
Characterization of Stable Pyrazole Derivatives of Curcumin with Improved Cytotoxicity on Osteosarcoma Cell Lines
 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of the Curcumin Analogs
2.2. Cell Cultures and Cytotoxicity Assay
2.3. Clonogenic Assay
2.4. Cell Cycle Analysis and Determination of Apoptotic Cell Death
2.5. Western Blot Analysis
2.6. UV–Visible Spectrum Analysis
2.7. Bioavailability Prediction Analysis
2.8. Statistical Analysis
3. Results
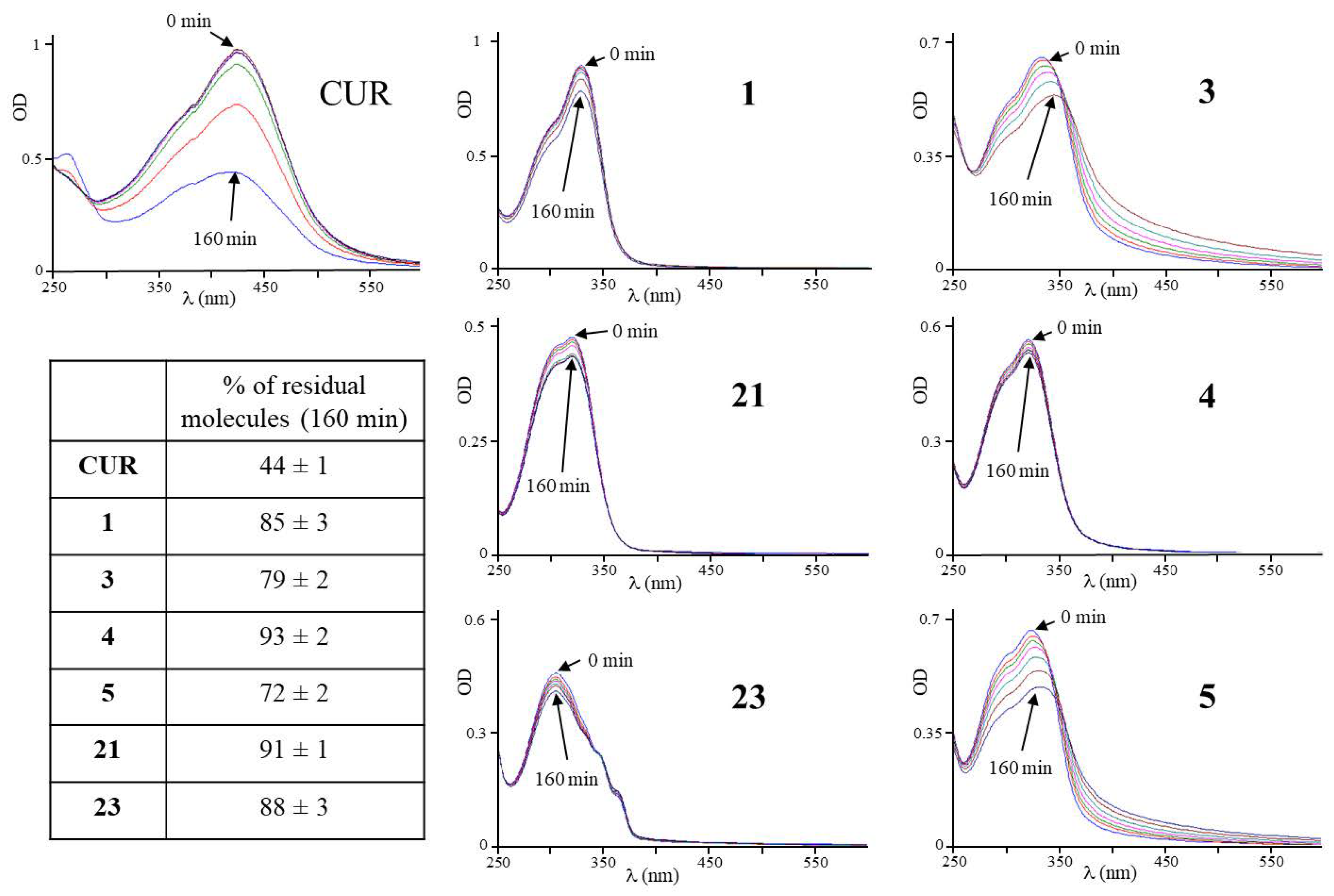
3.1. Design and Stability of the CUR-Derivatives in Solutions at Physiological pH
3.2. Cytotoxicity of CUR and Its Derivatives on Cancer Cell Lines
3.3. Effects of the Derivatives on the Cell Cycle and Induction of Apoptosis in OS Cells
3.4. Prediction of Oral Bioavailability of Curcumin Derivatives
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Picci, P. Osteosarcoma (Osteogenic sarcoma). Orphanet J. Rare Dis. 2007, 2, 6. [Google Scholar] [CrossRef]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, epidemiology, and end results program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef]
- Araujo, C.C.; Leon, L.L. Biological activities of Curcuma longa L. Mem. Inst. Oswaldo Cruz. 2001, 96, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Arslan, A.K.K.; Uzunhisarcıklı, E.; Yerer, M.B.; Bishayee, A. The golden spice curcumin in cancer: A perspective on finalized clinical trials during the last 10 years. J. Cancer Res. Ther. 2022, 18, 19–26. [Google Scholar]
- Zheng, M.; Ekmekcioglu, S.; Walch, E.T.; Tang, C.H.; Grimm, E.A. Inhibition of nuclear factor-kappaB and nitric oxide by curcumin induces G2/M cell cycle arrest and apoptosis in human melanoma cells. Melanoma Res. 2004, 14, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Nukina, N. Inhibition of proteasomal function by curcumin induces apoptosis through mitochondrial pathway. J. Biol. Chem. 2004, 279, 11680–11685. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Koh, W.; Aggarwal, B.B. Discovery of curcumin, a component of the golden spic, and its miraculous biological activities. Clin. Exp. Pharm. Physiol. 2013, 39, 283–299. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ (accessed on 1 September 2022).
- Cheng, A.L.; Hsu, C.H.; Lin, J.K.; Hsu, M.M.; Ho, Y.F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.R.; Ming-Shiang, W.; et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001, 21, 2895–2900. [Google Scholar]
