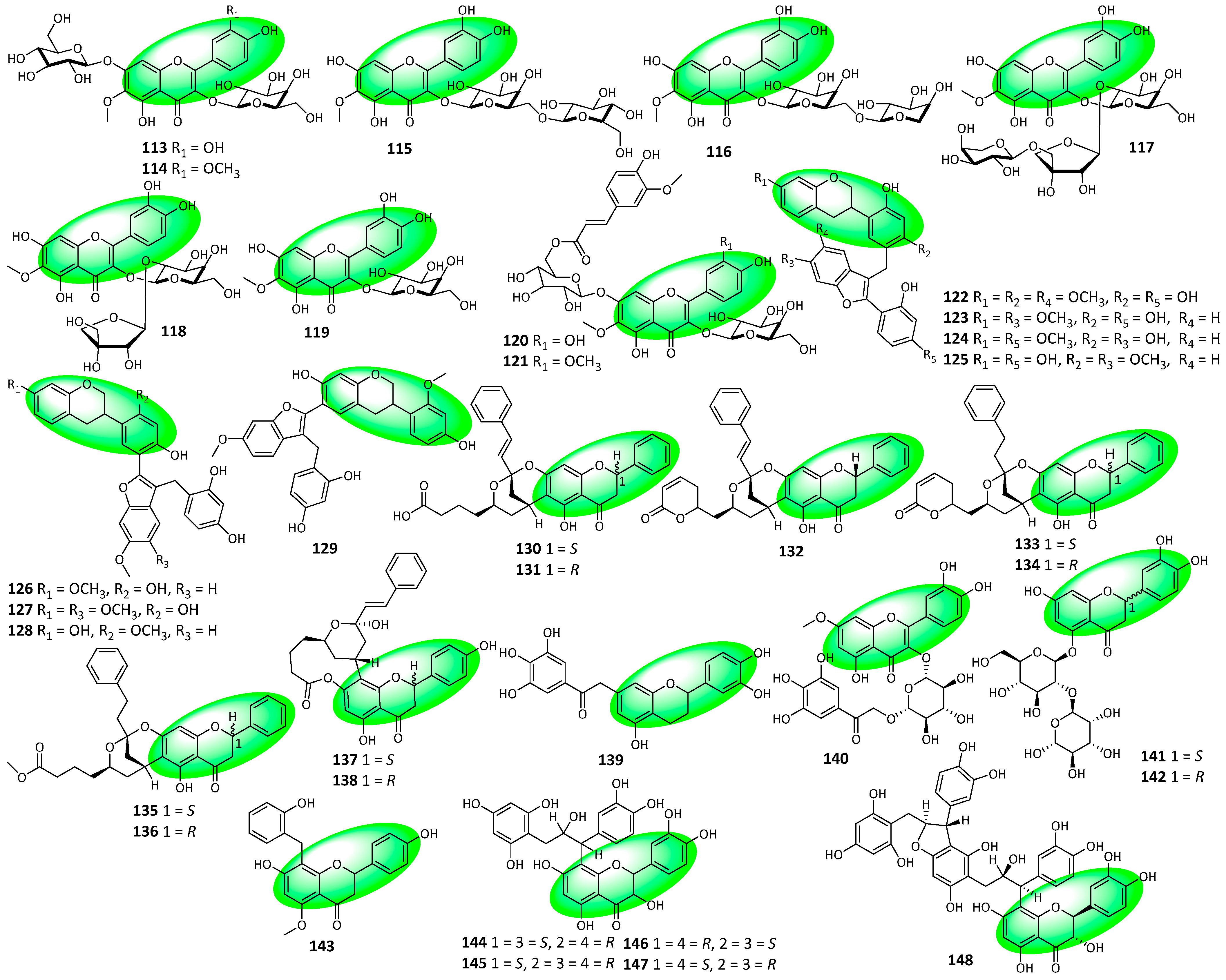
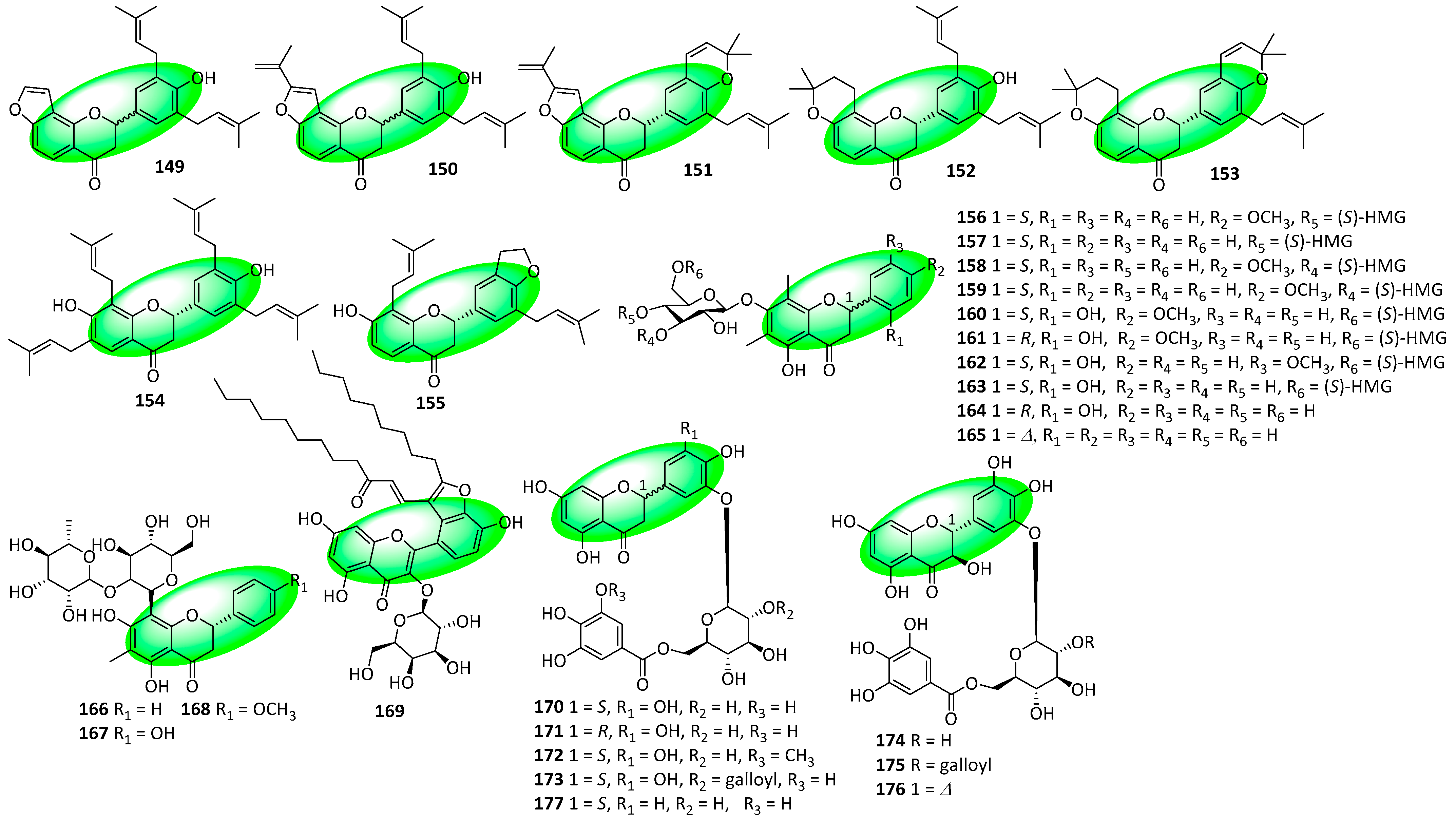
2.3. Structural Novelties
3. Synthesis of Flavonoids and Isoflavonoids
3.1. Total Synthesis
3.2. Semisynthesis
3.3. Synthesis of Derivatives
3.4. Synthetic Methods
3.4.1. Enzymes
3.4.2. Synthetic Methodology
3.5. Reappraisals
4. Bioactivities of Flavonoids, Isoflavonoids, and Neoflavonoids
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DIPEA | N,N-Diisopropylethylamine |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| mCPBA | meta-Chloroperoxybenzoic acid |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| DMSO | Dimethyl sulfoxide |
| PTT | Phenyltrimethylammonium tribromide |
| HSV-1 | Herpes simplex virus–1 |
| DMDO | Dimethyldioxirane |
| DIBAL-H | Diisobutylaluminium hydride |
| TMSI | Trimethylsilyl iodide |
| DMF-DMA | N,N-Dimethylformamide dimethyl acetal |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DMAP | 4-Dimethylaminopyridine |
References
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Dis. 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeresham, C. Natural products derived from plants as a source of drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200. [Google Scholar] [CrossRef] [PubMed]
- John, J.E. Natural products-based drug discovery: Some bottlenecks and considerations. Curr. Sci. 2009, 96, 753–754. [Google Scholar]
- Eddershaw, P.J.; Beresford, A.P.; Bayliss, M.K. ADME/PK as part of a rational approach to drug discovery. Drug Dis. 2000, 5, 409–414. [Google Scholar] [CrossRef]
- Appendino, G.; Fontana, G.; Pollastro, F. 3.08—Natural products drug discovery. Comprehensive Natural Products II Chemistry and Biology; Elsevier: Oxford, UK, 2010; pp. 205–236. [Google Scholar]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Weng, J.K.; Philippe, R.N.; Noel, J.P. The rise of chemodiversity in plants. Science 2012, 336, 1667–1670. [Google Scholar] [CrossRef] [Green Version]
- Mohr, J.T.; Krout, M.R.; Stoltz, B.M. Natural products as inspiration for the development of asymmetric catalysis. Nature 2008, 455, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Grigalunas, M.; Burhop, A.; Christoforow, A.; Waldmann, H. Pseudo-natural products and natural product-inspired methods in chemical biology and drug discovery. Curr. Opin. Chem. Biol. 2020, 56, 111–118. [Google Scholar] [CrossRef]
- Lahlou, M. The Success of Natural Products in Drug Discovery. Pharmacol. Pharm. 2013, 04, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Li, J.W.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, P. Harnessing Nature’s wisdom. Turning to Nature for inspiration and avoiding her follies. EMBO Rep. 2008, 9, 838–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, J.R. Natural Products: The Secondary Metabolites; Royal Society of Chemistry: London, UK, 2003; Volume 17. [Google Scholar]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouysegu, L. Plant polyphenols: Chemical properties, biological activities, and synthesis. Angew. Chem. Int. Ed. Engl. 2011, 50, 586–621. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E., Jr.; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [Green Version]
- Geetha, B.; Mahendran, S.; Subban, R.; Shrishailappa, B. Isolation and structural characterization of phytoconstituents from Strobilanthes kunthianus. Pharmacogn. J. 2020, 12. [Google Scholar]
- Nguyen, N.H.; Ta, Q.T.H.; Pham, Q.T.; Luong, T.N.H.; Phung, V.T.; Duong, T.H.; Vo, V.G. Anticancer Activity of Novel Plant Extracts and Compounds from Adenosma bracteosum (Bonati) in Human Lung and Liver Cancer Cells. Molecules 2020, 25, 2912. [Google Scholar] [CrossRef]
- Vestena, A.; Comerlato, L.; Bridi, H.; Guerini, L.; Ccana-Ccapatinta, G.V.; Vignoli-Silva, M.; Apel, M.A.; Fernandes, S.; Castro-Gamboa, I.; Zuanazzi, J.A.S. Chrysoeriol derivatives and other constituents from Glandularia selloi. Phytochem. Lett. 2019, 29, 30–34. [Google Scholar] [CrossRef]
- Waterman, M.J.; Nugraha, A.S.; Hendra, R.; Ball, G.E.; Robinson, S.A.; Keller, P.A. Antarctic Moss Biflavonoids Show High Antioxidant and Ultraviolet-Screening Activity. J. Nat. Prod. 2017, 80, 2224–2231. [Google Scholar] [CrossRef] [Green Version]
- Kamal, A.; Abdelhady, M.; Ben Hadda, T. Two Novel Flavone C-Glycosides Isolated From Afrocarpus Gracilior: Pom Analyses And In Vitro Cytotoxic Activity Aganist Hepatocellular Carcinoma. Int. J. Pharm. Pharm. Sci. 2019, 11, 57–62. [Google Scholar] [CrossRef]
- Kakar, M.; Amin, M.U.; Alghamdi, S.; Sahibzada, M.U.K.; Ahmad, N.; Ullah, N. Antimicrobial, cytotoxic, and antioxidant potential of a novel flavone “6, 7, 4′-Trimethyl Flavone” isolated from Wulfenia amherstiana. eCAM 2020, 2020, 3903682. [Google Scholar] [CrossRef]
- Yao, C.-Y.; Song, Z.-J.; Ruan, L.-J.; Yan, B.-X.; Wu, Q.-H.; He, L.-L.; Wu, Y.-Q.; Liu, X.-H.; Peng, Y.-D.; Miao, J.-H. A new methoxylated flavone from Lonicera hypoglauca and its chemotaxonomic significance. Biochem. Syst. Ecol. 2021, 97, 104279. [Google Scholar] [CrossRef]
- Lu, Y.; He, Y.; Zhu, S.; Zhong, X.; Chen, D.; Liu, Z. New acylglycosides flavones from fuzhuan brick tea and simulation analysis of their bioactive effects. Int. J. Mol. Sci. 2019, 20, 494. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kuang, X.D.; He, X.R.; Ren, G.; Wang, Y.; Xu, L.Y.; Feng, L.H.; Wang, B.; Zhou, Z.W. Prenylflavonoids from the Twigs of Artocarpus nigrifolius. Chem. Pharm. Bull. 2018, 66, 434–438. [Google Scholar] [CrossRef] [Green Version]
- You, C.X.; Zhang, K.; Li, X.; Liu, J.; Zhang, W.J.; Yu, X.X. Cytotoxic Flavonoids from the Leaves and Twigs of Murraya Tetramera. Molecules 2021, 26, 1284. [Google Scholar] [CrossRef]
