
Ion Channels and Ionotropic Receptors in Astrocytes: Physiological Functions and Alterations in Alzheimer’s Disease and Glioblastoma
 ,
,
Abstract
:1. Introduction
2. Ion Channels and Ionotropic Receptors in Astrocyte Physiology
2.1. Potassium Channels
2.2. Sodium Channels
2.3. Calcium Permeable Channels
2.4. Anion Channels
2.5. Ligand-Gated Ion Channels (Ionotropic Receptors)
2.6. Other Channels
2.6.1. Aquaporins
2.6.2. GAP Junctions and Hemichannels
3. Alzheimer’s Disease
3.1. Potassium Channels
3.2. Calcium Permeable Channels and Intracellular Calcium Activity
3.3. Anion Channels
3.4. Ionotropic Receptors
3.5. Aquaporins
4. Glioblastoma
4.1. Potassium Channels
4.2. Calcium Permeable Channels
4.2.1. TRP Channels
4.2.2. Piezo Channels
4.3. Anion Channels
4.4. Ligand Gated Ion Channels
4.4.1. GABAA Receptors
4.4.2. Glutamate Ionotropic Receptors
4.4.3. Nicotinic ACh Receptors
4.5. Other Channels
4.5.1. Aquaporins
4.5.2. Connexin 43
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Deneen, B. The Emerging Nature of Astrocyte Diversity. Annu. Rev. Neurosci. 2019, 42, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte–neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Lia, A.; Di Spiezio, A.; Speggiorin, M.; Zonta, M. Two decades of astrocytes in neurovascular coupling. Front. Netw. Physiol. 2023, 3, 1162757. [Google Scholar] [CrossRef]
- Semyanov, A.; Verkhratsky, A. Astrocytic processes: From tripartite synapses to the active milieu. Trends Neurosci. 2021, 44, 781–792. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.-M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Münch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Schubert, M.C.; Kuner, T.; Wick, W.; Winkler, F.; Venkataramani, V. Brain Tumor Networks in Diffuse Glioma. Neurotherapeutics 2022, 19, 1832–1843. [Google Scholar] [CrossRef]
- Kuffler, S.W.; Nicholls, J.G.; Orkand, R.K.; Sawyer, J.E.R.; Hennebry, J.E.; Revill, A.; Brown, A.M.; Du, Y.; Ma, B.; Kiyoshi, C.M.; et al. Physiological properties of glial cells in the central nervous system of amphibia. J. Neurophysiol. 1966, 29, 768–787. [Google Scholar] [CrossRef]
- Orkand, R.K.; Nicholls, J.G.; Kuffler, S.W.; Verkhratsky, A.; Nedergaard, M.; Sawyer, J.E.R.; Hennebry, J.E.; Revill, A.; Brown, A.M.; Wanke, E.; et al. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 1966, 29, 788–806. [Google Scholar] [CrossRef]
- Kuffler, S.W. The Ferrier Lecture–Neuroglial cells: Physiological properties and a potassium mediated effect of neuronal activity on the glial membrane potential. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1967, 168, 1–21. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Lyon, K.A.; Allen, N.J. From Synapses to Circuits, Astrocytes Regulate Behavior. Front. Neural Circuits 2022, 15, 786293. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, P.; Araque, A. Astrocytes and Behavior. Annu. Rev. Neurosci. 2021, 44, 49–67. [Google Scholar] [CrossRef]
- Ventura, R.; Harris, K.M. Three-Dimensional Relationships between Hippocampal Synapses and Astrocytes. J. Neurosci. 1999, 19, 6897–6906. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic Astrocytes in CA1 Stratum Radiatum Occupy Separate Anatomical Domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef]
- Lia, A.; Henriques, V.J.; Zonta, M.; Chiavegato, A.; Carmignoto, G.; Gómez-Gonzalo, M.; Losi, G. Calcium Signals in Astrocyte Microdomains, a Decade of Great Advances. Front. Cell. Neurosci. 2021, 15, 673433. [Google Scholar] [CrossRef]
- Ahmadpour, N.; Kantroo, M.; Stobart, J.L. Extracellular Calcium Influx Pathways in Astrocyte Calcium Microdomain Physiology. Biomolecules 2021, 11, 1467. [Google Scholar] [CrossRef]
- Armbruster, M.; Naskar, S.; Garcia, J.P.; Sommer, M.; Kim, E.; Adam, Y.; Haydon, P.G.; Boyden, E.S.; Cohen, A.E.; Dulla, C.G. Neuronal activity drives pathway-specific depolarization of peripheral astrocyte processes. Nat. Neurosci. 2022, 25, 607–616. [Google Scholar] [CrossRef]
- McNeill, J.; Rudyk, C.; Hildebrand, M.E.; Salmaso, N. Ion Channels and Electrophysiological Properties of Astrocytes: Implications for Emergent Stimulation Technologies. Front. Cell. Neurosci. 2021, 15, 644126. [Google Scholar] [CrossRef] [PubMed]
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Losi, G.; Cammarota, M.; Carmignoto, G. The Role of Astroglia in the Epileptic Brain. Front. Pharmacol. 2012, 3, 132. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Goldman, S.A. Astrocytic contributions to Huntington’s disease pathophysiology. Ann. N. Y. Acad. Sci. 2023, 1522, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-I.; Lee, H.-T.; Lin, H.-C.; Tsay, H.-J.; Tsai, F.-C.; Shyue, S.-K.; Lee, T.-S. Role of transient receptor potential ankyrin 1 channels in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 92. [Google Scholar] [CrossRef]
- Paumier, A.; Boisseau, S.; Jacquier-Sarlin, M.; Pernet-Gallay, K.; Buisson, A.; Albrieux, M. Astrocyte–neuron interplay is critical for Alzheimer’s disease pathogenesis and is rescued by TRPA1 channel blockade. Brain 2022, 145, 388–405. [Google Scholar] [CrossRef]
- Hu, J.; Chen, Q.; Zhu, H.; Hou, L.; Liu, W.; Yang, Q.; Shen, H.; Chai, G.; Zhang, B.; Chen, S.; et al. Microglial Piezo1 senses Aβ fibril stiffness to restrict Alzheimer’s disease. Neuron 2023, 111, 15–29.e8. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef]
- Reeves, B.C.; Karimy, J.K.; Kundishora, A.J.; Mestre, H.; Cerci, H.M.; Matouk, C.; Alper, S.L.; Lundgaard, I.; Nedergaard, M.; Kahle, K.T. Glymphatic System Impairment in Alzheimer’s Disease and Idiopathic Normal Pressure Hydrocephalus. Trends Mol. Med. 2020, 26, 285–295. [Google Scholar] [CrossRef]
- Nwaobi, S.E.; Cuddapah, V.A.; Patterson, K.C.; Randolph, A.C.; Olsen, M.L. The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 2016, 132, 1–21. [Google Scholar] [CrossRef]
- Wilcock, D.; Vitek, M.; Colton, C. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Huffels, C.F.M.; Osborn, L.M.; Hulshof, L.A.; Kooijman, L.; Henning, L.; Steinhäuser, C.; Hol, E.M. Amyloid-β plaques affect astrocyte Kir4.1 protein expression but not function in the dentate gyrus of APP/PS1 mice. Glia 2022, 70, 748–767. [Google Scholar] [CrossRef] [PubMed]
- Griffith, C.M.; Xie, M.-X.; Qiu, W.-Y.; Sharp, A.A.; Ma, C.; Pan, A.; Yan, X.-X.; Patrylo, P.R. Aberrant expression of the pore-forming KATP channel subunit Kir6.2 in hippocampal reactive astrocytes in the 3xTg-AD mouse model and human Alzheimer’s disease. Neuroscience 2016, 336, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pitta, M.; Lee, J.-H.; Ray, B.; Lahiri, D.K.; Furukawa, K.; Mughal, M.; Jiang, H.; Villarreal, J.; Cutler, R.G.; et al. The KATP Channel Activator Diazoxide Ameliorates Amyloid-β and Tau Pathologies and Improves Memory in the 3xTgAD Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 22, 443–457. [Google Scholar] [CrossRef]
- Macauley, S.L.; Stanley, M.; Caesar, E.E.; Yamada, S.A.; Raichle, M.E.; Perez, R.; Mahan, T.E.; Sutphen, C.L.; Holtzman, D.M. Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J. Clin. Investig. 2015, 125, 2463–2467. [Google Scholar] [CrossRef]
- Angulo, E.; Noe, V.; Casado, V.; Mallol, J.; Gomez-Isla, T.; Lluis, C.; Ferrer, I.; Ciudad, C.J.; Franco, R. Up-regulation of the Kv3.4 potassium channel subunit in early stages of Alzheimer’s disease. J. Neurochem. 2004, 91, 547–557. [Google Scholar] [CrossRef]
- Pannaccione, A.; Boscia, F.; Scorziello, A.; Adornetto, A.; Castaldo, P.; Sirabella, R.; Taglialatela, M.; Di Renzo, G.F.; Annunziato, L. Up-Regulation and Increased Activity of KV3.4 Channels and Their Accessory Subunit MinK-Related Peptide 2 Induced by Amyloid Peptide Are Involved in Apoptotic Neuronal Death. Mol. Pharmacol. 2007, 72, 665–673. [Google Scholar] [CrossRef]
- Boscia, F.; Pannaccione, A.; Ciccone, R.; Casamassa, A.; Franco, C.; Piccialli, I.; de Rosa, V.; Vinciguerra, A.; Di Renzo, G.; Annunziato, L. The expression and activity of K V 3.4 channel subunits are precociously upregulated in astrocytes exposed to Aβ oligomers and in astrocytes of Alzheimer’s disease Tg2576 mice. Neurobiol. Aging 2017, 54, 187–198. [Google Scholar] [CrossRef]
- Yi, M.; Yu, P.; Lu, Q.; Geller, H.M.; Yu, Z.; Chen, H. KCa3.1 constitutes a pharmacological target for astrogliosis associated with Alzheimer’s disease. Mol. Cell. Neurosci. 2016, 76, 21–32. [Google Scholar] [CrossRef]