- Chakraborti, S.; Dhar, G.; Dwivedi, V.; Das, A.; Poddar, A.; Chakraborti, G.; Basu, G.; Chakrabarti, P.; Surolia, A.; Bhattacharyya, B. Stable and potent analogues derived from the modification of the dicarbonyl moiety of curcumin. Biochemistry 2013, 52, 7449–7460. [Google Scholar] [CrossRef]
- Wahlström, B.; Blennow, G. A study on the fate of curcumin in the rat. Acta Pharmacol. Toxicol. 1978, 43, 86–92. [Google Scholar] [CrossRef]
- Devassy, J.G.; Nwachukwu, I.D.; Jones, P.J. Curcumin and cancer: Barriers to obtaining a health claim. Nutr. Rev. 2015, 73, 155–165. [Google Scholar] [CrossRef]
- Corrie, L.; Kaur, J.; Awasthi, A.; Vishwas, S.; Gulati, M.; Saini, S.; Kumar, B.; Pandey, N.K.; Gupta, G.; Dureja, H.; et al. Multivariate data analysis and central composite design-oriented optimization of solid carriers for formulation of curcumin-loaded solid SNEDDS: Dissolution and bioavailability assessment. Pharmaceutics 2022, 14, 2395. [Google Scholar] [CrossRef] [PubMed]
- Song, J.G.; Noh, H.M.; Lee, S.H.; Han, H.K. Lipid/clay-based solid dispersion formulation for improving the oral bioavailability of curcumin. Pharmaceutics 2022, 14, 2269. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.X.; Teo, M.Y.M.; Nordin, F.J.; Dewi, F.R.P.; Palanirajan, V.K.; In, L.L.A. Biophysical evaluation of water-soluble curcumin encapsulated in β-cyclodextrins on colorectal cancer cells. Int. J. Mol. Sci. 2022, 23, 12866. [Google Scholar] [CrossRef]
- Sharifi, S.; Abdolahinia, E.D.; Ghavimi, M.A.; Dizaj, S.M.; Aschner, M.; Saso, L.; Khan, H. Effect of curcumin-loaded mesoporous silica nanoparticles on the head and neck cancer cell line, HN5. Curr. Issues Mol. Biol. 2022, 44, 5247–5259. [Google Scholar] [CrossRef]
- Lu, K.H.; Lu, P.W.; Lin, C.W.; Yang, S.F. Curcumin in human osteosarcoma: From analogs to carriers. Drug Discov. Today 2022, 28, 103437. [Google Scholar] [CrossRef]
- Oetari, S.; Sudibyo, M.; Commandeur, J.N.; Samhoedi, R.; Vermeulen, N.P. Effects of curcumin on cytochrome P450 and glutathione S-transferase activities in rat liver. Biochem. Pharmacol. 1996, 51, 39–45. [Google Scholar] [CrossRef]
- Wang, Y.J.; Pan, M.H.; Cheng, A.L.; Lin, L.I.; Ho, Y.S.; Hsieh, C.Y.; Lin, J.K. Stability of curcumin in buffer solutions and characterization of its degradation products. J. Pharm. Biomed. Anal. 1997, 15, 1867–1876. [Google Scholar] [CrossRef]