- Li, J.; Tan, L.H.; Zou, H.; Zou, Z.X.; Long, H.P.; Wang, W.X.; Xu, P.S.; Liu, L.F.; Xu, K.P.; Tan, G.S. Palhinosides A-H: Flavone Glucosidic Truxinate Esters with Neuroprotective Activities from Palhinhaea cernua. J. Nat. Prod. 2020, 83, 216–222. [Google Scholar] [CrossRef]
- Seo, Y.H.; Kang, S.-Y.; Shin, J.-S.; Ryu, S.M.; Lee, A.Y.; Choi, G.; Moon, B.C.; Jang, D.-S.; Shim, S.H.; Lee, D. Chemical constituents from the aerial parts of Agastache rugosa and their inhibitory activities on prostaglandin E2 production in lipopolysaccharide-treated RAW 264.7 macrophages. J. Nat. Prod. 2019, 82, 3379–3385. [Google Scholar] [CrossRef]
- Rubin, D.; Sansom, C.E.; Lucas, N.T.; McAdam, C.J.; Simpson, J.; Lord, J.M.; Perry, N.B. O-Acylated Flavones in the Alpine Daisy Celmisia viscosa: Intraspecific Variation. J. Nat. Prod. 2022, 85, 1904–1911. [Google Scholar] [CrossRef]
- Seo, Y.H.; Trinh, T.A.; Ryu, S.M.; Kim, H.S.; Choi, G.; Moon, B.C.; Shim, S.H.; Jang, D.S.; Lee, D.; Kang, K.S. Chemical constituents from the aerial parts of Elsholtzia ciliata and their protective activities on glutamate-induced HT22 cell death. J. Nat. Prod. 2020, 83, 3149–3155. [Google Scholar] [CrossRef]
- Owor, R.O.; Bedane, K.G.; Zuhlke, S.; Derese, S.; Ong’amo, G.O.; Ndakala, A.; Spiteller, M. Anti-inflammatory Flavanones and Flavones from Tephrosia linearis. J. Nat. Prod. 2020, 83, 996–1004. [Google Scholar] [CrossRef]
- Qu, K.-J.; Wang, B.; Jiang, C.-S.; Xie, B.-G.; Liu, A.-H.; Li, S.-W.; Guo, Y.-W.; Li, J.; Mao, S.-C. Rearranged Diels–Alder Adducts and Prenylated Flavonoids as Potential PTP1B Inhibitors from Morus nigra. J. Nat. Prod. 2021, 84, 2303–2311. [Google Scholar] [CrossRef]
- Maeda, G.; Munissi, J.J.E.; Lindblad, S.; Duffy, S.; Pelletier, J.; Avery, V.M.; Nyandoro, S.S.; Erdelyi, M. A Meroisoprenoid, Heptenolides, and C-Benzylated Flavonoids from Sphaerocoryne gracilis ssp. gracilis. J. Nat. Prod. 2020, 83, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Ren, F.C.; Jiang, X.J.; Wen, S.Z.; Wang, L.X.; Li, X.M.; Wang, F. Prenylated 2-Phenoxychromones and Flavonoids from Epimedium brevicornum and Revised Structures of Epimedonins A and B. J. Nat. Prod. 2018, 81, 16–21. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, M.; Tang, M.H.; Xue, L.L.; Zhang, R.J.; Liu, L.; Ni, H.F.; Cai, X.Y.; Kuang, S.; Hong, F.; et al. Flavonoids with Inhibitory Effects on NLRP3 Inflammasome Activation from Millettia velutina. J. Nat. Prod. 2020, 83, 2950–2959. [Google Scholar] [CrossRef]
- Sugimoto, S.; Yamano, Y.; Desoukey, S.Y.; Katakawa, K.; Wanas, A.S.; Otsuka, H.; Matsunami, K. Isolation of Sesquiterpene-Amino Acid Conjugates, Onopornoids A-D, and a Flavonoid Glucoside from Onopordum alexandrinum. J. Nat. Prod. 2019, 82, 1471–1477. [Google Scholar] [CrossRef]
- Rocha, M.P.; Campana, P.R.V.; Padua, R.M.; Souza Filho, J.D.; Ferreira, D.; Braga, F.C. (3,3″)-Linked Biflavanones from Ouratea spectabilis and Their Effects on the Release of Proinflammatory Cytokines in THP-1 Cells. J. Nat. Prod. 2020, 83, 1891–1898. [Google Scholar] [CrossRef]
- Chang, Y.; Zhou, L.; Hou, X.; Zhu, T.; Pfeifer, B.A.; Li, D.; He, X.; Zhang, G.; Che, Q. Microbial Dimerization and Chlorination of Isoflavones by a Takla Makan Desert-Derived Streptomyces sp. HDN154127. J. Nat. Prod. 2023, 86, 34–44. [Google Scholar] [CrossRef]
- Cicek, S.S.; Galarza Perez, M.; Wenzel-Storjohann, A.; Bezerra, R.M.; Segovia, J.F.O.; Girreser, U.; Kanzaki, I.; Tasdemir, D. Antimicrobial Prenylated Isoflavones from the Leaves of the Amazonian Medicinal Plant Vatairea guianensis Aubl. J. Nat. Prod. 2022, 85, 927–935. [Google Scholar] [CrossRef]
- Ayoub, I.M.; Korinek, M.; Hwang, T.L.; Chen, B.H.; Chang, F.R.; El-Shazly, M.; Singab, A.N.B. Probing the Antiallergic and Anti-inflammatory Activity of Biflavonoids and Dihydroflavonols from Dietes bicolor. J. Nat. Prod. 2018, 81, 243–253. [Google Scholar] [CrossRef]
- Fan, J.-r.; Kuang, Y.; Dong, Z.-y.; Yi, Y.; Zhou, Y.-x.; Li, B.; Qiao, X.; Ye, M. Prenylated phenolic compounds from the aerial parts of Glycyrrhiza uralensis as PTP1B and α-glucosidase inhibitors. J. Nat. Prod. 2020, 83, 814–824. [Google Scholar] [CrossRef]
- Cho, H.-M.; Ha, T.-K.-Q.; Dang, L.-H.; Pham, H.-T.-T.; Tran, V.-O.; Huh, J.; An, J.-P.; Oh, W.-K. Prenylated phenolic compounds from the leaves of Sabia limoniacea and their antiviral activities against porcine epidemic diarrhea virus. J. Nat. Prod. 2019, 82, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, J.; Godevac, D.; Tesevic, V.; Dajic-Stevanovic, Z.; Ciric, A.; Sokovic, M.; Novakovic, M. Antibacterial and Antibiofilm Activity of Flavonoid and Saponin Derivatives from Atriplex tatarica against Pseudomonas aeruginosa. J. Nat. Prod. 2019, 82, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Kalenga, T.M.; Ndoile, M.M.; Atilaw, Y.; Gilissen, P.J.; Munissi, J.J.E.; Rudenko, A.; Bourgard, C.; Sunnerhagen, P.; Nyandoro, S.S.; Erdelyi, M. Biflavanones, Chalconoids, and Flavonoid Analogues from the Stem Bark of Ochna holstii. J. Nat. Prod. 2021, 84, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Kishore, N.; Twilley, D.; Blom van Staden, A.; Verma, P.; Singh, B.; Cardinali, G.; Kovacs, D.; Picardo, M.; Kumar, V.; Lall, N. Isolation of flavonoids and flavonoid glycosides from Myrsine africana and their inhibitory activities against mushroom tyrosinase. J. Nat. Prod. 2018, 81, 49–56. [Google Scholar] [CrossRef]