- Wei, T.; Yi, M.; Gu, W.; Hou, L.; Lu, Q.; Yu, Z.; Chen, H. The Potassium Channel KCa3.1 Represents a Valid Pharmacological Target for Astrogliosis-Induced Neuronal Impairment in a Mouse Model of Alzheimer’s Disease. Front. Pharmacol. 2016, 7, 528. [Google Scholar] [CrossRef]
- Yu, Z.; Dou, F.; Wang, Y.; Hou, L.; Chen, H. Ca2+-dependent endoplasmic reticulum stress correlation with astrogliosis involves upregulation of KCa3.1 and inhibition of AKT/mTOR signaling. J. Neuroinflamm. 2018, 15, 316. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Teaktong, T.; Graham, A.; Court, J.; Perry, R.; Jaros, E.; Johnson, M.; Hall, R.; Perry, E. Alzheimer’s disease is associated with a selective increase in ?7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia 2003, 41, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-F.; Guan, Z.-Z.; Bogdanovic, N.; Nordberg, A. High selective expression of α7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer’s disease and patients carrying Swedish APP 670/671 mutation: A possible association with neuritic plaques. Exp. Neurol. 2005, 192, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Lee, D.H.S.; Davis, C.B.; Shank, R.P. Amyloid Peptide Aβ1-42 Binds Selectively and with Picomolar Affinity to α7 Nicotinic Acetylcholine Receptors. J. Neurochem. 2002, 75, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Pirttimaki, T.M.; Codadu, N.K.; Awni, A.; Pratik, P.; Nagel, D.A.; Hill, E.J.; Dineley, K.T.; Parri, H.R. α7 Nicotinic Receptor-Mediated Astrocytic Gliotransmitter Release: Aβ Effects in a Preclinical Alzheimer’s Mouse Model. PLoS ONE 2013, 8, e81828. [Google Scholar] [CrossRef]
- Gulisano, W.; Melone, M.; Ripoli, C.; Tropea, M.R.; Puma, D.D.L.; Giunta, S.; Cocco, S.; Marcotulli, D.; Origlia, N.; Palmeri, A.; et al. Neuromodulatory Action of Picomolar Extracellular Aβ42 Oligomers on Presynaptic and Postsynaptic Mechanisms Underlying Synaptic Function and Memory. J. Neurosci. 2019, 39, 5986–6000. [Google Scholar] [CrossRef]
- Tropea, M.R.; Puma, D.D.L.; Melone, M.; Gulisano, W.; Arancio, O.; Grassi, C.; Conti, F.; Puzzo, D. Genetic deletion of α7 nicotinic acetylcholine receptors induces an age-dependent Alzheimer’s disease-like pathology. Prog. Neurobiol. 2021, 206, 102154. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Ryu, M.J.K.; Walker, D.G.; Choi, B.H.B. Upregulated Expression of Purinergic P2X7Receptor in Alzheimer Disease and Amyloid-β Peptide-Treated Microglia and in Peptide-Injected Rat Hippocampus. J. Neuropathol. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar] [CrossRef]
- Martínez-Frailes, C.; Di Lauro, C.; Bianchi, C.; de Diego-García, L.; Sebastián-Serrano, A.; Boscá, L.; Díaz-Hernández, M. Amyloid Peptide Induced Neuroinflammation Increases the P2X7 Receptor Expression in Microglial Cells, Impacting on Its Functionality. Front. Cell. Neurosci. 2019, 13, 143. [Google Scholar] [CrossRef]
- Jin, H.; Han, J.; Resing, D.; Liu, H.; Yue, X.; Miller, R.L.; Schoch, K.M.; Miller, T.M.; Perlmutter, J.S.; Egan, T.M.; et al. Synthesis and in vitro characterization of a P2X7 radioligand [123I]TZ6019 and its response to neuroinflammation in a mouse model of Alzheimer disease. Eur. J. Pharmacol. 2018, 820, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Amar, M.; Dalle, C.; Youssef, I.; Boucher, C.; Le Duigou, C.; Brückner, M.; Prigent, A.; Sazdovitch, V.; Halle, A.; et al. New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 2018, 24, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Caramia, M.; Sforna, L.; Franciolini, F.; Catacuzzeno, L. The Volume-Regulated Anion Channel in Glioblastoma. Cancers 2019, 11, 307. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Stauber, T. The Volume-Regulated Anion Channel LRRC8/VRAC Is Dispensable for Cell Proliferation and Migration. Int. J. Mol. Sci. 2019, 20, 2663. [Google Scholar] [CrossRef] [PubMed]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to P t-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef]
- Molenaar, R.J. Ion Channels in Glioblastoma. ISRN Neurol. 2011, 2011, e590249. [Google Scholar] [CrossRef]
- Brandalise, F.; Ratto, D.; Leone, R.; Olivero, F.; Roda, E.; Locatelli, C.A.; Bottone, M.G.; Rossi, P. Deeper and Deeper on the Role of BK and Kir4.1 Channels in Glioblastoma Invasiveness: A Novel Summative Mechanism? Front. Neurosci. 2020, 14, 595664. [Google Scholar] [CrossRef]
- Tan, G.; Sun, S.-Q.; Yuan, D.-L. Expression of Kir 4.1 in human astrocytic tumors: Correlation with pathologic grade. Biochem. Biophys. Res. Commun. 2008, 367, 743–747. [Google Scholar] [CrossRef]
- Madadi, A.; Wolfart, J.; Lange, F.; Brehme, H.; Linnebacher, M.; Bräuer, A.U.; Büttner, A.; Freiman, T.; Henker, C.; Einsle, A.; et al. Correlation between Kir4.1 expression and barium-sensitive currents in rat and human glioma cell lines. Neurosci. Lett. 2021, 741, 135481. [Google Scholar] [CrossRef]
- Brown, B.M.; Pressley, B.; Wulff, H. KCa3.1 Channel Modulators as Potential Therapeutic Compounds for Glioblastoma. Curr. Neuropharmacol. 2018, 16, 618–626. [Google Scholar] [CrossRef]
- Hausmann, D.; Hoffmann, D.C.; Venkataramani, V.; Jung, E.; Horschitz, S.; Tetzlaff, S.K.; Jabali, A.; Hai, L.; Kessler, T.; Azoŕin, D.D.; et al. Autonomous rhythmic activity in glioma networks drives brain tumour growth. Nature 2022, 613, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Franciolini, F. Role of KCa3.1 Channels in Modulating Ca2+ Oscillations during Glioblastoma Cell Migration and Invasion. Int. J. Mol. Sci. 2018, 19, 2970. [Google Scholar] [CrossRef] [PubMed]
- Preußat, K.; Beetz, C.; Schrey, M.; Kraft, R.; Wölfl, S.; Kalff, R.; Patt, S. Expression of voltage-gated potassium channels Kv1.3 and Kv1.5 in human gliomas. Neurosci. Lett. 2003, 346, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Arvind, S.; Arivazhagan, A.; Santosh, V.; Chandramouli, B.A. Differential expression of a novel voltage gated potassium channel–Kv 1.5 in astrocytomas and its impact on prognosis in glioblastoma. Br. J. Neurosurg. 2012, 26, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, A.; D’alessandro, G.; Di Castro, M.A.; Lauro, C.; Singh, V.; Pagani, F.; Sforna, L.; Grassi, F.; Di Angelantonio, S.; Catacuzzeno, L.; et al. Kv1.3 activity perturbs the homeostatic properties of astrocytes in glioma. Sci. Rep. 2018, 8, 7654. [Google Scholar] [CrossRef]
- Venturini, E.; Leanza, L.; Azzolini, M.; Kadow, S.; Mattarei, A.; Weller, M.; Tabatabai, G.; Edwards, M.J.; Zoratti, M.; Paradisi, C.; et al. Targeting the Potassium Channel Kv1.3 Kills Glioblastoma Cells. Neurosignals 2017, 25, 26–38. [Google Scholar] [CrossRef]
- Yang, W.; Wu, P.-F.; Ma, J.-X.; Liao, M.-J.; Xu, L.-S.; Yi, L. TRPV4 activates the Cdc42/N-wasp pathway to promote glioblastoma invasion by altering cellular protrusions. Sci. Rep. 2020, 10, 14151. [Google Scholar] [CrossRef]
- Huang, T.; Xu, T.; Wang, Y.; Zhou, Y.; Yu, D.; Wang, Z.; He, L.; Chen, Z.; Zhang, Y.; Davidson, D.; et al. Cannabidiol inhibits human glioma by induction of lethal mitophagy through activating TRPV4. Autophagy 2021, 17, 3592–3606. [Google Scholar] [CrossRef]
- Chen, X.; Wanggou, S.; Bodalia, A.; Zhu, M.; Dong, W.; Fan, J.J.; Yin, W.C.; Min, H.-K.; Hu, M.; Draghici, D.; et al. A Feedforward Mechanism Mediated by Mechanosensitive Ion Channel PIEZO1 and Tissue Mechanics Promotes Glioma Aggression. Neuron 2018, 100, 799–815.e7. [Google Scholar] [CrossRef]
- Varricchio, A.; Ramesh, S.A.; Yool, A.J. Novel Ion Channel Targets and Drug Delivery Tools for Controlling Glioblastoma Cell Invasiveness. Int. J. Mol. Sci. 2021, 22, 11909. [Google Scholar] [CrossRef]
- Vandebroek, A.; Yasui, M. Regulation of AQP4 in the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 1603. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.E.S.; Clementino-Neto, J.; Mendes, C.B.; Franzon, N.H.; Costa, E.d.O.; Moura-Neto, V.; Ximenes-Da-Silva, A. Evidence of Aquaporin 4 Regulation by Thyroid Hormone during Mouse Brain Development and in Cultured Human Glioblastoma Multiforme Cells. Front. Neurosci. 2019, 13, 317. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.; Jin, Z.; Elsir, T.; Pedder, H.; Nistér, M.; Alafuzoff, I.; Dimberg, A.; Edqvist, P.-H.; Pontén, F.; Aronica, E.; et al. GABA-A Channel Subunit Expression in Human Glioma Correlates with Tumor Histology and Clinical Outcome. PLoS ONE 2012, 7, e37041. [Google Scholar] [CrossRef]