- Zhu, J.; Sanidad, K.Z.; Sukamtoh, E.; Zhang, G. Potential roles of chemical degradation in the biological activities of curcumin. Food Funct. 2017, 8, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Priyadarsini, K.I. Photophysics, photochemistry and photobiology of curcumin: Studies from organic solutions, biomimetics and living cells. J. Photochem. Photobiol. 2009, 10, 81–89. [Google Scholar] [CrossRef]
- Zhao, C.; Yang, J.; Wang, Y.; Liang, D.; Yang, X.; Li, X.; Wu, J.; Wu, X.; Yang, S.; Li, X.; et al. Synthesis of mono-carbonyl analogues of curcumin and their effects on inhibition of cytokine release in LPS-stimulated RAW 264.7 macrophages. Bioorg. Med. Chem. 2010, 18, 2388–2393. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, Z.; Liang, G. Promising curcumin-based drug design: Mono-carbonyl analogues of curcumin (MACs). Curr. Pharm. Des. 2013, 19, 2114–2135. [Google Scholar]
- Simoni, D.; Rizzi, M.; Rondanin, R.; Baruchello, R.; Marchetti, P.; Invidiata, F.P.; Labbozzetta, M.; Poma, P.; Carina, V.; Notarbartolo, M.; et al. Antitumor effects of curcumin and structurally beta-diketone modified analogs on multidrug resistant cancer cells. Bioorg. Med. Chem. Lett. 2008, 18, 845–849. [Google Scholar] [CrossRef]
- Feriotto, G.; Tagliati, F.; Giriolo, R.; Casciano, F.; Tabolacci, C.; Beninati, S.; Khan, M.T.H.; Mischiati, C. Caffeic acid enhances the anti-leukemic effect of imatinib on chronic myeloid leukemia cells and triggers apoptosis in cells sensitive and resistant to imatinib. Int. J. Mol. Sci. 2021, 22, 1644. [Google Scholar] [CrossRef]
- Feriotto, G.; Calza, R.; Bergamini, C.M.; Griffin, M.; Wang, Z.; Beninati, S.; Ferretti, V.; Marzola, E.; Guerrini, R.; Pagnoni, A.; et al. Involvement of cell surface TG2 in the aggregation of K562 cells triggered by gluten. Amino Acids 2017, 49, 551–565. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Labbozzetta, M.; Baruchello, R.; Marchetti, P.; Gueli, M.C.; Poma, P.; Notarbartolo, M.; Simoni, D.; D’Alessandro, N. Lack of nucleophilic addition in the isoxazole and pyrazole diketone modified analogs of curcumin; implications for their antitumor and chemosensitizing activities. Chem. Biol. Interact. 2009, 181, 29–36. [Google Scholar] [CrossRef]