- Wu, Z.-G.; Wei, W.; Xu, H.-Y.; Zheng, L.-L.; Ma, C.-M.; Wang, Y.-C. Constituents from the leaves of Tetraena mongolica and their protective activity in HEK 293t cells damaged by CdCl2. J. Nat. Prod. 2019, 82, 2707–2712. [Google Scholar] [CrossRef]
- Norman, E.O.; Tuohey, H.; Pizzi, D.; Saidah, M.; Bell, R.; Brkljača, R.; White, J.M.; Gasser, R.B.; Taki, A.C.; Urban, S. Phytochemical Profiling and Biological Activity of the Australian Carnivorous Plant, Drosera magna. J. Nat. Prod. 2021, 84, 964–971. [Google Scholar] [CrossRef]
- Duong, T.H.; Beniddir, M.A.; Nguyen, V.K.; Aree, T.; Gallard, J.F.; Mac, D.H.; Nguyen, H.H.; Bui, X.H.; Boustie, J.; Nguyen, K.P.; et al. Sulfonic Acid-Containing Flavonoids from the Roots of Phyllanthus acidus. J. Nat. Prod. 2018, 81, 2026–2031. [Google Scholar] [CrossRef]
- Dias Silva, M.J.; Simonet, A.M.; Silva, N.C.; Dias, A.L.T.; Vilegas, W.; Macias, F.A. Bioassay-Guided Isolation of Fungistatic Compounds from Mimosa caesalpiniifolia Leaves. J. Nat. Prod. 2019, 82, 1496–1502. [Google Scholar] [CrossRef]
- Casanova, L.M.; Rodrigues, L.M.; de Aguiar, P.F.; Tinoco, L.W. An NMR-Based Chemometric Strategy to Identify Leishmania donovani Nucleoside Hydrolase Inhibitors from the Brazilian Tree Ormosia arborea. J. Nat. Prod. 2020, 83, 243–254. [Google Scholar] [CrossRef]
- Flores-Bocanegra, L.; Gonzalez-Andrade, M.; Bye, R.; Linares, E.; Mata, R. alpha-Glucosidase Inhibitors from Salvia circinata. J. Nat. Prod. 2017, 80, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, K.; Czerwinska, M.E.; Wilczek, M.; Strawa, J.; Tomczyk, M.; Granica, S. Anti-inflammatory Potential of Flavonoids from the Aerial Parts of Corispermum marschallii. J. Nat. Prod. 2018, 81, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Banzato, T.P.; Gubiani, J.R.; Bernardi, D.I.; Nogueira, C.R.; Monteiro, A.F.; Juliano, F.F.; de Alencar, S.M.; Pilli, R.A.; Lima, C.A.; Longato, G.B.; et al. Antiproliferative Flavanoid Dimers Isolated from Brazilian Red Propolis. J. Nat. Prod. 2020, 83, 1784–1793. [Google Scholar] [CrossRef]
- He, Q.; Li, S.; Fan, Y.; Liu, Y.; Su, Y.; Zhou, Z.; Zhang, Y.N.; Li, G.L.; Rao, L.; Zhang, C.R. Complex Flavanones from Cryptocarya metcalfiana and Structural Revision of Oboflavanone A. J. Nat. Prod. 2022, 85, 1617–1625. [Google Scholar] [CrossRef]
- Yu, J.S.; Park, M.; Pang, C.; Rashan, L.; Jung, W.H.; Kim, K.H. Antifungal Phenols from Woodfordia uniflora Collected in Oman. J. Nat. Prod. 2020, 83, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Danton, O.; Alexander, L.; Hunlun, C.; de Beer, D.; Hamburger, M.; Joubert, E. Bitter Taste Impact and Thermal Conversion of a Naringenin Glycoside from Cyclopia genistoides. J. Nat. Prod. 2018, 81, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Jaidee, W.; Andersen, R.J.; Chavez, M.A.G.; Wang, Y.A.; Patrick, B.O.; Pyne, S.G.; Muanprasat, C.; Borwornpinyo, S.; Laphookhieo, S. Amides and Flavonoids from the Fruit and Leaf Extracts of Melodorum siamensis. J. Nat. Prod. 2019, 82, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Alania, Y.; Reis, M.; Jing, S.X.; McAlpine, J.B.; Bedran-Russo, A.K.; Chen, S.N.; Ferreira, D.; Pauli, G.F. Seco B-Type Oligomers from Pinus massoniana Expand the Procyanidin Chemical Space and Exhibit Dental Bioactivity. J. Nat. Prod. 2022, 85, 2753–2768. [Google Scholar] [CrossRef]
- Ahn, J.; Kim, Y.-M.; Chae, H.-S.; Choi, Y.H.; Ahn, H.-C.; Yoo, H.; Kang, M.; Kim, J.; Chin, Y.-W. Prenylated flavonoids from the roots and rhizomes of Sophora tonkinensis and their effects on the expression of inflammatory mediators and proprotein convertase subtilisin/kexin type 9. J. Nat. Prod. 2019, 82, 309–317. [Google Scholar] [CrossRef]
- Huh, J.; Ha, T.K.Q.; Kang, K.B.; Kim, K.H.; Oh, W.K.; Kim, J.; Sung, S.H. C-methylated flavonoid glycosides from Pentarhizidium orientale rhizomes and their inhibitory effects on the H1N1 influenza virus. J. Nat. Prod. 2017, 80, 2818–2824. [Google Scholar] [CrossRef]
- Li, J.J.; Chen, G.D.; Fan, H.X.; Hu, D.; Zhou, Z.Q.; Lan, K.H.; Zhang, H.P.; Maeda, H.; Yao, X.S.; Gao, H. Houttuynoid M, an Anti-HSV Active Houttuynoid from Houttuynia cordata Featuring a Bis-houttuynin Chain Tethered to a Flavonoid Core. J. Nat. Prod. 2017, 80, 3010–3013. [Google Scholar] [CrossRef] [PubMed]
- Badral, D.; Odonbayar, B.; Murata, T.; Munkhjargal, T.; Tuvshintulga, B.; Igarashi, I.; Suganuma, K.; Inoue, N.; Brantner, A.H.; Odontuya, G.; et al. Flavonoid and Galloyl Glycosides Isolated from Saxifraga spinulosa and Their Antioxidative and Inhibitory Activities against Species That Cause Piroplasmosis. J. Nat. Prod. 2017, 80, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Campana, P.R.; Coleman, C.M.; Sousa, L.P.; Teixeira, M.M.; Ferreira, D.; Braga, F.C. Mansoins C-F, Oligomeric Flavonoid Glucosides Isolated from Mansoa hirsuta Fruits with Potential Anti-inflammatory Activity. J. Nat. Prod. 2016, 79, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.B.; Park, E.J.; Kim, J.; Sung, S.H. Berchemiosides A-C, 2-Acetoxy-omega-phenylpentaene Fatty Acid Triglycosides from the Unripe Fruits of Berchemia berchemiifolia. J. Nat. Prod. 2017, 80, 2778–2786. [Google Scholar] [CrossRef]