- Tantillo, E.; Scalera, M.; De Santis, E.; Meneghetti, N.; Cerri, C.; Menicagli, M.; Mazzoni, A.; Costa, M.; Mazzanti, C.M.; Vannini, E.; et al. Molecular changes underlying decay of sensory responses and enhanced seizure propensity in peritumoral neurons. Neuro-Oncology 2023, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Blanchart, A.; Fernando, R.; Häring, M.; Assaife-Lopes, N.; Romanov, R.A.; Andäng, M.; Harkany, T.; Ernfors, P. Endogenous GABAA receptor activity suppresses glioma growth. Oncogene 2017, 36, 777–786. [Google Scholar] [CrossRef]
- Radin, D.P.; Tsirka, S.E. Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment. Int. J. Mol. Sci. 2020, 21, 8476. [Google Scholar] [CrossRef]
- Zamora-Sánchez, C.J.; Bello-Alvarez, C.; Rodríguez-Dorantes, M.; Camacho-Arroyo, I. Allopregnanolone Promotes Migration and Invasion of Human Glioblastoma Cells through the Protein Tyrosine Kinase c-Src Activation. Int. J. Mol. Sci. 2022, 23, 4996. [Google Scholar] [CrossRef]
- Zamora-Sánchez, C.J.; Hansberg-Pastor, V.; Salido-Guadarrama, I.; Rodríguez-Dorantes, M.; Camacho-Arroyo, I. Allopregnanolone promotes proliferation and differential gene expression in human glioblastoma cells. Steroids 2017, 119, 36–42. [Google Scholar] [CrossRef]
- Feng, Y.-H.; Lim, S.-W.; Lin, H.-Y.; Wang, S.-A.; Hsu, S.-P.; Kao, T.-J.; Ko, C.-Y.; Hsu, T.-I. Allopregnanolone suppresses glioblastoma survival through decreasing DPYSL3 and S100A11 expression. J. Steroid Biochem. Mol. Biol. 2022, 219, 106067. [Google Scholar] [CrossRef]
- Thompson, E.G.; Sontheimer, H. Acetylcholine Receptor Activation as a Modulator of Glioblastoma Invasion. Cells 2019, 8, 1203. [Google Scholar] [CrossRef]
- Pucci, S.; Bolchi, C.; Bavo, F.; Pallavicini, M.; De Palma, C.; Renzi, M.; Fucile, S.; Benfante, R.; Di Lascio, S.; Lattuada, D.; et al. Evidence of a dual mechanism of action underlying the anti-proliferative and cytotoxic effects of ammonium-alkyloxy-stilbene-based α7- and α9-nicotinic ligands on glioblastoma cells. Pharmacol. Res. 2022, 175, 105959. [Google Scholar] [CrossRef]
- Pucci, S.; Zoli, M.; Clementi, F.; Gotti, C. α9-Containing Nicotinic Receptors in Cancer. Front. Cell. Neurosci. 2021, 15, 805123. [Google Scholar] [CrossRef]
- Spina, R.; Voss, D.M.; Asnaghi, L.; Sloan, A.; Bar, E.E. Atracurium Besylate and other neuromuscular blocking agents promote astroglial differentiation and deplete glioblastoma stem cells. Oncotarget 2016, 7, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Bavo, F.; Pallavicini, M.; Pucci, S.; Appiani, R.; Giraudo, A.; Oh, H.; Kneisley, D.L.; Eaton, B.; Lucero, L.; Gotti, C.; et al. Subnanomolar Affinity and Selective Antagonism at α7 Nicotinic Receptor by Combined Modifications of 2-Triethylammonium Ethyl Ether of 4-Stilbenol (MG624). J. Med. Chem. 2023, 66, 306–332. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Alfonso, J.; Monyer, H.; Wick, W.; Winkler, F. Neuronal signatures in cancer. Int. J. Cancer 2020, 147, 3281–3291. [Google Scholar] [CrossRef]
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine Glutamate Signaling Promotes Glioma Cell Invasion. Cancer Res. 2007, 67, 9463–9471. [Google Scholar] [CrossRef]
- Nandakumar, D.; Ramaswamy, P.; Prasad, C.; Srinivas, D.; Goswami, K. Glioblastoma invasion and NMDA receptors: A novel prospect. Imaging 2019, 106, 250–260. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ye, X.; Chamberlain, M.; Mikkelsen, T.; Batchelor, T.; Desideri, S.; Piantadosi, S.; Fisher, J.; Fine, H.A. Talampanel With Standard Radiation and Temozolomide in Patients With Newly Diagnosed Glioblastoma: A Multicenter Phase II Trial. J. Clin. Oncol. 2009, 27, 4155–4161. [Google Scholar] [CrossRef]
- Cacciatore, I.; Fornasari, E.; Marinelli, L.; Eusepi, P.; Ciulla, M.; Ozdemir, O.; Tatar, A.; Turkez, H.; Di Stefano, A. Memantine-derived drugs as potential antitumor agents for the treatment of glioblastoma. Eur. J. Pharm. Sci. 2017, 109, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Tsuji, S.; Nakamura, S.; Egashira, Y.; Shimazawa, M.; Nakayama, N.; Yano, H.; Iwama, T.; Hara, H. Riluzole enhances the antitumor effects of temozolomide via suppression of MGMT expression in glioblastoma. J. Neurosurg. 2020, 134, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, G.; Konac, E.; Dere, U.A.; Emmez, H. Targeting cancer cell metabolism with metformin, dichloroacetate and memantine in glioblastoma (gbm). Turk. Neurosurg. 2021, 31, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Blyufer, A.; Lhamo, S.; Tam, C.; Tariq, I.; Thavornwatanayong, T.; Mahajan, S.S. Riluzole: A neuroprotective drug with potential as a novel anti-cancer agent (Review). Int. J. Oncol. 2021, 59, 95. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Kuner, T.; Wick, W.; Winkler, F. Synaptic input to brain tumors: Clinical implications. Neuro-Oncology 2021, 23, 23–33. [Google Scholar] [CrossRef]
- Caltabiano, R.; Torrisi, A.; Condorelli, D.; Albanese, V.; Lanzafame, S. High levels of connexin 43 mRNA in high grade astrocytomas. Study of 32 cases with in situ hybridization. Acta Histochem. 2010, 112, 529–535. [Google Scholar] [CrossRef]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef]
- Uzu, M.; Sin, W.C.; Shimizu, A.; Sato, H. Conflicting Roles of Connexin43 in Tumor Invasion and Growth in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 1159. [Google Scholar] [CrossRef]
- McCutcheon, S.; Spray, D.C. Glioblastoma–Astrocyte Connexin 43 Gap Junctions Promote Tumor Invasion. Mol. Cancer Res. 2022, 20, 319–331. [Google Scholar] [CrossRef]
- Dong, H.; Zhou, X.-W.; Wang, X.; Yang, Y.; Luo, J.-W.; Liu, Y.-H.; Mao, Q. Complex role of connexin 43 in astrocytic tumors and possible promotion of glioma-associated epileptic discharge (Review). Mol. Med. Rep. 2017, 16, 7890–7900. [Google Scholar] [CrossRef]
- Xing, L.; Yang, T.; Cui, S.; Chen, G. Connexin Hemichannels in Astrocytes: Role in CNS Disorders. Front. Mol. Neurosci. 2019, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Gielen, P.R.; Aftab, Q.; Ma, N.; Chen, V.C.; Hong, X.; Lozinsky, S.; Naus, C.C.; Sin, W.C. Connexin43 confers Temozolomide resistance in human glioma cells by modulating the mitochondrial apoptosis pathway. Neuropharmacology 2013, 75, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Ransom, B.R.; Goldring, S.; Sawyer, J.E.R.; Hennebry, J.E.; Revill, A.; Brown, A.M.; Kohn, A.; Metz, C.; Tommerdahl, M.A.; Whitsel, B.L.; et al. Ionic determinants of membrane potential of cells presumed to be glia in cerebral cortex of cat. J. Neurophysiol. 1973, 36, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Seifert, G.; Henneberger, C.; Steinhäuser, C. Diversity of astrocyte potassium channels: An update. Brain Res. Bull. 2018, 136, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, P.; Newman, E.A. Potassium buffering in the central nervous system. Neuroscience 2004, 129, 1043–1056. [Google Scholar] [CrossRef]
- Shih, P.-Y.; Savtchenko, L.P.; Kamasawa, N.; Dembitskaya, Y.; McHugh, T.J.; Rusakov, D.A.; Shigemoto, R.; Semyanov, A. Retrograde Synaptic Signaling Mediated by K+ Efflux through Postsynaptic NMDA Receptors. Cell Rep. 2013, 5, 941–951. [Google Scholar] [CrossRef]
- Tyurikova, O.; Shih, P.; Dembitskaya, Y.; Savtchenko, L.P.; McHugh, T.J.; Rusakov, D.A.; Semyanov, A. K+ efflux through postsynaptic NMDA receptors suppresses local astrocytic glutamate uptake. Glia 2022, 70, 961–974. [Google Scholar] [CrossRef]
- Kaila, K.; Lamsa, K.; Smirnov, S.; Taira, T.; Voipio, J. Long-Lasting GABA-Mediated Depolarization Evoked by High-Frequency Stimulation in Pyramidal Neurons of Rat Hippocampal Slice Is Attributable to a Network-Driven, Bicarbonate-Dependent K+Transient. J. Neurosci. 1997, 17, 7662–7672. [Google Scholar] [CrossRef]
- Voipio, J.; Kaila, K. GABAergic excitation and K+-mediated volume transmission in the hippocampus. Prog. Brain Res. 2000, 125, 329–338. [Google Scholar] [CrossRef]
- Viitanen, T.; Ruusuvuori, E.; Kaila, K.; Voipio, J. The K+-Cl− cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J. Physiol. 2010, 588, 1527–1540. [Google Scholar] [CrossRef]
- Olsen, M.L.; Sontheimer, H. Functional implications for Kir4.1 channels in glial biology: From K+ buffering to cell differentiation. J. Neurochem. 2008, 107, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Brasko, C.; Hawkins, V.; De La Rocha, I.C.; Butt, A.M. Expression of Kir4.1 and Kir5.1 inwardly rectifying potassium channels in oligodendrocytes, the myelinating cells of the CNS. Anat. Embryol. 2017, 222, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Fujita, A.; Iwai, K.; Yamada, M.; Kurachi, Y. Differential Assembly of Inwardly Rectifying K+ Channel Subunits, Kir4.1 and Kir5.1, in Brain Astrocytes. J. Biol. Chem. 2004, 279, 44065–44073. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y.; Yamamura, H.; Suzuki, Y.; Yamamura, H.; Asai, K.; et al. Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-L.; Hu, G. ATP-sensitive potassium channels: A promising target for protecting neurovascular unit function in stroke. Clin. Exp. Pharmacol. Physiol. 2010, 37, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Sun, T.; Lu, M.; Ding, J.-H.; Du, R.-H.; Hu, G. Kir6.1/K-ATP channel on astrocytes protects against dopaminergic neurodegeneration in the MPTP mouse model of Parkinson’s disease via promoting mitophagy. Brain Behav. Immun. 2019, 81, 509–522. [Google Scholar] [CrossRef]