- Pan, M.H.; Huang, T.M.; Lin, J.K. Biotransformation of curcumin through reduction and glucuronidation in mice. Drug Metab. Dispos. 1999, 27, 486–494. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Colo38 | TT | SK-ChA-1 | Mz-ChA-1 | MG63 | TE85 | |
|---|---|---|---|---|---|---|
| CUR | 13.6 ± 0.7 | 13.5 ± 0.9 | 4.6 ± 0.3 | 7.6 ± 0.6 | 8.7 ± 0.7 | 9.5 ± 0.7 |
| 1 | 6.3 ± 0.4 | 6.0 ± 0.5 | 8.2 ± 0.3 | 8.8 ± 0.3 | 2.7 ± 0.2 | 0.5 ± 0.03 |
| 3 | 8.7 ± 0.6 | 9.1 ± 0.7 | 10.9 ± 0.9 | 12.4 ± 1.0 | 11.8 ± 0.7 | 7.6 ± 0.4 |
| 4 | 20.2 ± 1.6 | 16.7 ± 1.3 | 21.1 ± 1.1 | 16.7 ± 1.1 | 23.5 ± 1.2 | 16.7 ± 1 |
| 5 | 10.3 ± 1.8 | 7.2 ± 0.4 | 8.2 ± 0.5 | 6.6 ± 0.5 | 8.8 ± 0.7 | 9.5 ± 0.8 |
| 21 | 29.6 ± 1.7 | 15.8 ± 0.9 | 27.9 ± 2.2 | 28.3 ± 1.4 | 26.3 ± 1.3 | 6.9 ± 0.4 |
| 23 | 15.4 ± 0.9 | 10.6 ± 0.5 | 24.2 ± 1.4 | 18.7 ± 1.1 | 28.2 ± 1.7 | 20.2 ± 1.5 |
| Limit Values | CUR | 1 | 3 | 4 | 5 | 21 | 23 | |
|---|---|---|---|---|---|---|---|---|
| ≤5 | LogP | 2.3 | 4.23 | 7.15 | 3.75 | 6.12 | 4.60 | 4.82 |
| ≤500 | MW | 368.38 | 364.40 | 550.61 | 426.47 | 510.63 | 304.35 | 452.51 |
| ≤10 | nON | 6 | 6 | 8 | 8 | 8 | 4 | 8 |
| ≤5 | nOHnH | 2 | 3 | 2 | 2 | 2 | 3 | 1 |
| ≤7 | nrotb | 8 | 6 | 12 | 10 | 12 | 4 | 10 |
| <140 Å | PSA (Å) | 93.07 | 87.61 | 102.12 | 102.12 | 102.12 | 69.14 | 84.09 |
| ≤1 | violations | 1 | 0 | 3 | 1 | 3 | 0 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feriotto, G.; Rondanin, R.; Marchetti, P.; Tagliati, F.; Beninati, S.; Tabolacci, C.; Grusi, E.; Aguzzi, S.; Mischiati, C. Characterization of Stable Pyrazole Derivatives of Curcumin with Improved Cytotoxicity on Osteosarcoma Cell Lines. Life 2023, 13, 431. https://doi.org/10.3390/life13020431
Feriotto G, Rondanin R, Marchetti P, Tagliati F, Beninati S, Tabolacci C, Grusi E, Aguzzi S, Mischiati C. Characterization of Stable Pyrazole Derivatives of Curcumin with Improved Cytotoxicity on Osteosarcoma Cell Lines. Life. 2023; 13(2):431. https://doi.org/10.3390/life13020431
Chicago/Turabian StyleFeriotto, Giordana, Riccardo Rondanin, Paolo Marchetti, Federico Tagliati, Simone Beninati, Claudio Tabolacci, Elisa Grusi, Serena Aguzzi, and Carlo Mischiati. 2023. "Characterization of Stable Pyrazole Derivatives of Curcumin with Improved Cytotoxicity on Osteosarcoma Cell Lines" Life 13, no. 2: 431. https://doi.org/10.3390/life13020431