- Milella, L.; Milazzo, S.; De Leo, M.; Vera Saltos, M.B.; Faraone, I.; Tuccinardi, T.; Lapillo, M.; De Tommasi, N.; Braca, A. alpha-Glucosidase and alpha-Amylase Inhibitors from Arcytophyllum thymifolium. J. Nat. Prod. 2016, 79, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- Sendker, J.; Boker, I.; Lengers, I.; Brandt, S.; Jose, J.; Stark, T.; Hofmann, T.; Fink, C.; Abdel-Aziz, H.; Hensel, A. Phytochemical Characterization of Low Molecular Weight Constituents from Marshmallow Roots (Althaea officinalis) and Inhibiting Effects of the Aqueous Extract on Human Hyaluronidase-1. J. Nat. Prod. 2017, 80, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Li, Z.; Song, W.; Wang, Y.; Liang, W.; Li, K.; Tang, S.; Wang, Q.; Qiao, X.; Zhou, D.; et al. Bioactive Constituents of Glycyrrhiza uralensis (Licorice): Discovery of the Effective Components of a Traditional Herbal Medicine. J. Nat. Prod. 2016, 79, 281–292. [Google Scholar] [CrossRef]
- Kim, C.S.; Bae, M.; Oh, J.; Subedi, L.; Suh, W.S.; Choi, S.Z.; Son, M.W.; Kim, S.Y.; Choi, S.U.; Oh, D.C.; et al. Anti-Neurodegenerative Biflavonoid Glycosides from Impatiens balsamina. J. Nat. Prod. 2017, 80, 471–478. [Google Scholar] [CrossRef]
- Yue, S.J.; Qu, C.; Zhang, P.X.; Tang, Y.P.; Jin, Y.; Jiang, J.S.; Yang, Y.N.; Zhang, P.C.; Duan, J.A. Carthorquinosides A and B, Quinochalcone C-Glycosides with Diverse Dimeric Skeletons from Carthamus tinctorius. J. Nat. Prod. 2016, 79, 2644–2651. [Google Scholar] [CrossRef]
- Kuo, P.C.; Hung, H.Y.; Hwang, T.L.; Du, W.K.; Ku, H.C.; Lee, E.J.; Tai, S.H.; Chen, F.A.; Wu, T.S. Anti-inflammatory Flavan-3-ol-dihydroretrochalcones from Daemonorops draco. J. Nat. Prod. 2017, 80, 783–789. [Google Scholar] [CrossRef]
- Muharini, R.; Diaz, A.; Ebrahim, W.; Mandi, A.; Kurtan, T.; Rehberg, N.; Kalscheuer, R.; Hartmann, R.; Orfali, R.S.; Lin, W.; et al. Antibacterial and Cytotoxic Phenolic Metabolites from the Fruits of Amorpha fruticosa. J. Nat. Prod. 2017, 80, 169–180. [Google Scholar] [CrossRef]
- Hanakova, Z.; Hosek, J.; Kutil, Z.; Temml, V.; Landa, P.; Vanek, T.; Schuster, D.; Dall’Acqua, S.; Cvacka, J.; Polansky, O.; et al. Anti-inflammatory Activity of Natural Geranylated Flavonoids: Cyclooxygenase and Lipoxygenase Inhibitory Properties and Proteomic Analysis. J. Nat. Prod. 2017, 80, 999–1006. [Google Scholar] [CrossRef]
- Ma, G.L.; Xiong, J.; Yang, G.X.; Pan, L.L.; Hu, C.L.; Wang, W.; Fan, H.; Zhao, Q.H.; Zhang, H.Y.; Hu, J.F. Biginkgosides A-I, Unexpected Minor Dimeric Flavonol Diglycosidic Truxinate and Truxillate Esters from Ginkgo biloba Leaves and Their Antineuroinflammatory and Neuroprotective Activities. J. Nat. Prod. 2016, 79, 1354–1364. [Google Scholar] [CrossRef]
- Kang, K.B.; Kim, H.W.; Kim, J.W.; Oh, W.K.; Kim, J.; Sung, S.H. Catechin-Bound Ceanothane-Type Triterpenoid Derivatives from the Roots of Zizyphus jujuba. J. Nat. Prod. 2017, 80, 1048–1054. [Google Scholar] [CrossRef]
- Odonbayar, B.; Murata, T.; Batkhuu, J.; Yasunaga, K.; Goto, R.; Sasaki, K. Antioxidant Flavonols and Phenolic Compounds from Atraphaxis frutescens and Their Inhibitory Activities against Insect Phenoloxidase and Mushroom Tyrosinase. J. Nat. Prod. 2016, 79, 3065–3071. [Google Scholar] [CrossRef]
- Rak Lee, S.; Schalk, F.; Schwitalla, J.W.; Benndorf, R.; Vollmers, J.; Kaster, A.K.; de Beer, Z.W.; Park, M.; Ahn, M.J.; Jung, W.H.; et al. Polyhalogenation of Isoflavonoids by the Termite-Associated Actinomadura sp. RB99. J. Nat. Prod. 2020, 83, 3102–3110. [Google Scholar] [CrossRef]
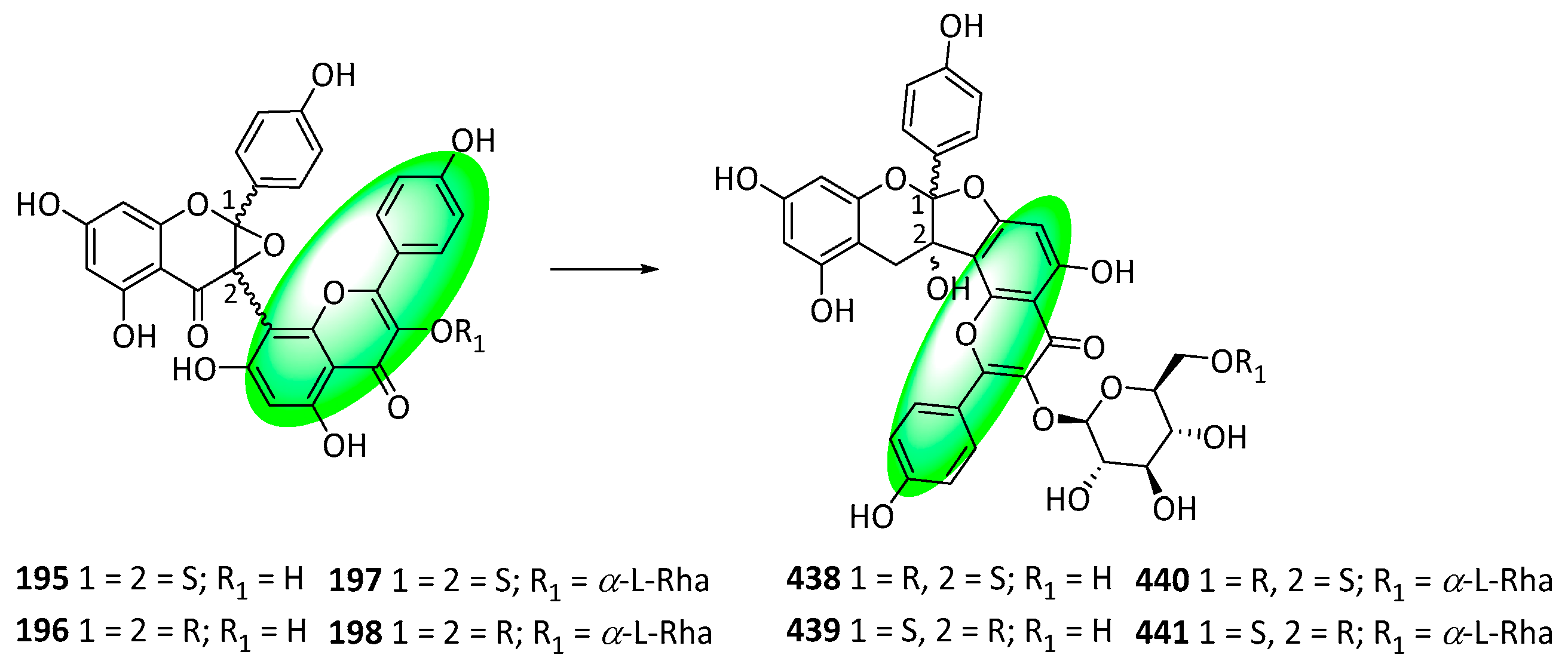
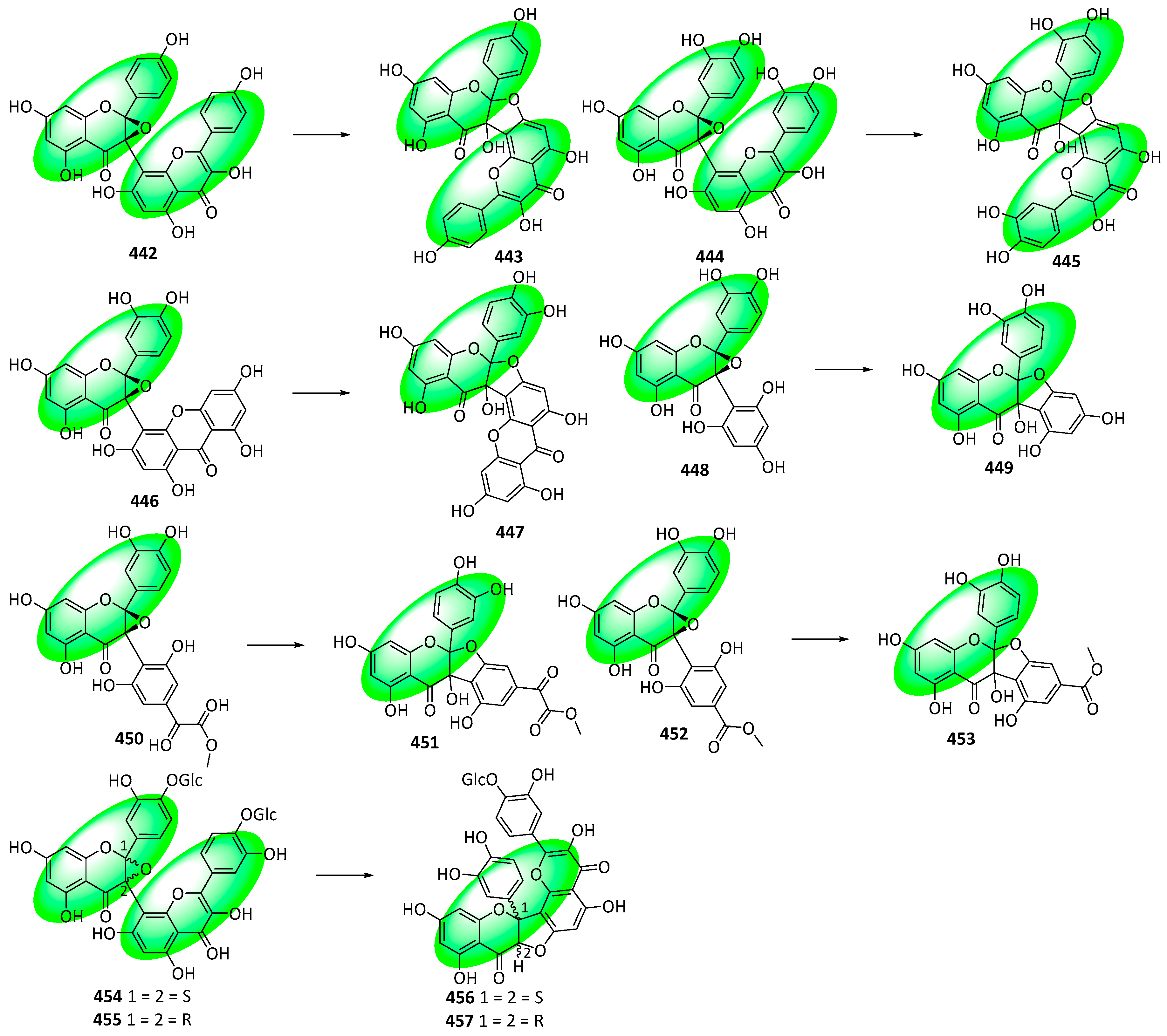
- Lee, T.H.; Ham, S.L.; Lee, D.Y.; Lee, J.R.; Kim, J.; Kim, C.S. Structure Revision of Balsamisides A-D and Establishment of an Empirical Rule for Distinguishing Four Classes of Biflavonoids. J. Nat. Prod. 2022, 85, 2461–2467. [Google Scholar] [CrossRef]
- Dong, H.; Wu, M.; Xiang, S.; Song, T.; Li, Y.; Long, B.; Feng, C.; Shi, Z. Total Syntheses and Antibacterial Evaluations of Neocyclomorusin and Related Flavones. J. Nat. Prod. 2022, 85, 2217–2225. [Google Scholar] [CrossRef]
- Tanaka, H.; Etoh, H.; Watanabe, N.; Shimizu, H.; Ahmad, M.; Rizwani, G.H. Erysubins C-F, four isoflavonoids from Erythrina suberosa var. glabrescences. Phytochemistry 2001, 56, 769–773. [Google Scholar] [CrossRef]
- Rukachaisirikul, T.; Saekee, A.; Tharibun, C.; Watkuolham, S.; Suksamrarn, A. Biological activities of the chemical constituents of Erythrina stricta and Erythrina subumbrans. Arch. Pharm. Res. 2007, 30, 1398–1403. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Sharma, G.; Dao, T.T.; Uddin, M.N.; Kang, K.W.; Ndinteh, D.T.; Mbafor, J.T.; Oh, W.K. New prenylated isoflavonoids as protein tyrosine phosphatase 1B (PTP1B) inhibitors from Erythrina addisoniae. Bioorg. Med. Chem. 2012, 20, 6459–6464. [Google Scholar] [CrossRef]
- Kwesiga, G.; Kelling, A.; Kersting, S.; Sperlich, E.; von Nickisch-Rosenegk, M.; Schmidt, B. Total Syntheses of Prenylated Isoflavones from Erythrina sacleuxii and Their Antibacterial Activity: 5-Deoxy-3′-prenylbiochanin A and Erysubin F. J. Nat. Prod. 2020, 83, 3445–3453. [Google Scholar] [CrossRef]
- Ahmad, G.; Yadav, P.P.; Maurya, R. Furanoflavonoid glycosides from Pongamia pinnata fruits. Phytochemistry 2004, 65, 921–924. [Google Scholar] [CrossRef]
- Guo, H.; Bai, Z.; Xu, Y.; Wu, X.; Li, N.; Zhu, Y.; Wang, X.; Zhang, P. Anti-inflammation compounds from the seedpods of Pongamia pinnata (L.) Pierre guided by the bioactivity and UPLC-HRESIMS. Arch. Pharmacal Res. 2017, 40, 818–824. [Google Scholar] [CrossRef]
- Wen, R.; Lv, H.N.; Jiang, Y.; Tu, P.F. Anti-inflammatory flavone and chalcone derivatives from the roots of Pongamia pinnata (L.) Pierre. Phytochemistry 2018, 149, 56–63. [Google Scholar] [CrossRef]
- Dong, H.; Wu, M.; Li, Y.; Lu, L.; Qin, J.; He, Y.; Shi, Z. Total Syntheses and Anti-inflammatory Evaluations of Pongamosides A-C, Natural Furanoflavonoid Glucosides from Fruit of Pongamia pinnata (L.) Pierre. J. Nat. Prod. 2022, 85, 1118–1127. [Google Scholar] [CrossRef]