- Chen, M.-M.; Hu, Z.-L.; Ding, J.-H.; Du, R.-H.; Hu, G. Astrocytic Kir6.1 deletion aggravates neurodegeneration in the lipopolysaccharide-induced mouse model of Parkinson’s disease via astrocyte-neuron cross talk through complement C3-C3R signaling. Brain Behav. Immun. 2021, 95, 310–320. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, G.; Xie, M.; Zhang, X.; Schools, G.P.; Ma, L.; Kimelberg, H.K.; Chen, H. TWIK-1 and TREK-1 Are Potassium Channels Contributing Significantly to Astrocyte Passive Conductance in Rat Hippocampal Slices. J. Neurosci. 2009, 29, 8551–8564. [Google Scholar] [CrossRef]
- Hwang, E.M.; Kim, E.; Yarishkin, O.; Woo, D.H.; Han, K.-S.; Park, N.; Bae, Y.; Woo, J.; Kim, D.; Park, M.; et al. A disulphide-linked heterodimer of TWIK-1 and TREK-1 mediates passive conductance in astrocytes. Nat. Commun. 2014, 5, 3227. [Google Scholar] [CrossRef]
- Du, Y.; Kiyoshi, C.M.; Wang, Q.; Wang, W.; Ma, B.; Alford, C.C.; Zhong, S.; Wan, Q.; Chen, H.; Lloyd, E.E.; et al. Genetic Deletion of TREK-1 or TWIK-1/TREK-1 Potassium Channels does not Alter the Basic Electrophysiological Properties of Mature Hippocampal Astrocytes In Situ. Front. Cell. Neurosci. 2016, 10, 13. [Google Scholar] [CrossRef]
- Price, D.L.; Ludwig, J.W.; Mi, H.; Schwarz, T.L.; Ellisman, M.H. Distribution of rSlo Ca2+-activated K+ channels in rat astrocyte perivascular endfeet. Brain Res. 2002, 956, 183–193. [Google Scholar] [CrossRef]
- Filosa, J.A.; Bonev, A.D.; Straub, S.V.; Meredith, A.L.; Wilkerson, M.K.; Aldrich, R.W.; Nelson, M.T. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat. Neurosci. 2006, 9, 1397–1403. [Google Scholar] [CrossRef]
- Carmignoto, G.; Gómez-Gonzalo, M. The contribution of astrocyte signalling to neurovascular coupling. Brain Res. Rev. 2010, 63, 138–148. [Google Scholar] [CrossRef]
- Filosa, J.A.; Iddings, J.A.; Shih, E.K.; Robinson, M.B.; Sun, S.; Li, H.; Chen, J.; Qian, Q.; Merchant, S.; Medow, M.S.; et al. Astrocyte regulation of cerebral vascular tone. Am. J. Physiol. Circ. Physiol. 2013, 305, H609–H619. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, L.W.; Black, J.A.; Waxman, S.G. Sodium channels in astroglia and microglia. Glia 2016, 64, 1628–1645. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Newcombe, J.; Waxman, S.G. Astrocytes within multiple sclerosis lesions upregulate sodium channel Nav1.5. Brain 2010, 133, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhao, Y.; Wu, H.; Jiang, N.; Wang, Z.; Lin, W.; Jin, J.; Ji, Y. Remarkable alterations of Nav1.6 in reactive astrogliosis during epileptogenesis. Sci. Rep. 2016, 6, 38108. [Google Scholar] [CrossRef] [PubMed]
- Grosche, J.; Matyash, V.; Möller, T.; Verkhratsky, A.; Reichenbach, A.; Kettenmann, H. Microdomains for neuron–glia interaction: Parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 1999, 2, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Panatier, A.; Vallée, J.; Haber, M.; Murai, K.K.; Lacaille, J.-C.; Robitaille, R. Astrocytes Are Endogenous Regulators of Basal Transmission at Central Synapses. Cell 2011, 146, 785–798. [Google Scholar] [CrossRef]
- Di Castro, M.A.; Chuquet, J.; Liaudet, N.; Bhaukaurally, K.; Santello, M.; Bouvier, D.; Tiret, P.; Volterra, A. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci. 2011, 14, 1276–1284. [Google Scholar] [CrossRef]
- Shigetomi, E.; Bushong, E.A.; Haustein, M.D.; Tong, X.; Jackson-Weaver, O.; Kracun, S.; Xu, J.; Sofroniew, M.V.; Ellisman, M.H.; Khakh, B.S. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J. Gen. Physiol. 2013, 141, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Sekiya, H.; Xu, M.; Satoh, K.; Kitajima, N.; Yoshida, K.; Okubo, Y.; Sasaki, T.; Moritoh, S.; Hasuwa, H.; et al. In Vivo Visualization of Subtle, Transient, and Local Activity of Astrocytes Using an Ultrasensitive Ca2+ Indicator. Cell Rep. 2014, 8, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Huang, B.S.; Venugopal, S.; Johnston, A.D.; Chai, H.; Zeng, H.; Golshani, P.; Khakh, B.S. Ca2+ signaling in astrocytes from Ip3r2−/− mice in brain slices and during startle responses in vivo. Nat. Neurosci. 2015, 18, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Bindocci, E.; Savtchouk, I.; Liaudet, N.; Becker, D.; Carriero, G.; Volterra, A. Three-dimensional Ca2+ imaging advances understanding of astrocyte biology. Science 2017, 356, eaai8185. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, L.; Losi, G.; Lia, A.; Melone, M.; Chiavegato, A.; Gómez-Gonzalo, M.; Sessolo, M.; Bovetti, S.; Forli, A.; Zonta, M.; et al. Interneuron-specific signaling evokes distinctive somatostatin-mediated responses in adult cortical astrocytes. Nat. Commun. 2018, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Ferrari, K.D.; Barrett, M.J.P.; Glück, C.; Stobart, M.J.; Zuend, M.; Weber, B. Cortical Circuit Activity Evokes Rapid Astrocyte Calcium Signals on a Similar Timescale to Neurons. Neuron 2018, 98, 726–735.e4. [Google Scholar] [CrossRef]
- Arizono, M.; Inavalli, V.V.G.K.; Panatier, A.; Pfeiffer, T.; Angibaud, J.; Levet, F.; Ter Veer, M.J.T.; Stobart, J.; Bellocchio, L.; Mikoshiba, K.; et al. Structural basis of astrocytic Ca2+ signals at tripartite synapses. Nat. Commun. 2020, 11, 1906. [Google Scholar] [CrossRef]
- Shigetomi, E.; Saito, K.; Sano, F.; Koizumi, S. Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders. Int. J. Mol. Sci. 2019, 20, 996. [Google Scholar] [CrossRef]
- Nagai, J.; Yu, X.; Papouin, T.; Cheong, E.; Freeman, M.R.; Monk, K.R.; Hastings, M.H.; Haydon, P.G.; Rowitch, D.; Shaham, S.; et al. Behaviorally consequential astrocytic regulation of neural circuits. Neuron 2021, 109, 576–596. [Google Scholar] [CrossRef]
- Mariotti, L.; Losi, G.; Sessolo, M.; Marcon, I.; Carmignoto, G. The inhibitory neurotransmitter GABA evokes long-lasting Ca2+ oscillations in cortical astrocytes. Glia 2015, 64, 363–373. [Google Scholar] [CrossRef]
- Durkee, C.A.; Covelo, A.; Lines, J.; Kofuji, P.; Aguilar, J.; Araque, A. Gi/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia 2019, 67, 1076–1093. [Google Scholar] [CrossRef]
- Caudal, L.C.; Gobbo, D.; Scheller, A.; Kirchhoff, F. The Paradox of Astroglial Ca2+ Signals at the Interface of Excitation and Inhibition. Front. Cell. Neurosci. 2020, 14, 609947. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Ziemens, D.; Verkhratsky, A. On the special role of NCX in astrocytes: Translating Na+-transients into intracellular Ca2+ signals. Cell Calcium 2020, 86, 102154. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V. Store-operated calcium entry in neuroglia. Neurosci. Bull. 2013, 30, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Yoast, R.E.; Emrich, S.M.; Trebak, M. The anatomy of native CRAC channel(s). Curr. Opin. Physiol. 2020, 17, 89–95. [Google Scholar] [CrossRef]
- Kwon, J.; An, H.; Sa, M.; Won, J.; Shin, J.; Lee, C.J. Orai1 and Orai3 in Combination with Stim1 Mediate the Majority of Store-operated Calcium Entry in Astrocytes. Exp. Neurobiol. 2017, 26, 42–54. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Reyes, R.C.; Parpura, V. TRP Channels Coordinate Ion Signalling in Astroglia. Ergeb. Physiol. 2014, 166, 1–22. [Google Scholar] [CrossRef]
- Shigetomi, E.; Jackson-Weaver, O.; Huckstepp, R.T.; O’Dell, T.J.; Khakh, B.S. TRPA1 Channels Are Regulators of Astrocyte Basal Calcium Levels and Long-Term Potentiation via Constitutive D-Serine Release. J. Neurosci. 2013, 33, 10143–10153. [Google Scholar] [CrossRef]
- Shigetomi, E.; Tong, X.; Kwan, K.Y.; Corey, D.P.; Khakh, B.S. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat. Neurosci. 2011, 15, 70–80. [Google Scholar] [CrossRef]
- Dunn, K.M.; Hill-Eubanks, D.C.; Liedtke, W.B.; Nelson, M.T. TRPV4 channels stimulate Ca2+ -induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc. Natl. Acad. Sci. USA 2013, 110, 6157–6162. [Google Scholar] [CrossRef]
- Sucha, P.; Hermanova, Z.; Chmelova, M.; Kirdajova, D.; Garcia, S.C.; Marchetti, V.; Vorisek, I.; Tureckova, J.; Shany, E.; Jirak, D.; et al. The absence of AQP4/TRPV4 complex substantially reduces acute cytotoxic edema following ischemic injury. Front. Cell. Neurosci. 2022, 16, 1054919. [Google Scholar] [CrossRef] [PubMed]
- Tureckova, J.; Hermanova, Z.; Marchetti, V.; Anderova, M. Astrocytic TRPV4 Channels and Their Role in Brain Ischemia. Int. J. Mol. Sci. 2023, 24, 7101. [Google Scholar] [CrossRef]
- Lewis, A.H.; Grandl, J. Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. eLife 2015, 4, e12088. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, V.; Caprini, M.; Dovizio, M.; Mylonakou, M.N.; Ferroni, S.; Ottersen, O.P.; Amiry-Moghaddam, M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, N.R.; Hermanson, O.; Heimrich, B.; Shastri, V.P. Stochastic nanoroughness modulates neuron–astrocyte interactions and function via mechanosensing cation channels. Proc. Natl. Acad. Sci. USA 2014, 111, 16124–16129. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.; Cui, Y.; Wang, H.; Jiang, J.; Zhang, T.; Sun, S.; Zhou, Z.; Zhong, Y.; Xiao, B. Astrocytic Piezo1-mediated mechanotransduction determines adult neurogenesis and cognitive functions. Neuron 2022, 110, 2984–2999.e8. [Google Scholar] [CrossRef]
- Elorza-Vidal, X.; Gaitán-Peñas, H.; Estévez, R. Chloride Channels in Astrocytes: Structure, Roles in Brain Homeostasis and Implications in Disease. Int. J. Mol. Sci. 2019, 20, 1034. [Google Scholar] [CrossRef]
- Makara, J.K.; Rappert, A.; Matthias, K.; Steinhäuser, C.; Spät, A.; Kettenmann, H. Astrocytes from mouse brain slices express ClC-2-mediated Cl− currents regulated during development and after injury. Mol. Cell. Neurosci. 2003, 23, 521–530. [Google Scholar] [CrossRef]
- Jeworutzki, E.; López-Hernández, T.; Capdevila-Nortes, X.; Sirisi, S.; Bengtsson, L.; Montolio, M.; Zifarelli, G.; Arnedo, T.; Müller, C.S.; Schulte, U.; et al. GlialCAM, a Protein Defective in a Leukodystrophy, Serves as a ClC-2 Cl− Channel Auxiliary Subunit. Neuron 2012, 73, 951–961. [Google Scholar] [CrossRef]
- Qiu, Z.; Dubin, A.E.; Mathur, J.; Tu, B.; Reddy, K.; Miraglia, L.J.; Reinhardt, J.; Orth, A.P.; Patapoutian, A. SWELL1, a Plasma Membrane Protein, Is an Essential Component of Volume-Regulated Anion Channel. Cell 2014, 157, 447–458. [Google Scholar] [CrossRef]
- Voss, F.K.; Ullrich, F.; Münch, J.; Lazarow, K.; Lutter, D.; Mah, N.; Andrade-Navarro, M.A.; von Kries, J.P.; Stauber, T.; Jentsch, T.J. Identification of LRRC8 Heteromers as an Essential Component of the Volume-Regulated Anion Channel VRAC. Science 2014, 344, 634–638. [Google Scholar] [CrossRef]
- Mongin, A.A. Volume-regulated anion channel—A frenemy within the brain. Pflügers Archiv-Eur. J. Physiol. 2016, 468, 421–441. [Google Scholar] [CrossRef]
- Osei-Owusu, J.; Yang, J.; Vitery, M.D.C.; Qiu, Z. Molecular Biology and Physiology of Volume-Regulated Anion Channel (VRAC). Curr. Top Membr. 2018, 81, 177–203. [Google Scholar] [PubMed]
- Yang, J.; Vitery, M.d.C.; Chen, J.; Osei-Owusu, J.; Chu, J.; Qiu, Z. Glutamate-Releasing SWELL1 Channel in Astrocytes Modulates Synaptic Transmission and Promotes Brain Damage in Stroke. Neuron 2019, 102, 813–827.e6. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gonzalo, M.; Zehnder, T.; Requie, L.M.; Bezzi, P.; Carmignoto, G. Insights into the release mechanism of astrocytic glutamate evoking in neurons NMDA receptor-mediated slow depolarizing inward currents. Glia 2018, 66, 2188–2199. [Google Scholar] [CrossRef]
- Fellin, T.; Pascual, O.; Gobbo, S.; Pozzan, T.; Haydon, P.G.; Carmignoto, G. Neuronal Synchrony Mediated by Astrocytic Glutamate through Activation of Extrasynaptic NMDA Receptors. Neuron 2004, 43, 729–743. [Google Scholar] [CrossRef]
- Gómez-Gonzalo, M.; Losi, G.; Chiavegato, A.; Zonta, M.; Cammarota, M.; Brondi, M.; Vetri, F.; Uva, L.; Pozzan, T.; de Curtis, M.; et al. An Excitatory Loop with Astrocytes Contributes to Drive Neurons to Seizure Threshold. PLOS Biol. 2010, 8, e1000352. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, J.; Liu, Y.; Chen, K.H.; Baraban, J.M.; Qiu, Z. Ventral tegmental area astrocytes modulate cocaine reward by tonically releasing GABA. Neuron 2023, 111, 1104–1117.e6. [Google Scholar] [CrossRef]
- Lalo, U.; Palygin, O.; Rasooli-Nejad, S.; Andrew, J.; Haydon, P.G.; Pankratov, Y. Exocytosis of ATP From Astrocytes Modulates Phasic and Tonic Inhibition in the Neocortex. PLoS Biol. 2014, 12, e1001747. [Google Scholar] [CrossRef]
- Sabirov, R.Z.; Islam, R.; Okada, T.; Merzlyak, P.G.; Kurbannazarova, R.S.; Tsiferova, N.A.; Okada, Y. The ATP-Releasing Maxi-Cl Channel: Its Identity, Molecular Partners, and Physiological/Pathophysiological Implications. Life 2021, 11, 509. [Google Scholar] [CrossRef]
- Kanai, N.; Lu, R.; Satriano, J.A.; Bao, Y.; Wolkoff, A.W.; Schuster, V.L. Identification and Characterization of a Prostaglandin Transporter. Science 1995, 268, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Sabirov, R.Z.; Merzlyak, P.G.; Okada, T.; Islam, R.; Uramoto, H.; Mori, T.; Makino, Y.; Matsuura, H.; Xie, Y.; Okada, Y. The organic anion transporter SLCO2A1 constitutes the core component of the Maxi-Cl channel. EMBO J. 2017, 36, 3309–3324. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-T.; Sabirov, R.Z.; Okada, Y. Oxygen-glucose deprivation induces ATP release via maxi-anion channels in astrocytes. Purinergic Signal. 2008, 4, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tashmukhamedov, B.A.; Inoue, H.; Okada, Y.; Sabirov, R.Z. Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 2006, 54, 343–357. [Google Scholar] [CrossRef]
- Woo, D.H.; Han, K.-S.; Shim, J.W.; Yoon, B.-E.; Kim, E.; Bae, J.Y.; Oh, S.-J.; Hwang, E.M.; Marmorstein, A.D.; Bae, Y.C.; et al. TREK-1 and Best1 Channels Mediate Fast and Slow Glutamate Release in Astrocytes upon GPCR Activation. Cell 2012, 151, 25–40. [Google Scholar] [CrossRef]
- Park, H.; Han, K.-S.; Oh, S.-J.; Jo, S.; Woo, J.; Yoon, B.-E.; Lee, C.J. High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Mol. Brain 2013, 6, 54. [Google Scholar] [CrossRef]
- Lee, S.; Yoon, B.-E.; Berglund, K.; Oh, S.-J.; Park, H.; Shin, H.-S.; Augustine, G.J.; Lee, C.J. Channel-Mediated Tonic GABA Release from Glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef]
- Yoon, B.-E.; Jo, S.; Woo, J.; Lee, J.-H.; Kim, T.; Kim, D.; Lee, C.J. The amount of astrocytic GABA positively correlates with the degree of tonic inhibition in hippocampal CA1 and cerebellum. Mol. Brain 2011, 4, 42. [Google Scholar] [CrossRef]
- Yoon, B.-E.; Woo, J.; Lee, C.J. Astrocytes as GABA-ergic and GABA-ceptive Cells. Neurochem. Res. 2012, 37, 2474–2479. [Google Scholar] [CrossRef]
- Park, H.; Han, K.-S.; Seo, J.; Lee, J.; Dravid, S.M.; Woo, J.; Chun, H.; Cho, S.; Bae, J.Y.; An, H.; et al. Channel-mediated astrocytic glutamate modulates hippocampal synaptic plasticity by activating postsynaptic NMDA receptors. Mol. Brain 2015, 8, 7. [Google Scholar] [CrossRef]
- Kwak, H.; Koh, W.; Kim, S.; Song, K.; Shin, J.-I.; Lee, J.M.; Lee, E.H.; Bae, J.Y.; Ha, G.E.; Oh, J.-E.; et al. Astrocytes Control Sensory Acuity via Tonic Inhibition in the Thalamus. Neuron 2020, 108, 691–706.e10. [Google Scholar] [CrossRef] [PubMed]
- Höft, S.; Griemsmann, S.; Seifert, G.; Steinhäuser, C. Heterogeneity in expression of functional ionotropic glutamate and GABA receptors in astrocytes across brain regions: Insights from the thalamus. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130602. [Google Scholar] [CrossRef]
- Mölders, A.; Koch, A.; Menke, R.; Klöcker, N. Heterogeneity of the astrocytic AMPA-receptor transcriptome. Glia 2018, 66, 2604–2616. [Google Scholar] [CrossRef]
- López-Bayghen, E.; Espinoza-Rojo, M.; Ortega, A. Glutamate down-regulates GLAST expression through AMPA receptors in Bergmann glial cells. Mol. Brain Res. 2003, 115, 1–9. [Google Scholar] [CrossRef]
- Saab, A.S.; Neumeyer, A.; Jahn, H.M.; Cupido, A.; Šimek, A.A.M.; Boele, H.-J.; Scheller, A.; Le Meur, K.; Götz, M.; Monyer, H.; et al. Bergmann Glial AMPA Receptors Are Required for Fine Motor Coordination. Science 2012, 337, 749–753. [Google Scholar] [CrossRef]
- Vargas, J.R.; Takahashi, D.K.; Thomson, K.E.; Wilcox, K.S. The Expression of Kainate Receptor Subunits in Hippocampal Astrocytes After Experimentally Induced Status Epilepticus. J. Neuropathol. Exp. Neurol. 2013, 72, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Kirchhoff, F. NMDA Receptors in Glia. Neuroscientist 2007, 13, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Skowrońska, K.; Obara-Michlewska, M.; Zielińska, M.; Albrecht, J. NMDA Receptors in Astrocytes: In Search for Roles in Neurotransmission and Astrocytic Homeostasis. Int. J. Mol. Sci. 2019, 20, 309. [Google Scholar] [CrossRef]