- Chen, S.D.; Gao, H.; Zhu, Q.C.; Wang, Y.Q.; Li, T.; Mu, Z.Q.; Wu, H.L.; Peng, T.; Yao, X.S. Houttuynoids A-E, anti-herpes simplex virus active flavonoids with novel skeletons from Houttuynia cordata. Org. Lett. 2012, 14, 1772–1775. [Google Scholar] [CrossRef]
- Jian, J.; Fan, J.; Yang, H.; Lan, P.; Li, M.; Liu, P.; Gao, H.; Sun, P. Total Synthesis of the Flavonoid Natural Product Houttuynoid A. J. Nat. Prod. 2018, 81, 371–377. [Google Scholar] [CrossRef]
- Prachyawarakorn, V.; Mahidol, C.; Ruchirawat, S. Longeracemosones A–F, aromatase inhibitors from Dunbaria longeracemosa. Eur. J. Org. Chem. 2011, 2011, 3803–3808. [Google Scholar] [CrossRef]
- Kim, E.S.; Jang, H.; Chang, S.Y.; Baek, S.H.; Bae, O.N.; Kim, H. Total Synthesis and Biological Evaluation of Sericetin for Protection against Cisplatin-Induced Acute Kidney Injury. J. Nat. Prod. 2018, 81, 2647–2653. [Google Scholar] [CrossRef]
- Yokosuka, A.; Haraguchi, M.; Usui, T.; Kazami, S.; Osada, H.; Yamori, T.; Mimaki, Y. Glaziovianin A, a new isoflavone, from the leaves of Ateleia glazioviana and its cytotoxic activity against human cancer cells. Bioorg. Med. Chem. Lett. 2007, 17, 3091–3094. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, I.; Shioda, S.; Chinen, T.; Hatanaka, T.; Ebisu, H.; Sakakura, A.; Usui, T.; Kigoshi, H. Discovery of O6-benzyl glaziovianin A, a potent cytotoxic substance and a potent inhibitor of α, β-tubulin polymerization. Bioorg. Med. Chem. 2016, 24, 5639–5645. [Google Scholar] [CrossRef] [Green Version]
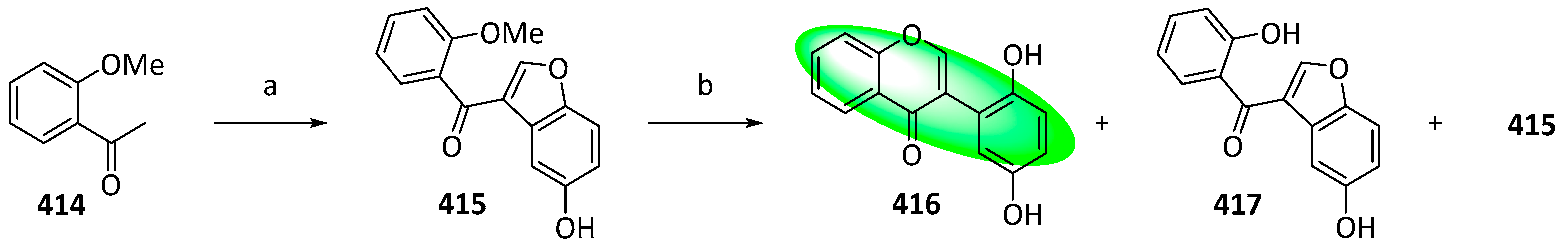
- Kunyane, P.; Sonopo, M.S.; Selepe, M.A. Synthesis of Isoflavones by Tandem Demethylation and Ring-Opening/Cyclization of Methoxybenzoylbenzofurans. J. Nat. Prod. 2019, 82, 3074–3082. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-L.; Chen, Y.-L.; Kuo, Y.-H. Three new flavonoids, 3′-methoxylupinifolin, laxifolin, and isolaxifolin from the roots of Derris laxiflora Benth. Chem. Pharm. Bull. 1991, 39, 3132–3135. [Google Scholar] [CrossRef] [Green Version]
- Sreelatha, T.; Hymavathi, A.; Rama Subba Rao, V.; Devanand, P.; Usha Rani, P.; Madhusudana Rao, J.; Suresh Babu, K. A new benzil derivative from Derris scandens: Structure-insecticidal activity study. Bioorg. Med. Chem. Lett. 2010, 20, 549–553. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, W.N.; Hu, N.; Banwell, M.G.; Ma, C.; Gardiner, M.G.; Lan, P. Cytotoxicity and Anti-inflammatory Properties of Apigenin-Derived Isolaxifolin. J. Nat. Prod. 2019, 82, 2451–2459. [Google Scholar] [CrossRef]
- Thakur, K.; Zhang, Y.-Y.; Mocan, A.; Zhang, F.; Zhang, J.-G.; Wei, Z.-J. 1-Deoxynojirimycin, its potential for management of non-communicable metabolic diseases. Trends Food Sci. Technol. 2019, 89, 88–99. [Google Scholar] [CrossRef]
- Shahana, S.; Nikalje, A.P.G. Phytochemistry and bioactivity of (Mulberry) plant: A comprehensive Morus alba. Asian J. Pharm. Pharmacol. 2019, 5, 207–217. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Y.; Xin, X.; Huang, G.; Zhang, N.; Zeng, Q.; Tang, L.; Attaribo, T.; Lee, K.S.; Jin, B.R.; et al. Dual-Targeting Antiproliferation Hybrids Derived from 1-Deoxynojirimycin and Kaempferol Induce MCF-7 Cell Apoptosis through the Mitochondria-Mediated Pathway. J. Nat. Prod. 2021, 84, 1534–1543. [Google Scholar] [CrossRef]
- Gazak, R.; Walterova, D.; Kren, V. Silybin and silymarin--new and emerging applications in medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [CrossRef]
- Džubák, P.; Hajdúch, M.; Gažák, R.; Svobodová, A.; Psotová, J.; Walterová, D.; Sedmera, P.; Křen, V. New derivatives of silybin and 2, 3-dehydrosilybin and their cytotoxic and P-glycoprotein modulatory activity. Bioorg. Med. Chem. 2006, 14, 3793–3810. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Lin, W.W.; Chen, H.L.; Chang, Y.H.; Ou, H.C.; Kuo, J.S.; Hong, J.S.; Jeng, K.C. Silymarin protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity by inhibiting microglia activation. Eur. J. Neurosci. 2002, 16, 2103–2112. [Google Scholar] [CrossRef] [PubMed]
- Thongphasuk, P.; Stremmel, W.; Chamulitrat, W. Potent direct or TNF-alpha-promoted anticancer effects of 2,3-dehydrosilybin: Comparison study with silybin. Chemotherapy 2008, 54, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Thongphasuk, P.; Erben, G.; Lehmann, W.-D.; Tuma, S.; Stremmel, W.; Chamulitrat, W. Significantly greater antioxidant anticancer activities of 2, 3-dehydrosilybin than silybin. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 837–847. [Google Scholar] [CrossRef]
- Gazak, R.; Valentova, K.; Fuksova, K.; Marhol, P.; Kuzma, M.; Medina, M.A.; Oborna, I.; Ulrichova, J.; Kren, V. Synthesis and antiangiogenic activity of new silybin galloyl esters. J. Med. Chem. 2011, 54, 7397–7407. [Google Scholar] [CrossRef]
- Karas, D.; Gazak, R.; Valentova, K.; Chambers, C.S.; Pivodova, V.; Biedermann, D.; Krenkova, A.; Oborna, I.; Kuzma, M.; Cvacka, J.; et al. Effects of 2,3-Dehydrosilybin and Its Galloyl Ester and Methyl Ether Derivatives on Human Umbilical Vein Endothelial Cells. J. Nat. Prod. 2016, 79, 812–820. [Google Scholar] [CrossRef]
- Yan, Y.; Mo, T.; Huang, W.; Xu, X.; Tian, W.; Wang, Y.; Song, Y.; Li, J.; Shi, S.; Liu, X.; et al. Glycosylation of Aromatic Glycosides by a Promiscuous Glycosyltransferase UGT71BD1 from Cistanche tubulosa. J. Nat. Prod. 2022, 85, 1826–1836. [Google Scholar] [CrossRef]
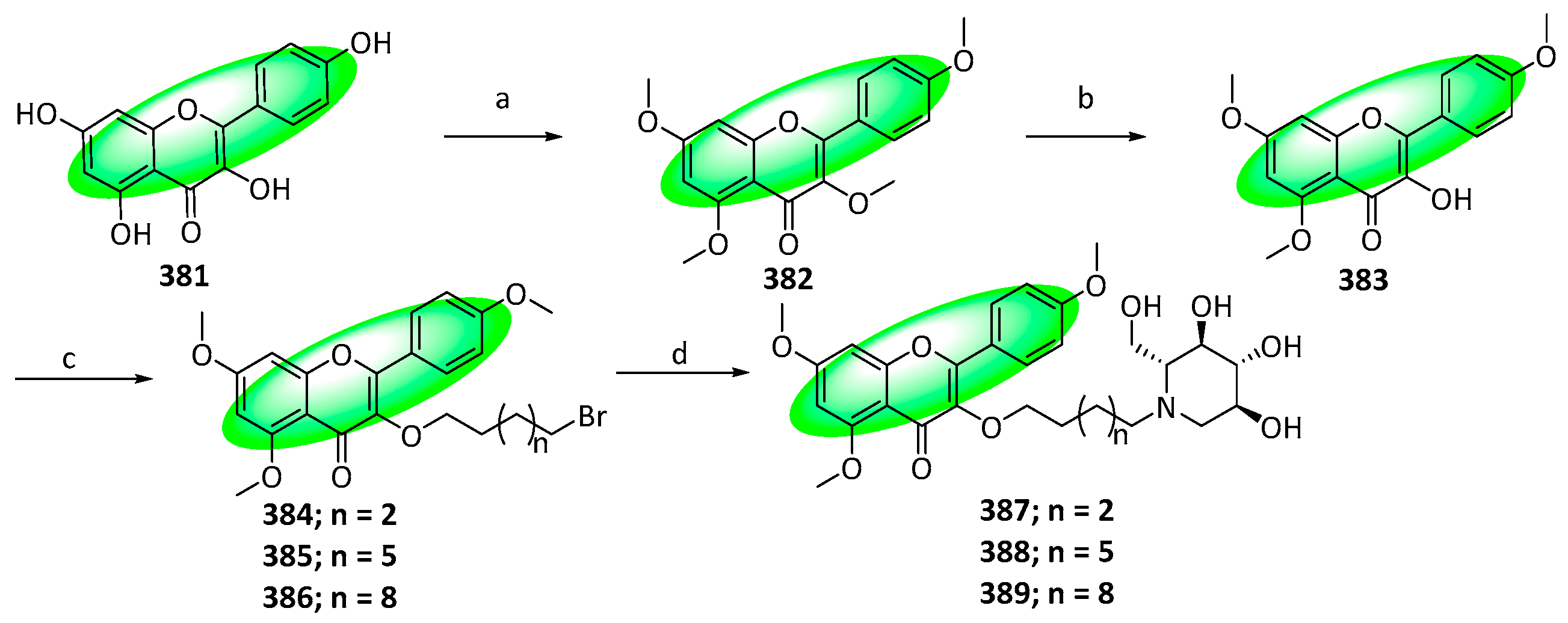
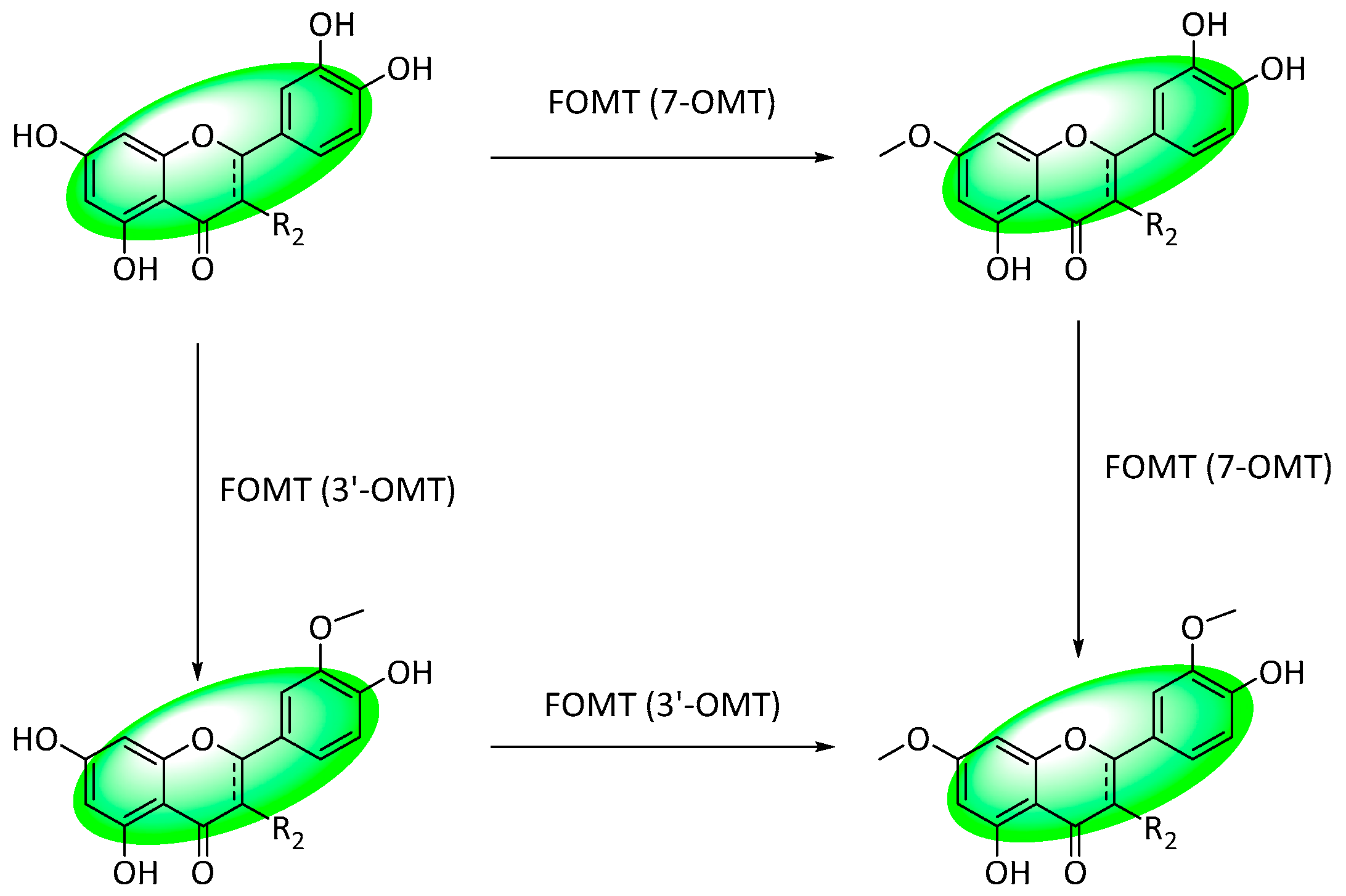
- Lee, D.; Park, H.L.; Lee, S.W.; Bhoo, S.H.; Cho, M.H. Biotechnological Production of Dimethoxyflavonoids Using a Fusion Flavonoid O-Methyltransferase Possessing Both 3′- and 7-O-Methyltransferase Activities. J. Nat. Prod. 2017, 80, 1467–1474. [Google Scholar] [CrossRef]