- Labrakakis, C.; Patt, S.; Hartmann, J.; Kettenmann, H. Functional GABAA receptors on human glioma cells. Eur. J. Neurosci. 1998, 10, 231–238. [Google Scholar] [CrossRef]
- Muller, T.; Fritschy, J.; Grosche, J.; Pratt, G.; Mohler, H.; Kettenmann, H. Developmental regulation of voltage-gated K+ channel and GABAA receptor expression in Bergmann glial cells. J. Neurosci. 1994, 14, 2503–2514. [Google Scholar] [CrossRef]
- Fraser, D.; Duffy, S.; Angelides, K.; Perez-Velazquez, J.; Kettenmann, H.; MacVicar, B. GABAA/benzodiazepine receptors in acutely isolated hippocampal astrocytes. J. Neurosci. 1995, 15, 2720–2732. [Google Scholar] [CrossRef]
- Egawa, K.; Yamada, J.; Furukawa, T.; Yanagawa, Y.; Fukuda, A. Cl− homeodynamics in gap junction-coupled astrocytic networks on activation of GABAergic synapses. J. Physiol. 2013, 591, 3901–3917. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.-F.; Xie, M.-J.; Zhou, M. Bicarbonate efflux via GABAA receptors depolarizes membrane potential and inhibits two-pore domain potassium channels of astrocytes in rat hippocampal slices. Glia 2012, 60, 1761–1772. [Google Scholar] [CrossRef]
- Puia, G.; Belelli, D. Neurosteroids on our minds. Trends Pharmacol. Sci. 2001, 22, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Belelli, D.; Lambert, J.J. Neurosteroids: Endogenous regulators of the GABAA receptor. Nat. Rev. Neurosci. 2005, 6, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S. Neurosteroids. Progress Brain Res. 2010, 186, 113–137. [Google Scholar] [CrossRef]
- Gotti, C.; Zoli, M.; Clementi, F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. [Google Scholar] [CrossRef]
- Shen, J.-X.; Yakel, J.L. Functional α7 Nicotinic ACh Receptors on Astrocytes in Rat Hippocampal CA1 Slices. J. Mol. Neurosci. 2012, 48, 14–21. [Google Scholar] [CrossRef]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef]
- Koukouli, F.; Changeux, J.-P. Do Nicotinic Receptors Modulate High-Order Cognitive Processing? Trends Neurosci. 2020, 43, 550–564. [Google Scholar] [CrossRef]
- Wang, X.; Lippi, G.; Carlson, D.M.; Berg, D.K. Activation of α7-containing nicotinic receptors on astrocytes triggers AMPA receptor recruitment to glutamatergic synapses. J. Neurochem. 2013, 127, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Dunphy, J.M.; Tolman, M.; Dineley, K.T.; Haydon, P.G. Septal Cholinergic Neuromodulation Tunes the Astrocyte-Dependent Gating of Hippocampal NMDA Receptors to Wakefulness. Neuron 2017, 94, 840–854.e7. [Google Scholar] [CrossRef] [PubMed]
- Lezmy, J.; Arancibia-Cárcamo, I.L.; Quintela-López, T.; Sherman, D.L.; Brophy, P.J.; Attwell, D. Astrocyte Ca2+-evoked ATP release regulates myelinated axon excitability and conduction speed. Science 2021, 374, eabh2858. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, N.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D.; Burnstock, G. Purinergic signalling in neuron–glia interactions. Nat. Rev. Neurosci. 2006, 7, 423–436. [Google Scholar] [CrossRef]
- Burnstock, G. Purine and purinergic receptors. Brain Neurosci. Adv. 2018, 2, 2398212818817494. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xie, N.; Illes, P.; Di Virgilio, F.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.-G.; Huang, C.; et al. From purines to purinergic signalling: Molecular functions and human diseases. Signal Transduct. Target. Ther. 2021, 6, 162. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Coutinho-Silva, R. Purinergic signaling, DAMPs, and inflammation. Am. J. Physiol. Physiol. 2020, 318, C832–C835. [Google Scholar] [CrossRef]
- Engel, T.; Jiménez-Mateos, E.M.; Diaz-Hernandez, M. Purinergic Signalling and Inflammation-Related Diseases. Cells 2022, 11, 3748. [Google Scholar] [CrossRef]
- Ishibashi, K.; Morishita, Y.; Tanaka, Y. The Evolutionary Aspects of Aquaporin Family. Adv. Exp. Med. Biol. 2017, 969, 35–50. [Google Scholar] [CrossRef]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized Membrane Domains for Water Transport in Glial Cells: High-Resolution Immunogold Cytochemistry of Aquaporin-4 in Rat Brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, M.; Kakita, H.; Kato, S.; Tomita, M.; Asai, K. Region-specific expression of a water channel protein, aquaporin 4, on brain astrocytes. J. Neurosci. Res. 2012, 90, 2272–2280. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of Glymphatic Pathway Function Promotes Tau Pathology after Traumatic Brain Injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef]
- Vella, J.; Zammit, C.; Di Giovanni, G.; Muscat, R.; Valentino, M. The central role of aquaporins in the pathophysiology of ischemic stroke. Front. Cell. Neurosci. 2015, 9, 108. [Google Scholar] [CrossRef]
- Filippidis, A.S.; Carozza, R.B.; Rekate, H.L. Aquaporins in Brain Edema and Neuropathological Conditions. Int. J. Mol. Sci. 2016, 18, 55. [Google Scholar] [CrossRef]
- Clément, T.; Rodriguez-Grande, B.; Badaut, J. Aquaporins in brain edema. J. Neurosci. Res. 2018, 98, 9–18. [Google Scholar] [CrossRef]
- Pannasch, U.; Rouach, N. Emerging role for astroglial networks in information processing: From synapse to behavior. Trends Neurosci. 2013, 36, 405–417. [Google Scholar] [CrossRef]
- Giaume, C.; Leybaert, L.; Naus, C.C.; Sáez, J.C. Connexin and pannexin hemichannels in brain glial cells: Properties, pharmacology, and roles. Front. Pharmacol. 2013, 4, 88. [Google Scholar] [CrossRef]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007, 94, 120–143. [Google Scholar] [CrossRef]
- Houades, V.; Koulakoff, A.; Ezan, P.; Seif, I.; Giaume, C. Gap Junction-Mediated Astrocytic Networks in the Mouse Barrel Cortex. J. Neurosci. 2008, 28, 5207–5217. [Google Scholar] [CrossRef]
- Decrock, E.; De Bock, M.; Wang, N.; Bultynck, G.; Giaume, C.; Naus, C.C.; Green, C.R.; Leybaert, L. Connexin and pannexin signaling pathways, an architectural blueprint for CNS physiology and pathology? Cell. Mol. Life Sci. 2015, 72, 2823–2851. [Google Scholar] [CrossRef] [PubMed]
- Charvériat, M.; Naus, C.C.; Leybaert, L.; Sáez, J.C.; Giaume, C. Connexin-Dependent Neuroglial Networking as a New Therapeutic Target. Front. Cell. Neurosci. 2017, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Mylvaganam, S.; Ramani, M.; Krawczyk, M.; Carlen, P.L. Roles of gap junctions, connexins, and pannexins in epilepsy. Front. Physiol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Li, Q.; Li, Q.-Q.; Jia, J.-N.; Liu, Z.-Q.; Zhou, H.-H.; Mao, X.-Y. Targeting gap junction in epilepsy: Perspectives and challenges. Biomed. Pharmacother. 2019, 109, 57–65. [Google Scholar] [CrossRef]
- Guo, A.; Zhang, H.; Li, H.; Chiu, A.; García-Rodríguez, C.; Lagos, C.F.; Sáez, J.C.; Lau, C.G. Inhibition of connexin hemichannels alleviates neuroinflammation and hyperexcitability in temporal lobe epilepsy. Proc. Natl. Acad. Sci. USA 2022, 119, e2213162119. [Google Scholar] [CrossRef]
- Sinyuk, M.; Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J. Cancer Connectors: Connexins, Gap Junctions, and Communication. Front. Oncol. 2018, 8, 646. [Google Scholar] [CrossRef]
- Zhou, M.; Zheng, M.; Zhou, X.; Tian, S.; Yang, X.; Ning, Y.; Li, Y.; Zhang, S. The roles of connexins and gap junctions in the progression of cancer. Cell Commun. Signal. 2023, 21, 8. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Lee, C.J. Reactive astrocytes in Alzheimer’s disease: A double-edged sword. Neurosci. Res. 2018, 126, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Viejo, L.; Noori, A.; Merrill, E.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Systematic review of human post-mortem immunohistochemical studies and bioinformatics analyses unveil the complexity of astrocyte reaction in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12753. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef]
- Nanclares, C.; Baraibar, A.M.; Araque, A.; Kofuji, P. Dysregulation of Astrocyte–Neuronal Communication in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7887. [Google Scholar] [CrossRef]
- Abdelhak, A.; Foschi, M.; Abu-Rumeileh, S.; Yue, J.K.; D’anna, L.; Huss, A.; Oeckl, P.; Ludolph, A.C.; Kuhle, J.; Petzold, A.; et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat. Rev. Neurol. 2022, 18, 158–172. [Google Scholar] [CrossRef]
- Asken, B.M.; Elahi, F.M.; La Joie, R.; Strom, A.; Staffaroni, A.M.; Lindbergh, C.A.; Apple, A.C.; You, M.; Weiner-Light, S.; Brathaban, N.; et al. Plasma Glial Fibrillary Acidic Protein Levels Differ Along the Spectra of Amyloid Burden and Clinical Disease Stage1. J. Alzheimer’s Dis. 2020, 78, 265–276. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Ballard, C. Overlap Between Pathology of Alzheimer Disease and Vascular Dementia. Alzheimer Dis. Assoc. Disord. 1999, 13, S115–S123. [Google Scholar] [CrossRef]
- Yi, M.; Wei, T.; Wang, Y.; Lu, Q.; Chen, G.; Gao, X.; Geller, H.M.; Chen, H.; Yu, Z. The potassium channel KCa3.1 constitutes a pharmacological target for astrogliosis associated with ischemia stroke. J. Neuroinflamm. 2017, 14, 203. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef]
- Lines, J.; Baraibar, A.M.; Fang, C.; Martin, E.D.; Aguilar, J.; Lee, M.K.; Araque, A.; Kofuji, P. Astrocyte-neuronal network interplay is disrupted in Alzheimer’s disease mice. Glia 2022, 70, 368–378. [Google Scholar] [CrossRef]
- Åbjørsbråten, K.S.; Skaaraas, G.H.S.; Cunen, C.; Bjørnstad, D.M.; Binder, K.M.G.; Bojarskaite, L.; Jensen, V.; Nilsson, L.N.; Rao, S.B.; Tang, W.; et al. Impaired astrocytic Ca2+ signaling in awake-behaving Alzheimer’s disease transgenic mice. eLife 2022, 11, e75055. [Google Scholar] [CrossRef] [PubMed]
- Lia, A.; Sansevero, G.; Chiavegato, A.; Sbrissa, M.; Pendin, D.; Mariotti, L.; Pozzan, T.; Berardi, N.; Carmignoto, G.; Fasolato, C.; et al. Rescue of astrocyte activity by the calcium sensor STIM1 restores long-term synaptic plasticity in female mice modelling Alzheimer’s disease. Nat. Commun. 2023, 14, 1590. [Google Scholar] [CrossRef] [PubMed]
- Bosson, A.; Paumier, A.; Boisseau, S.; Jacquier-Sarlin, M.; Buisson, A.; Albrieux, M. TRPA1 channels promote astrocytic Ca2+ hyperactivity and synaptic dysfunction mediated by oligomeric forms of amyloid-β peptide. Mol. Neurodegener. 2017, 12, 53. [Google Scholar] [CrossRef]
- Satoh, K.; Hata, M.; Takahara, S.; Tsuzaki, H.; Yokota, H.; Akatsu, H.; Yamamoto, T.; Kosaka, K.; Yamada, T. A novel membrane protein, encoded by the gene covering KIAA0233, is transcriptionally induced in senile plaque-associated astrocytes. Brain Res. 2006, 1108, 19–27. [Google Scholar] [CrossRef]
- Velasco-Estevez, M.; Mampay, M.; Boutin, H.; Chaney, A.; Warn, P.; Sharp, A.; Burgess, E.; Moeendarbary, E.; Dev, K.K.; Sheridan, G.K. Infection Augments Expression of Mechanosensing Piezo1 Channels in Amyloid Plaque-Reactive Astrocytes. Front. Aging Neurosci. 2018, 10, 332. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of Microglia by Amyloid β Requires P2X7 Receptor Expression. J. Immunol. 2009, 182, 4378–4385. [Google Scholar] [CrossRef]
- Orellana, J.A.; Shoji, K.F.; Abudara, V.; Ezan, P.; Amigou, E.; Sáez, P.J.; Jiang, J.X.; Naus, C.C.; Sáez, J.C.; Giaume, C. Amyloid beta-Induced Death in Neurons Involves Glial and Neuronal Hemichannels. J. Neurosci. 2011, 31, 4962–4977. [Google Scholar] [CrossRef]
- Storck, S.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 transports amyloid-β1–42 across the blood-brain barrier. J. Clin. Investig. 2016, 126, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Gallwitz, L.; Schmidt, L.; Marques, A.R.; Tholey, A.; Cassidy, L.; Ulku, I.; Multhaup, G.; Di Spiezio, A.; Saftig, P. Cathepsin D: Analysis of its potential role as an amyloid beta degrading protease. Neurobiol. Dis. 2022, 175, 105919. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K.; et al. Impaired glymphatic function and clearance of tau in an Alzheimer’s disease model. Brain 2020, 143, 2576–2593. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Prim. 2015, 1, 15017. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hänggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro-Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef]
- Li, X.; Spelat, R.; Bartolini, A.; Cesselli, D.; Ius, T.; Skrap, M.; Caponnetto, F.; Manini, I.; Yang, Y.; Torre, V. Mechanisms of malignancy in glioblastoma cells are linked to MCU upregulation and higher intracellular calcium level. J. Cell Sci. 2020, 133, jcs237503. [Google Scholar] [CrossRef]
- Venkataramani, V.; Schneider, M.; Giordano, F.A.; Kuner, T.; Wick, W.; Herrlinger, U.; Winkler, F. Disconnecting multicellular networks in brain tumours. Nat. Rev. Cancer 2022, 22, 481–491. [Google Scholar] [CrossRef]
- Lo, M.; Wang, Y.-Z.; Gout, P.W. The xc−cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.M.; Buckingham, S.C.; Campbell, S.L.; Robel, S.; Holt, K.T.; Ogunrinu-Babarinde, T.; Warren, P.P.; White, D.M.; Reid, M.A.; Eschbacher, J.M.; et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl. Med. 2015, 7, 289ra86. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Goethe, E.A.; Deneen, B.; Noebels, J.; Rao, G. The Role of Hyperexcitability in Gliomagenesis. Int. J. Mol. Sci. 2023, 24, 749. [Google Scholar] [CrossRef]
- Elias, A.F.; Lin, B.C.; Piggott, B.J. Ion Channels in Gliomas—From Molecular Basis to Treatment. Int. J. Mol. Sci. 2023, 24, 2530. [Google Scholar] [CrossRef] [PubMed]
- Litan, A.; Langhans, S.A. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef]
- Wawrzkiewicz-Jałowiecka, A.; Trybek, P.; Dworakowska, B.; Machura, Ł. Multifractal Properties of BK Channel Currents in Human Glioblastoma Cells. J. Phys. Chem. B 2020, 124, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Chanteloup, G.; Boucher, J.; Pernet, N.; Boudesco, C.; Jego, G.; Chatelier, A.; Bois, P.; Gobbo, J.; Cronier, L.; et al. Modulation of the inwardly rectifying potassium channel Kir4.1 by the pro-invasive miR-5096 in glioblastoma cells. Oncotarget 2017, 8, 37681–37693. [Google Scholar] [CrossRef]
- D’alessandro, G.; Monaco, L.; Catacuzzeno, L.; Antonangeli, F.; Santoro, A.; Esposito, V.; Franciolini, F.; Wulff, H.; Limatola, C. Radiation Increases Functional KCa3.1 Expression and Invasiveness in Glioblastoma. Cancers 2019, 11, 279. [Google Scholar] [CrossRef]
- Aissaoui, D.; Mlayah-Bellalouna, S.; Jebali, J.; Abdelkafi-Koubaa, Z.; Souid, S.; Moslah, W.; Othman, H.; Luis, J.; ElAyeb, M.; Marrakchi, N.; et al. Functional role of Kv1.1 and Kv1.3 channels in the neoplastic progression steps of three cancer cell lines, elucidated by scorpion peptides. Int. J. Biol. Macromol. 2018, 111, 1146–1155. [Google Scholar] [CrossRef]
- Chinigò, G.; Castel, H.; Chever, O.; Gkika, D. TRP Channels in Brain Tumors. Front. Cell Dev. Biol. 2021, 9, 617801. [Google Scholar] [CrossRef]
- Rubino, S.; Bach, M.D.; Schober, A.L.; Lambert, I.H.; Mongin, A.A. Downregulation of Leucine-Rich Repeat-Containing 8A Limits Proliferation and Increases Sensitivity of Glioblastoma to Temozolomide and Carmustine. Front. Oncol. 2018, 8, 142. [Google Scholar] [CrossRef]
- Synowitz, M.; Ahmann, P.; Matyash, M.; Kuhn, S.A.; Hofmann, B.; Zimmer, C.; Kirchhoff, F.; Kiwit, J.C.W.; Kettenmann, H. GABAA-receptor expression in glioma cells is triggered by contact with neuronal cells. Eur. J. Neurosci. 2001, 14, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Pallud, J.; Le Van Quyen, M.; Bielle, F.; Pellegrino, C.; Varlet, P.; Labussiere, M.; Cresto, N.; Dieme, M.-J.; Baulac, M.; Duyckaerts, C.; et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 2014, 6, 244ra89. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; Robel, S.; Cuddapah, V.A.; Robert, S.; Buckingham, S.C.; Kahle, K.T.; Sontheimer, H. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 2015, 63, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Babateen, O.; Jin, Z.; Bhandage, A.; Korol, S.V.; Westermark, B.; Nilsson, K.F.; Uhrbom, L.; Smits, A.; Birnir, B. Etomidate, propofol and diazepam potentiate GABA-evoked GABAA currents in a cell line derived from human glioblastoma. Eur. J. Pharmacol. 2015, 748, 101–107. [Google Scholar] [CrossRef]
- Huberfeld, G.; Vecht, C.J. Seizures and gliomas—Towards a single therapeutic approach. Nat. Rev. Neurol. 2016, 12, 204–216. [Google Scholar] [CrossRef]
- Puia, G.; Gullo, F.; Dossi, E.; Lecchi, M.; Wanke, E. Novel modulatory effects of neurosteroids and benzodiazepines on excitatory and inhibitory neurons excitability: A multi-electrode array recording study. Front. Neural Circuits 2012, 6, 94. [Google Scholar] [CrossRef]
- Pinacho-Garcia, L.M.; Valdez, R.A.; Navarrete, A.; Cabeza, M.; Segovia, J.; Romano, M.C. The effect of finasteride and dutasteride on the synthesis of neurosteroids by glioblastoma cells. Steroids 2019, 155, 108556. [Google Scholar] [CrossRef]