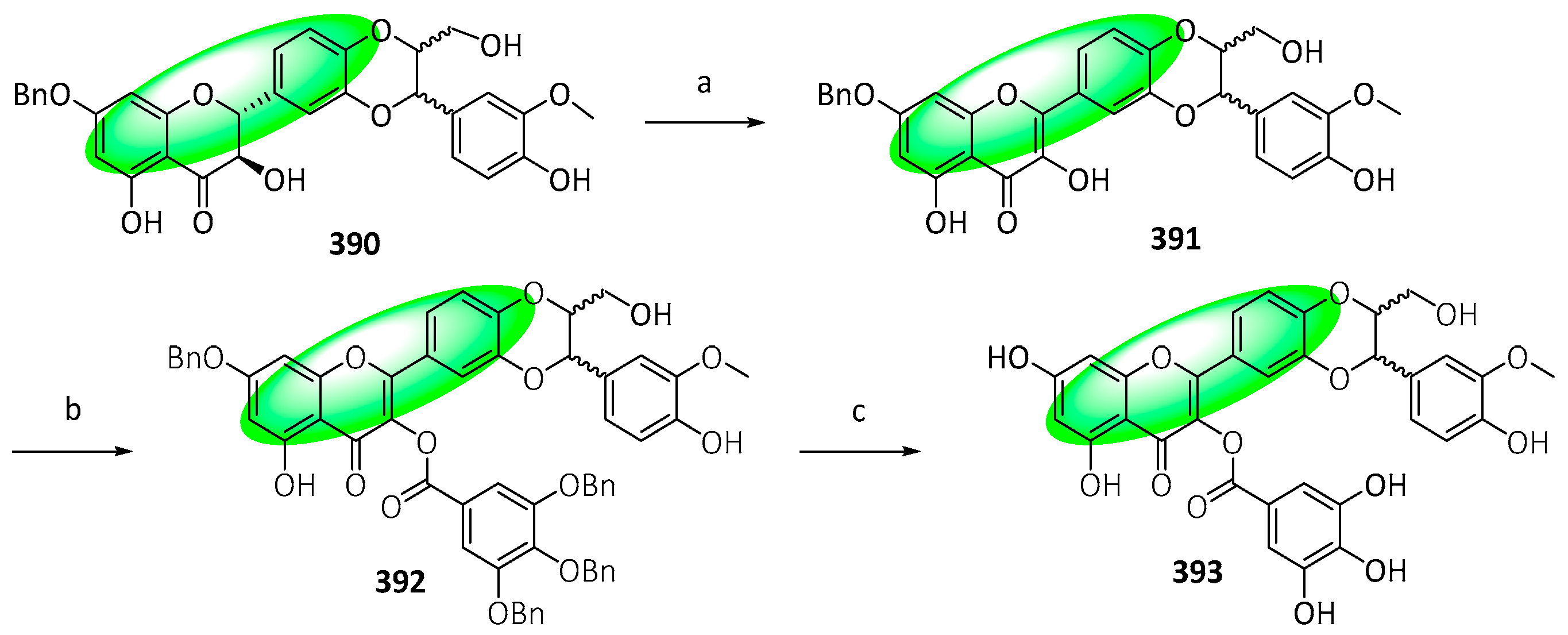
- Pilkington, L.I.; Wagoner, J.; Kline, T.; Polyak, S.J.; Barker, D. 1,4-Benzodioxane Lignans: An Efficient, Asymmetric Synthesis of Flavonolignans and Study of Neolignan Cytotoxicity and Antiviral Profiles. J. Nat. Prod. 2018, 81, 2630–2637. [Google Scholar] [CrossRef]
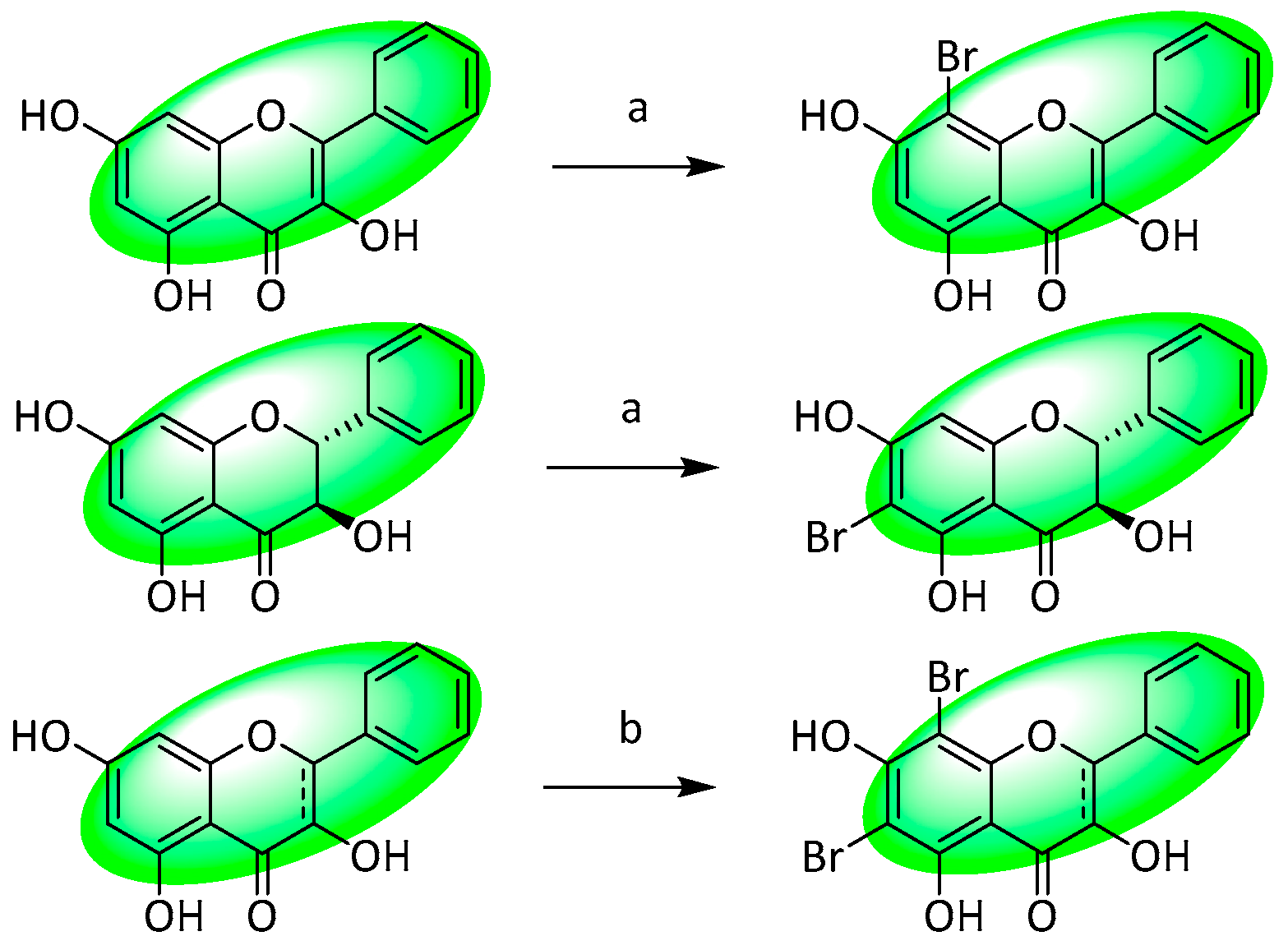
- Hurtova, M.; Biedermann, D.; Kuzma, M.; Kren, V. Mild and Selective Method of Bromination of Flavonoids. J. Nat. Prod. 2020, 83, 3324–3331. [Google Scholar] [CrossRef]
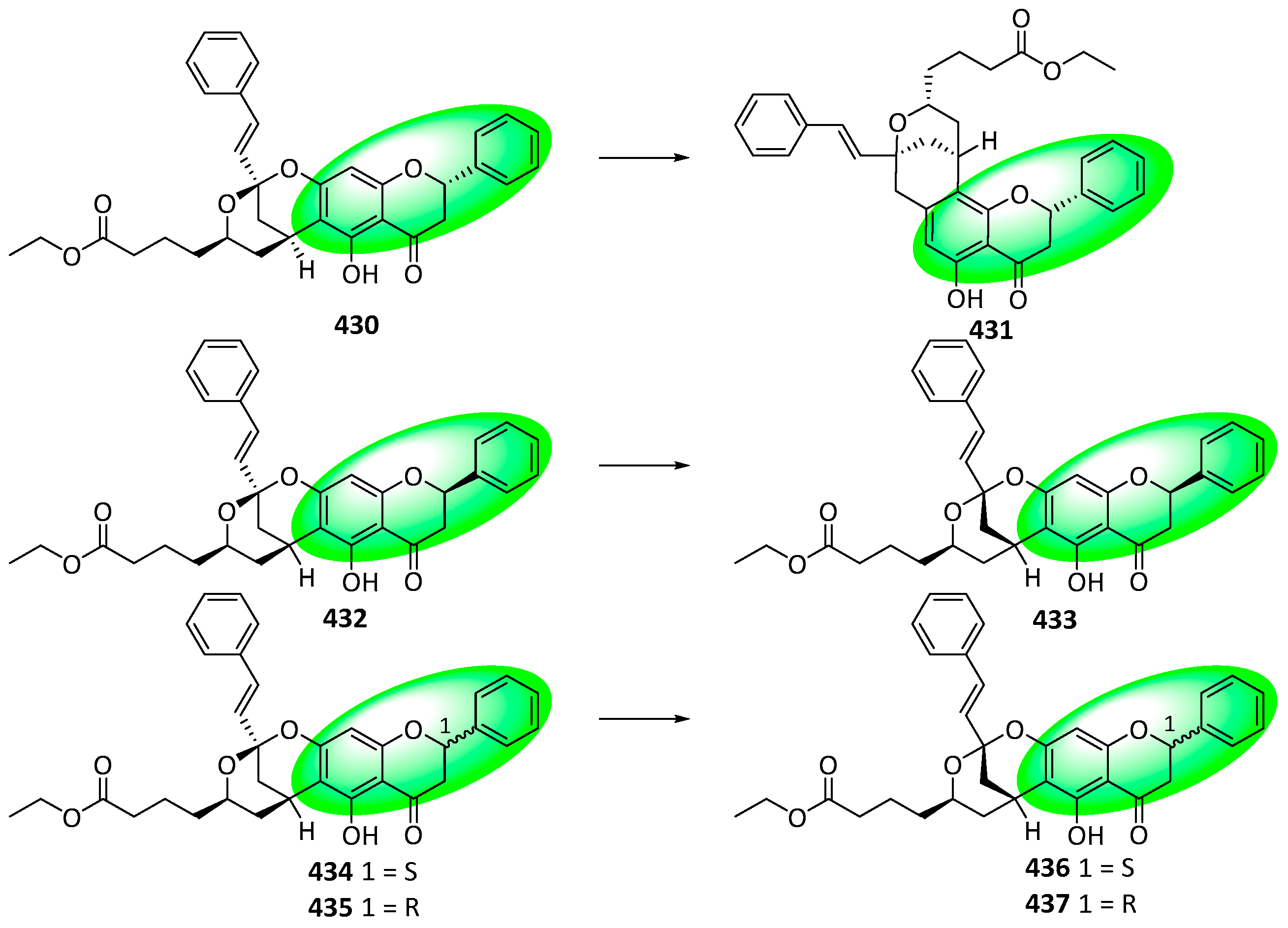
- Kawazoe, R.; Matsuo, Y.; Saito, Y.; Tanaka, T. Computationally Assisted Structural Revision of Flavoalkaloids with a Seven-Membered Ring: Aquiledine, Isoaquiledine, and Cheliensisine. J. Nat. Prod. 2020, 83, 3347–3353. [Google Scholar] [CrossRef] [PubMed]







































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
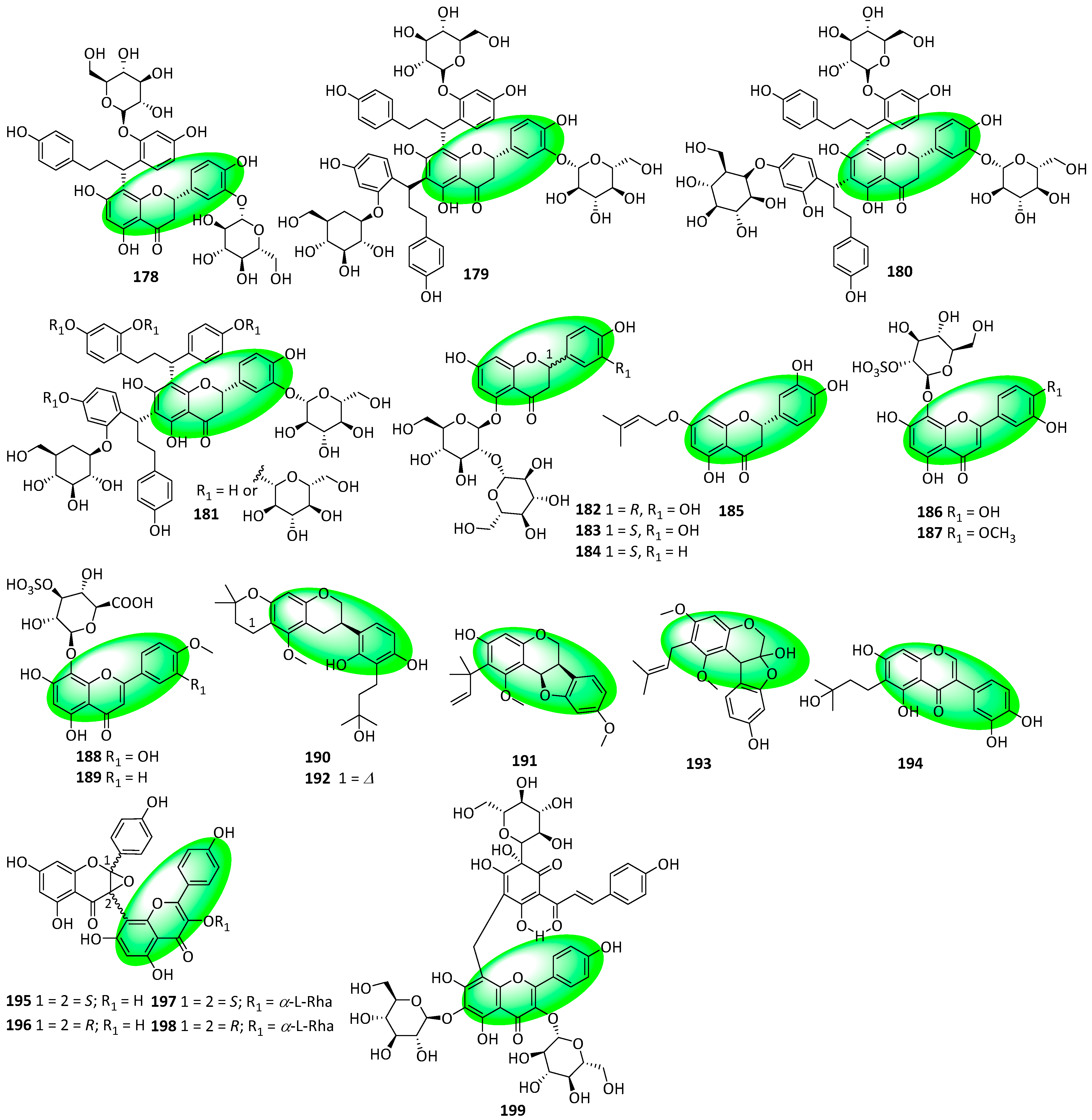{kind=link}
{kind=link}
{kind=link}
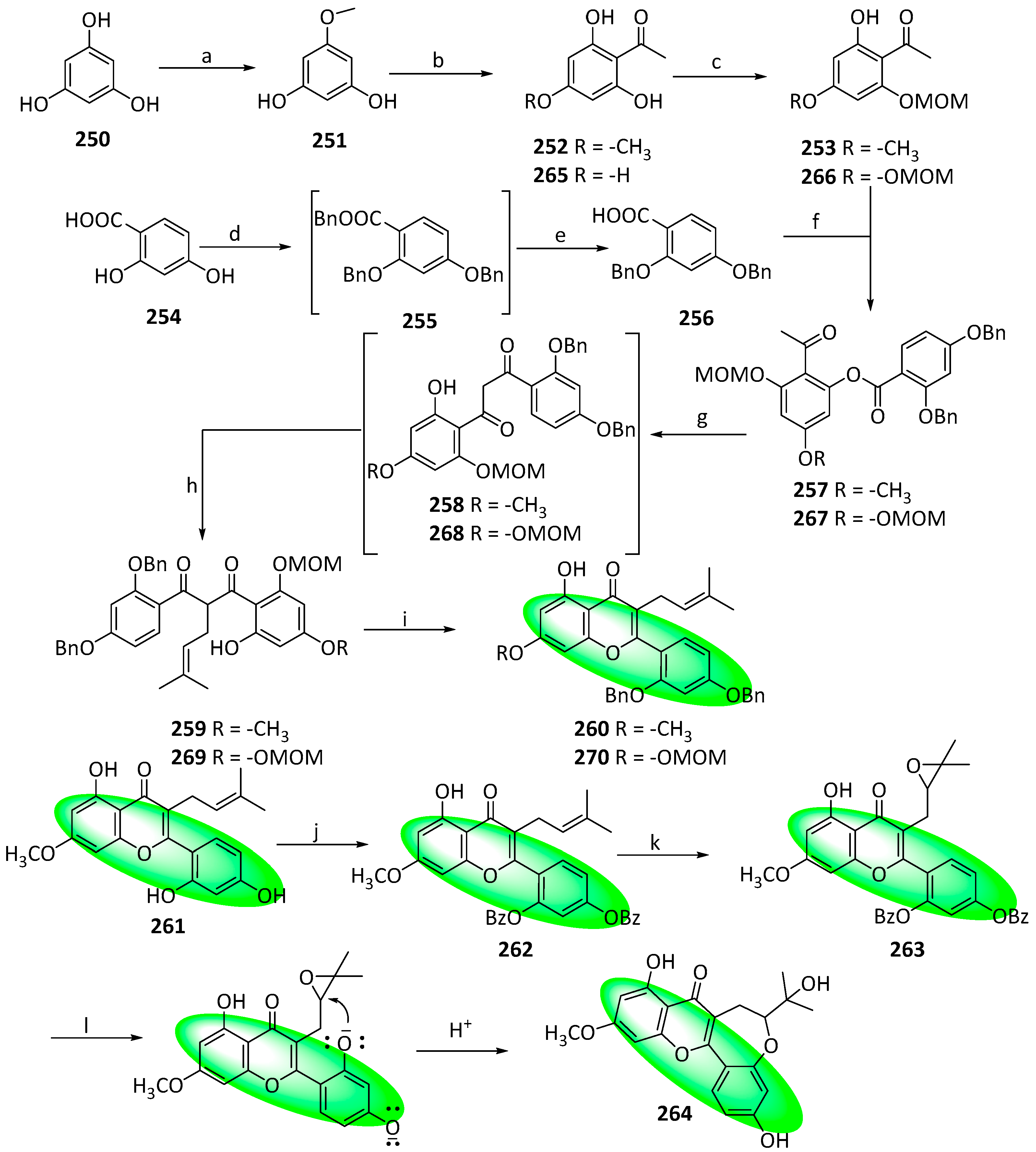{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
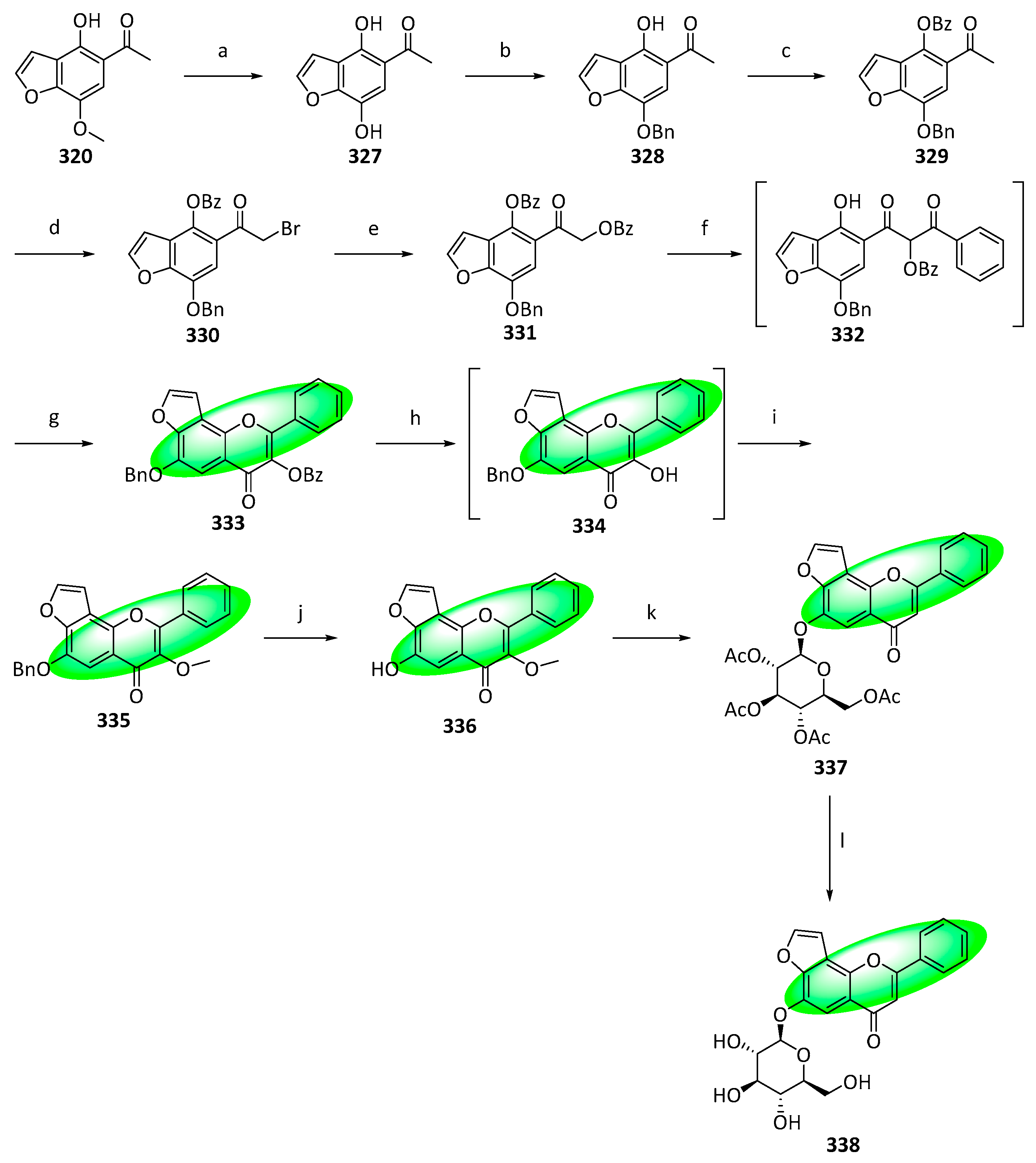{kind=link}
{kind=link}
{kind=link}
{kind=link}
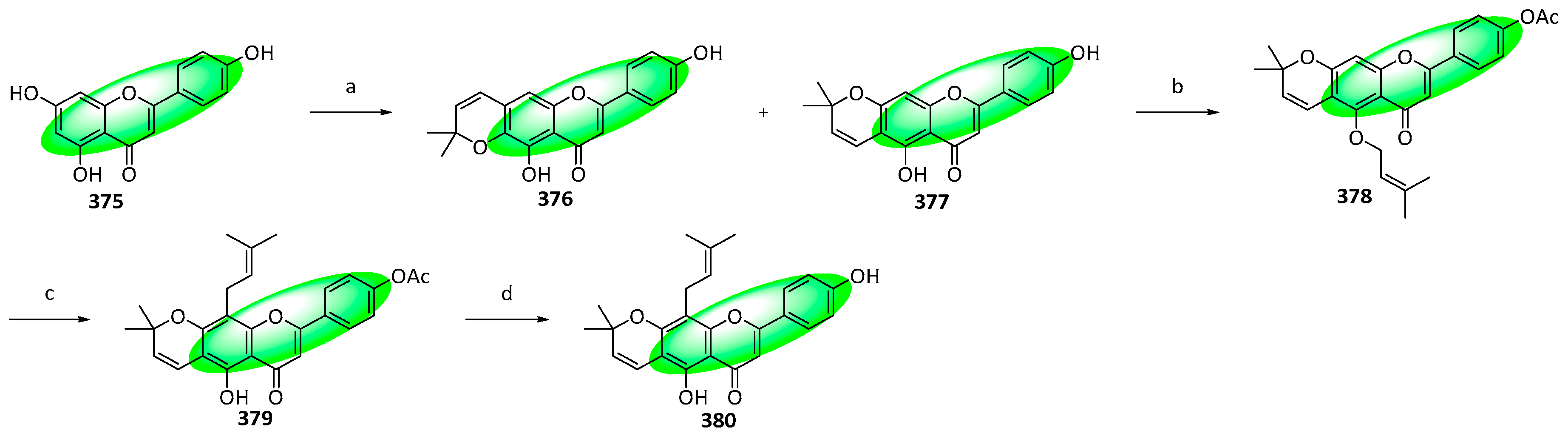{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
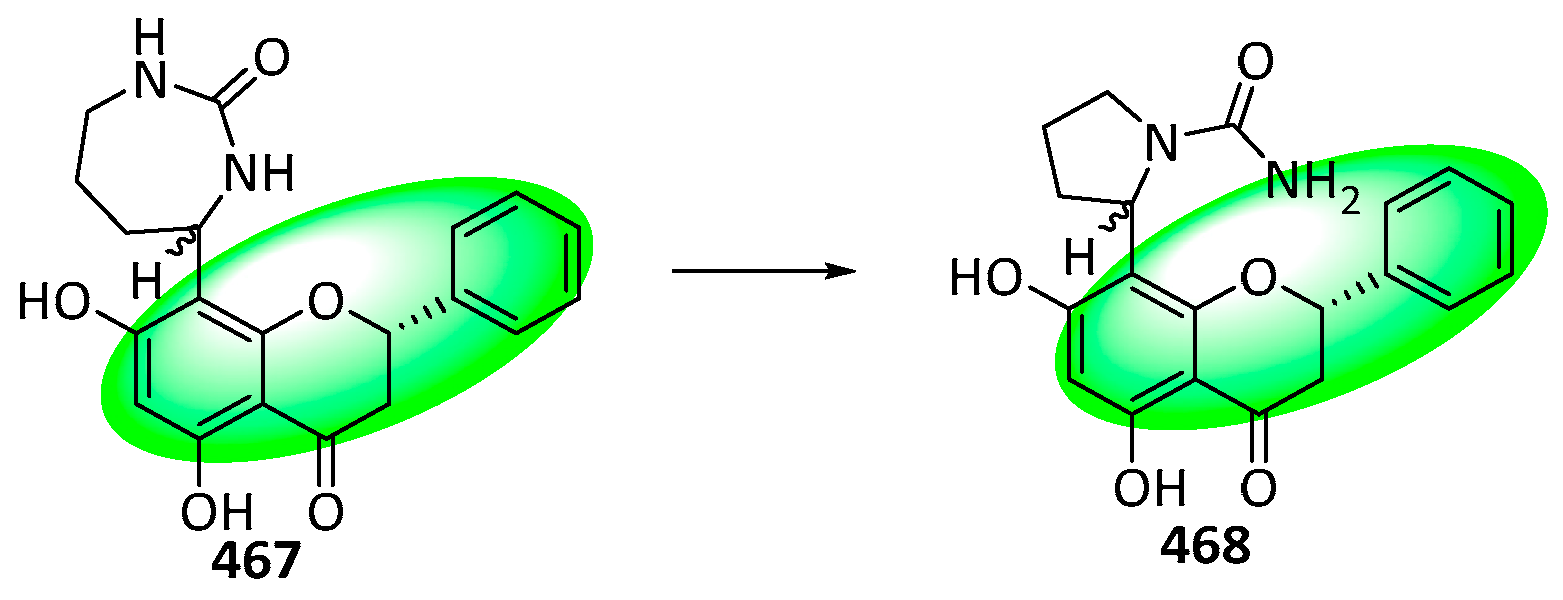{kind=link}
| Compound Serial | Source | Isolation Method | Ref |
|---|---|---|---|
| 1 | Strobilanthes kunthianus | Extracted successively with petroleum ether, chloroform, ethyl acetate, and methanol | [19] |
| 2 | Adenosma bracteosum | Extracted with ethanol; concentrated and partitioned with water, n-hexane, chloroform, ethyl acetate, and n-butanol. | [20] |
| 3–4 | Glandularia selloi | Extracted with methanol | [21] |
| 5–6 | Ceratodon purpureus | Extracted with methanol | [22] |
| 7–8 | Afrocarpus gracilior | Extracted five times with hot 80% methanol; washed with chloroform | [23] |
| 9 | Wulfenia amherstiana | Extracted with ethanol | [24] |
| 10 | Lonicera hypoglauca | Extracted thrice with 85% ethanol; concentrated and extracted with petroleum ether and ethyl acetate | [25] |
| 11–14 | Fuzhuan brick tea | Extracted with ethanol; concentrated and resuspended in water; washed with petroleum ether | [26] |
| 15–16 | Artocarpus nigrifolius | Extracted with ethanol; concentrated and redissolved in ethyl acetate | [27] |
| 17 | Murraya tetramera | Extracted with methanol | [28] |
| 18–25 | Palhinhaea cernua | Extracted thrice with 75% ethanol; concentrated and resuspended in water; partitioned with petroleum ether, ethyl acetate, and n-butanol | [29] |
| 26 | Agastache rugosa | Extracted with 70% ethanol; concentrated and resuspended in water; partitioned with n-hexane, ethyl acetate, and n-butanol | [30] |
| 27–34 | Celmisia viscosa | Separately extracted with chloroform and ethanol | [31] |
| 35–37 | Elsholtzia ciliata | Extracted with 70% ethanol; concentrated and resuspended in water; partitioned with n-hexane, ethyl acetate, and n-butanol | [32] |
| 38–44 | Tephrosia linearis | Extracted thrice with dichloromethane and methanol; concentrated and partitioned with water and n-hexane; aqueous layer was further partitioned with ethyl acetate | [33] |
| 45–48 | Morus nigra | Extracted thrice with 95% ethanol; concentrated and resuspended in water; partitioned with petroleum ether and ethyl acetate | [34] |
| 49–50 | Sphaerocoryne gracilis ssp. Gracilis | Extracted with methanol | [35] |
| 51–52 | Epimedium brevicornum | Extracted with 95% ethanol | [36] |
| 53–54 | Millettia velutina | Extracted with 95% ethanol; concentrated and resuspended in water; extracted with petroleum ether and ethyl acetate | [37] |
| 55 | Onopordum alexandrinum | Extracted thrice with methanol, concentrated, and partitioned with ethyl acetate and water; the aqueous phase was further partitioned with butanol | [38] |
| 56–59 | Ouratea spectabilis | Extracted with 96% ethanol | [39] |
| 60–75 | Streptomyces sp. HDN154127 | Extracted with ethyl acetate | [40] |
| 76–81 | Vatairea guianenis aubl. | Extracted five times with methanol; concentrated and resuspended in water; extracted with dichloromethane and ethyl acetate | [41] |
| 82 | Dietes bicolor | Extracted with 80% methanol; concentrated and resuspended in water and methanol (1:4); partitioned with n-hexane, dichloromethane, and n-butanol | [42] |
| 83–91 | Glycyrrhiza uralensis | Extracted with 90% and 70% ethanol; concentrated and resuspended in water; partitioned with petroleum ether, ethyl acetate, and n-butanol. | [43] |
| 92 | Sabia limoniacea | Extracted with 70% ethanol; concentrated and suspended in water; partitioned with ethyl acetate | [44] |
| 93 | Atriplex tatarica | Extracted with methanol; concentrated and resuspended in water; rinsed with dichloromethane and extracted with n-butanol; concentrated and dissolved in methanol | [45] |
| 94–95 | Ochna holstii | Extracted with methanol and dichloromethane (7:3) | [46] |
| 96 | Myrsine africana | Extracted with methanol; concentrated and resuspended in water; partitioned with petroleum ether, chloroform, ethyl acetate, and n-butanol | [47] |
| 97 | Tetraena mongolica | Extracted thrice with methanol; concentrated and resuspended in water; partitioned with ethyl acetate and n-butanol | [48] |
| 98–101 | Drosera magna | Extracted with methanol and dichloromethane (3:1); concentrated and partitioned with methanol and dichloromethane | [49] |
| 102–104 | Phyllanthus acidus | Extracted with ethanol | [50] |
| 105–109 | Mimosa caesalpiniifolia | Extracted with ethanol and water; concentrated and resuspended in water and ethyl acetate (1:1); extracted with ethyl acetate. | [51] |
| 110–111 | Ormosia arborea | Extracted with ethanol and water | [52] |
| 112 | Salvia circinata | Separately extracted with boiling water and a dichloromethane–methanol (1:1) mixture | [53] |
| 113–121 | Corispermum marschallii | Extracted with acetone–methanol–water (3:1:1); concentrated and partitioned with chloroform, diethyl ether, ethyl acetate, and n-butanol | [54] |
| 122–129 | Brazilian red propolis | Extracted with 80% ethanol; washed with hexane and concentrated; resuspended in water and ethyl acetate (3:5); partitioned with water and ethyl acetate. | [55] |
| 130–138 | Cryptocarya metcalfiana | Extracted with 95% ethanol; concentrated and resuspended in water; partitioned with ethyl acetate | [56] |
| 139–140 | Woodfordia uniflora | Extracted with ethanol, methanol, 90% methanol, and acetone; concentrated and resuspended in water; partitioned with ethyl acetate and n-butanol | [57] |
| 141–142 | Cyclopia genistoides | Extracted with hot water; concentrated and resuspended in ethanol | [58] |
| 143 | Melodorum siamensis | Dried fruits were extracted with ethyl acetate and methanol; dried leaves were extracted with acetone | [59] |
| 144–148 | Pinus massoniana | Extracted with 70% acetone | [60] |
| 149–155 | Sophora tonkinensis | Extracted with ethanol and water (1:1); concentrated and partitioned with water-saturated n-butanol | [61] |
| 156–168 | Pentarhizidium orientale | Extracted thrice with 80% methanol; concentrated and resuspended in water; partitioned with dichloromethane and n-butanol | [62] |
| 169 | Houttuynia cordata | Extracted twice with ethanol and water (3:2) | [63] |
| 170–177 | Saxifraga spinulosa | Extracted with acetone and water (4:1); concentrated and resuspended in water; extracted with diethyl ether and separately combined with the aqueous phase | [64] |
| 178–181 | Mansoa hirsuta | Extracted with ethanol | [65] |
| 182–184 | Berchemia berchemiifolia | Extracted with methanol; concentrated and resuspended in water; partitioned with chloroform, ethyl acetate, and n-butanol | [66] |
| 185 | Arcytophyllum thymifolium | Extracted with n-hexane, chloroform, and methanol | [67] |
| 186–189 | Althaea officinalis | Extracted with methanol and water (1:1) and centrifuged; the resulting pellet was again extracted with methanol and water (1:1); concentrated and high molecular weight compounds were precipitated by adding the extract into cold ethanol; the suspension was centrifuged, and ethanol was removed from the supernatant | [68] |
| 190–194 | Glycyrrhiza uralensis | Extracted with 95% and 70% ethanol; concentrated and resuspended in water; partitioned with ethyl acetate and n-butanol | [69] |
| 195–198 | Impatiens balsamina | Extracted with 80% methanol; concentrated and resuspended in water; partitioned with n-hexane, chloroform, ethyl acetate, and n-butanol | [70] |
| 199 | Carthamus tinctorius | Extracted with 95% and 70% ethanol; concentrated and resuspended in water; partitioned with petroleum ether and ethyl acetate | [71] |
| 200–203 | Daemonorops draco | Extracted with chloroform; partitioned with water | [72] |
| 204–214 | Amorpha fruticosa | Extracted thrice with dichloromethane and methanol (1:1) | [73] |
| 215–216 | Paulownia tomentosa | Extracted in 96% ethanol; concentrated and resuspended in 10% ethanol; extracted with chloroform; concentrated and resuspended in 90% methanol; washed with hexane | [74] |
| 217–225 | Ginkgo biloba | Extracted with acetone and water | [75] |
| 226–228 | Zizyphus jujuba | Extracted with methanol; concentrated and resuspended in water; partitioned with chloroform, ethyl acetate, and butanol | [76] |
| 229–234 | Atraphaxis frutescens | Extracted with acetone and water (4:1); concentrated and resuspended in water; partitioned with diethyl ether | [77] |
| 235–247 | Actinomadura sp. RB99 | Extracted with methanol; concentrated and resuspended in water; partitioned with ethyl acetate | [78] |
| 248–249 | Impatiens balsamina | Extracted with 80% methanol; concentrated and resuspended in water; partitioned with n-hexane, chloroform, ethyl acetate, and n-butanol | [79] |
| Compound | Cytotoxic (GI50; [Cell Line]; μM) | Ref |
|---|---|---|
| 2 a | 4.57 [NCI-H460], 5.67 [HepG2 cells] | [20] |
| 7 a | 9.02 [Hep-G2] | [23] |
| 8 a | 15.61 [Hep-G2] | [23] |
| 15 | 48.7 [SiHa], 55.0 [SGC-7901] | [27] |
| 16 | 20.9 [SiHa], 21.3 [SGC-7901] | [27] |
| 51 | 4.3 [HL-60], 7.1 [A-549], 5.4 [MCF-7], 5.1 [SW-480] | [36] |
| 123 | 17.2 [U-251], 27 [MCF7], 19.1 [NCI-ADR/RES], 19.1 [PC-3] | [55] |
| 126 | 25 [U-251], 34.6 [MCF7], 29.9 [NCI-ADR/RES], 21.9 [PC-3] | [55] |
| 132 | 7.2 [HCT-116] | [56] |
| 133 | 6.1 [HCT-116] | [56] |
| 134 | 5.8 [HCT-116] | [56] |
| 137 | 4.2 [HCT-116] | [56] |
| 138 | 4.5 [HCT-116] | [56] |
| 211 | 7.6 [L5178Y] | [73] |
| 226 | 43.5 [HSC-T6] | [76] |
| 388 | 8.8 [HCT-116], 7.6 [A549] | [101] |
| 389 | 3.6 [MCF-7], 7.1 [HCC-1937], 7.7 [HepG-2], 7.8 [HCT-116], 4.5 [BGC-823], 5.1 [A549] | [101] |
| 393 | 18.9 [HUVEC] | [108] |
| 396 | 3.4 [HUVEC] | [108] |
| 399 | 4.3 [HUVEC] | [108] |
| 404 | 11.5 [HUVEC] | [108] |
| Antibacterial (MIC; μM) | ||
| 9 a | 256 [S. pneumoniae], 256 [B. subtilis], 128 [S. aureus], 512 [S. flexneri], 512 [P. aeruginosa], 256 [S. typhi] | [24] |
| 60 | 6.2 [B. cereus], 50.0 [P. species], 12.0 [M. phlei], 25.0 [B. subtilis], 50.0 [V. parahemolyticus] | [40] |
| 61 | 12.0 [B. cereus], 25.0 [P. species], 25.0 [M. phlei], 12.0 [B. subtilis], 50.0 [V. parahemolyticus], 12.0 [MRSA] | [40] |
| 62 | 1.6 [B. cereus], 12.0 [P. species], 3.1 [M. phlei], 6.2 [B. subtilis], 6.2 [V. parahemolyticus] | [40] |
| 63 | 12.0 [B. cereus], 12.0 [P. species], 12.0 [M. phlei], 12.0 [B. subtilis] | [40] |
| 64 | 3.1 [B. cereus], 50.0 [P. species], 6.2 [M. phlei], 6.2 [B. subtilis], 12.0 [V. parahemolyticus] | [40] |
| 65 | 3.1 [B. cereus], 0.8 [P. species], 0.8 [M. phlei], 0.8 [B. subtilis], 0.8 [V. parahemolyticus], 0.8 [MRSA] | [40] |
| 66 | 25.0 [B. cereus], 25.0 [M. phlei], 25.0 [B. subtilis] | [40] |
| 67 | 12.0 [B. cereus], 12.0 [P. species], 12.0 [M. phlei], 12.0 [B. subtilis], 50.0 [V. parahemolyticus] | [40] |
| 69 | 0.4 [B. subtilis] | [40] |
| 70 | 0.8 [B. subtilis], 3.1 [V. parahemolyticus], 1.6 [M. albicans] | [40] |
| 72 | 0.8 [B. subtilis], 0.8 [V. parahemolyticus], 1.6 [M. albicans] | [40] |
| 73 | 1.6 [B. subtilis], 3.1 [V. parahemolyticus], 1.6 [M. albicans] | [40] |
| 75 | 1.6 [V. parahemolyticus], 1.6 [M. albicans] | [40] |
| 76 b | 29.6 [MRSA] | [41] |
| 77 b | 37.0 [MRSA], 80.6 [E. faecium] | [41] |
| 78 b | 49.0 [MRSA] | [41] |
| 93 | 295.5 [M. flavus], 886.5 [L. monocytogenes], 295.5 [P. aeruginosa], 591.0 [E. coli] | [45] |
| 94 | 14.0 [B. subtilis] | [46] |
| 204 | 100 [S. aureus], 100 [MRSA], 50 [E. faecalis], 100 [E. faecalis] c, 100 [E. faecium], 100 [E. faecium] c | [73] |
| 205 | 100 [S. aureus], 100 [MRSA], 50 [E. faecalis], 100 [E. faecalis] c, 25 [E. faecium], 25 [E. faecium] c | [73] |
| 206 | 100 [S. aureus], 100 [MRSA], 25 [E. faecalis], 50 [E. faecalis] c, 25 [E. faecium], 12.5 [E. faecium] c | [73] |
| 241 a | 35.1 [H. pylori] | [78] |
| 245 a | 58.1 [H. pylori] | [78] |
| 264 a | 128 [S. aureus] | [80] |
| 276 a | 8 [S. aureus], 8 [S. epidermidis], 16 [B. subtilis] | [80] |
| 277 a | 4 [S. aureus], 16 [S. epidermidis], 16 [B. subtilis] | [80] |
| 278 a | 64 [S. aureus], 64 [B. subtilis] | [80] |
| 299 | 15.4 [MRSA] | [84] |
| 301 | 20.5 [MRSA] | [84] |
| Antioxidant (EC50; μM) | ||
| 2 a | 4.04 | [20] |
| 5 | 33.5 | [22] |
| 6 | 127.8 | [22] |
| 9 | 20 | [24] |
| 170 | 53.1 | [64] |
| 171 | 58.8 | [64] |
| 172 | 64.9 | [64] |
| 173 | 42.3 | [64] |
| 174 | 29.3 | [64] |
| 175 | 42.5 | [64] |
| 176 | 44.7 | [64] |
| 177 | 72.9 | [64] |
| 229 | 26.2 | [77] |
| 230 | 12.9 | [77] |
| 231 | 9.9 | [77] |
| 232 | 13.6 | [77] |
| 233 | 15.4 | [77] |
| Anti-Inflammatory (IC50; μM) | ||
| 26 | 16.8 [PGE2] | [30] |
| 59 | 3.1 [CCL2] | [39] |
| 113 | 26.9 [ROS], 66.0 [IL-8] | [54] |
| 114 | 31.5 [ROS], 25.7 [IL-8], 16.3 [TNF-α] | [54] |
| 115 | 5.7 [ROS], 28.9 [IL-8], 9.3 [TNF-α] | [54] |
| 116 | 5.7 [ROS], 25.5 [IL-8] | [54] |
| 117 | 5.4 [ROS], 24.5 [IL-8], 9.7 [TNF-α] | [54] |
| 118 | 7.1 [ROS], 7.2 [IL-8], 12.8 [TNF-α] | [54] |
| 119 | 38.5 [ROS], 51.9 [IL-8], 16.3 [TNF-α] | [54] |
| 120 | 36.6 [ROS], 46.7 [IL-8], 23.2 [TNF-α] | [54] |
| 121 | 6.0 [ROS], 20.7 [TNF-α] | [54] |
| 181 | 19.3 [TNF-α] | [65] |
| 193 | 24.6 [NF-kB] | [69] |
| 200 | 3.1 [O2− inhibition], 4.5 [elastase inhibition] | [72] |
| 201 | 1.3 [O2− inhibition], 3.1 [elastase inhibition] | [72] |
| Antifungal (MIC; μM) | ||
| 9 a | 256 [T. longifusis], 256 [A. flavus], 512 [M. canis], 512 [F. solani], 128 [C. albicans], 256 [C. glabrata] | [24] |
| 107 a | 15 [C. krusei] | [51] |
| 139 | 1.9 [C. neoformans], 14.8 [C. albicans] | [57] |
| 140 | 10 [C. neoformans] | [57] |
| Antiparasitic (EC50; μM) | ||
| 110 | 28.2 [LdNH] | [52] |
| 111 | 25.6 [LdNH] | [52] |
| 170 | 9.4 [B. bovisa], 19.9 [B. bigeminaa] | [64] |
| 171 | 12.1 [B. bovisa], 22.7 [B. bigeminaa] | [64] |
| Antiviral (EC50; μM) | ||
| 169 | 17.72 [HSV] | [63] |
| Inhibition of NO Production (IC50; μM) | ||
| 195 | 33.33 | [70] |
| 196 | 56.86 | [70] |
| 197 | 39.16 | [70] |
| 198 | 31.02 | [70] |
| 221 | 2.91 | [75] |
| 224 | 17.23 | [75] |
| Inhibition of PTP1B (IC50; μM) | ||
| 45 | 12.5 | [34] |
| 46 | 7.7 | [34] |
| 47 | 5.3 | [34] |
| 84 | 5.9 | [43] |
| 88 | 6.7 | [43] |
| Inhibition of Tyrosinase (IC50; mM) | ||
| 229 | 0.9 | [77] |
| 230 | 4.7 | [77] |
| 231 | 1.2 | [77] |
| Inhibition of α-glucosidase (IC50; μM) | ||
| 84 | 20.1 | [43] |
| 112 | 39 | [53] |
| 185 | 28.1 | [67] |
| 387 | 40.56 | [101] |
| 388 | 1.78 | [101] |
| 389 | 0.24 | [101] |
| Inhibition of Oxygenases (IC50; μM) | ||
| 215 | 0.37 [5-LOX] | [74] |
| 216 | 26.3 [COX-1], 9.5 [COX-2] | [74] |
| Neuroprotective Activity (EC50; μM) | ||
| 35 | 69.7 [HT22] | [32] |
| Inhibition of Xanthine Oxidase (IC50; μM) | ||
| 184 | 95.4 | [66] |
| Glucose uptake rate in an insulin resistant HepG2 cell model | ||
| 86 | 95% | [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umer, S.M.; Shamim, S.; Khan, K.M.; Saleem, R.S.Z. Perplexing Polyphenolics: The Isolations, Syntheses, Reappraisals, and Bioactivities of Flavonoids, Isoflavonoids, and Neoflavonoids from 2016 to 2022. Life 2023, 13, 736. https://doi.org/10.3390/life13030736
Umer SM, Shamim S, Khan KM, Saleem RSZ. Perplexing Polyphenolics: The Isolations, Syntheses, Reappraisals, and Bioactivities of Flavonoids, Isoflavonoids, and Neoflavonoids from 2016 to 2022. Life. 2023; 13(3):736. https://doi.org/10.3390/life13030736
Chicago/Turabian StyleUmer, Syed Muhammad, Shahbaz Shamim, Khalid Mohammed Khan, and Rahman Shah Zaib Saleem. 2023. "Perplexing Polyphenolics: The Isolations, Syntheses, Reappraisals, and Bioactivities of Flavonoids, Isoflavonoids, and Neoflavonoids from 2016 to 2022" Life 13, no. 3: 736. https://doi.org/10.3390/life13030736