- Maas, S.; Patt, S.; Schrey, M.; Rich, A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc. Natl. Acad. Sci. USA 2001, 98, 14687–14692. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Lin, J.H.-C.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001, 7, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Corsi, L.; Mescola, A.; Alessandrini, A. Glutamate Receptors and Glioblastoma Multiforme: An Old “Route” for New Perspectives. Int. J. Mol. Sci. 2019, 20, 1796. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Tamura, M.; et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef]
- Piao, Y.; Lu, L.; de Groot, J. AMPA receptors promote perivascular glioma invasion via β1 integrin–dependent adhesion to the extracellular matrix. Neuro-Oncol. 2009, 11, 260–273. [Google Scholar] [CrossRef]
- Längle, M.; Lutz, H.; Hehlgans, S.; Rödel, F.; Rau, K.; Laube, B. NMDA Receptor-Mediated Signaling Pathways Enhance Radiation Resistance, Survival and Migration in Glioblastoma Cells—A Potential Target for Adjuvant Radiotherapy. Cancers 2019, 11, 503. [Google Scholar] [CrossRef]
- Ramaswamy, P.; Devi, N.A.; Fathima, K.H.; Nanjaiah, N.D. Activation of NMDA receptor of glutamate influences MMP-2 activity and proliferation of glioma cells. Neurol. Sci. 2014, 35, 823–829. [Google Scholar] [CrossRef]
- Lange, F.; Hörnschemeyer, J.; Kirschstein, T. Glutamatergic Mechanisms in Glioblastoma and Tumor-Associated Epilepsy. Cells 2021, 10, 1226. [Google Scholar] [CrossRef]
- Stepulak, A.; Luksch, H.; Gebhardt, C.; Uckermann, O.; Marzahn, J.; Sifringer, M.; Rzeski, W.; Staufner, C.; Brocke, K.S.; Turski, L.; et al. Expression of glutamate receptor subunits in human cancers. Histochem. Cell Biol. 2009, 132, 435–445. [Google Scholar] [CrossRef]
- Yu, L.J.; Wall, B.A.; Wangari-Talbot, J.; Chen, S. Metabotropic glutamate receptors in cancer. Neuropharmacology 2017, 115, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Salmaggi, A.; Corno, C.; Maschio, M.; Donzelli, S.; D’urso, A.; Perego, P.; Ciusani, E. Synergistic Effect of Perampanel and Temozolomide in Human Glioma Cell Lines. J. Pers. Med. 2021, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.A.; Jameson, M.J.; Broaddus, W.C.; Lin, P.S.; Chung, T.D. Nicotine enhances proliferation, migration, and radioresistance of human malignant glioma cells through EGFR activation. Brain Tumor Pathol. 2013, 30, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A. The Mystery and Magic of Glia: A Perspective on Their Roles in Health and Disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef]
- Endo, F.; Kasai, A.; Soto, J.S.; Yu, X.; Qu, Z.; Hashimoto, H.; Gradinaru, V.; Kawaguchi, R.; Khakh, B.S. Molecular basis of astrocyte diversity and morphology across the CNS in health and disease. Science 2022, 378, eadc9020. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.; et al. Uniquely Hominid Features of Adult Human Astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Anat. Embryol. 2017, 222, 2017–2029. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Alzheimer’s Disease | ||
|---|---|---|
| Channel or Receptor | Main Findings | References |
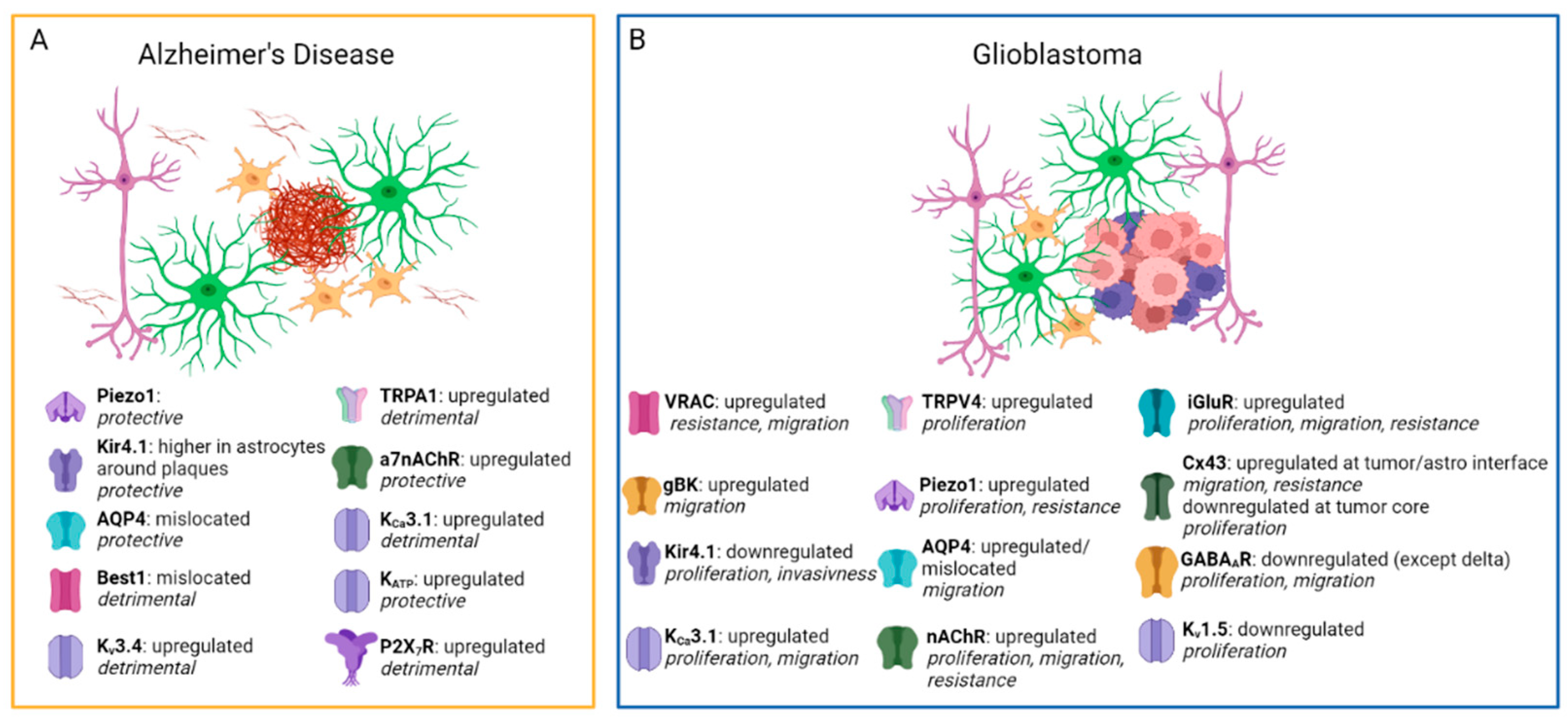
| TRPA1 | Upregulated in the late AD phase. | Lee et al. (2016) [35] |
| Pharmacological inhibition or genetic KO ameliorates AD outcomes in mice. | Lee et al. (2016) [35], Paumier et al. (2022) [36] | |
| Piezo1 | Pharmacological activation reduces Aβ accumulation and improves plasticity and memory. | Hu et al. (2023) [37] |
| Selective KO in microglia exacerbates AD pathology. | ||
| AQP4 | AQP4-KO in AD mice increases AB deposition and cognitive deficits. | Xu et al. (2015) [38] |
| AQP4 mislocalization has been found in AD patients. | Reeves et al. (2020) [39] | |
| Kir4.1 | Kir4.1 KO mice show neuronal hyperexcitability associated with AD. | Nwaobi et al. (2016) [40] |
| Kir4.1 is reduced in postmortem AD brains. | Wilcock et al. (2009) [41] | |
| Dentate gyrus astrocytes around Aβ plaque show higher Kir4.1 | Huffels et al. (2022) [42] | |
| KATP | Kir6.2 subunit is upregulated in AD mice and postmortem AD brains. | Griffith et al. (2016) [43] |
| Pharmacological activation of KATP reduces AD hallmarks and cognitive deficits in AD mice. | Liu et al. (2010) [44] | |
| Pharmacological inhibition of KATP increases Aβ deposition in mice. | Macauley et al. (2015) [45] | |
| Kv3.4 | Kv3.4 is upregulated in AD human and mouse brains. | Angulo et al. (2004) [46], Pannaccione et al. (2007) [47], Boscia et al. (2017) [48] |
| Kv3.4 silencing reduces GFAP expression and Aβ loading in mice. | Boscia et al. (2017) [48] | |
| KCa3.1 | KCa3.1 is upregulated in AD patients. | Yi et al. (2016) [49] |
| KCa3.1 blockade attenuates neuroinflammation and ameliorates cognitive deficits in AD mice. | Wei et al. (2016) [50], Yi et al. (2016) [49], Yu et al. (2018) [51] | |
| Best1 | Mediates abnormal GABA release in AD mice hippocampus and affects synaptic plasticity. | Jo et al. (2014) [52] |
| Altered localization in astrocytes from AD mice. | ||
| α7nAChR | Higher expression in astrocytes from AD patients. | Teaktong et al. (2003) [53], Yu et al. (2005) [54] |
| Activated by Aβ at physiological or pathological concentrations, affecting synaptic plasticity. | Wang et al. (2002) [55], Pirttimaki et al. (2013) [56], Gulisano et al. (2019) [57] | |
| α7nAChRsKO mice develop an AD-like pathology | Tropea et al. (2021) [58] | |
| P2X7R | Upregulated in microglia from both AD mice and postmortem AD brains. | McLarnon et al. (2006) [59]; Martínez-Frailes et al. (2019) [60] |
| Upregulated in astrocytes from AD mice. | Jin et al. (2018) [61], Martin et al. (2019) [62] | |
| P2X7R KO in AD mice reduces cognitive deficits and Aβ plaques without affecting microglia. | Martin et al. (2019) [62] | |
| Glioblastoma | ||
|---|---|---|
| Channel or Receptor | Main Findings | References |
| VRAC | Highly expressed in GBM cells. | Caramia et al. (2019) [63] |
| Promotes migration and resistance to apoptosis but is not necessary for tumor development. | Caramia et al. (2019) [63]; Liu and Stauber (2019) [64] | |
| May transport anticancer drugs like cisplatin and carboplatin. | Planells-Cases et al. (2015) [65] | |
| gBK | Overexpressed in GBM and contributes to aggressive tumor growth and migration. | Molenaar (2011) [66] |
| Inhibition reduces tumor migration only when GBM cells are in their active state. | Brandalise et al. (2020) [67] | |
| Kir4.1 | Downregulated in GBM cells. | Tan et al. (2008) [68]; Brandalise et al. (2020) [67] |
| The remaining portion of functional Kir4.1 channels may cooperate with gBK channels to promote tumor invasion. | Brandalise et al. (2020) [67] | |
| Kir4.1 loss depolarizes GBM cells and increases tumor proliferation, while Kir4.1 reintroduction reduces proliferation. | Madadi et al. (2021) [69] | |
| KCa3.1 | Elevated levels in GBM cells correlate with poor survival. | Brown et al. (2018) [70]; Hausmann et al. (2023) [71] |
| Contribute to GBM cell migration by influencing Ca2+ signaling. | Catacuzzeno and Franciolini (2018) [72] | |
| Silencing and inhibition reduce tumor infiltration and improve survival in mouse models. | Brown et al. (2018) [70] | |
| Kv1.3, Kv1.5 | Inverse correlation between Kv1.5 expression and glioma malignancy (no such association was observed for Kv1.3). | Preußat et al. (2003) [73]; Arvind et al. (2012) [74] |
| Kv1.3 inhibition in glial populations regulates astrocyte and microglia reactivity, reducing tumor growth and invasiveness. | Grimaldi et al. (2018) [75] | |
| Kv1.3 inhibitors can induce cell death in glioma cells. | Venturini et al. (2017) [76] | |
| TRPV4 | Overexpressed in malignant gliomas like GBM. Negatively correlated with the prognosis. | Yang et al. (2020) [77] |
| CBD treatment causes TRPV4-mediated calcium influx triggered mitophagy and consequent glioma cell death. | Huang et al. (2021) [78] | |
| PIEZO1 | Overexpressed in malignant gliomas like GBM. Negatively correlates with the prognosis. | Chen et al. (2018) [79] |
| ECM stiffness activates PIEZO1 and increases its expression, promoting GBM invasiveness. | ||
| AQPs | AQPs facilitate tumor mobility, survival, and growth through diverse mechanisms. | Varricchio et al. (2021) [80] |
| AQP4 overexpression and redistribution in GBM are linked to increased cell migration. | Vandebroek and Yasui (2020) [81] | |
| T3 hormone may decrease AQP4 in GBM cells, leading to reduced tumor growth and migration. | Costa et al. (2019) [82] | |
| GABAAR | The majority of GABAAR subunits are downregulated in GBM, except for the δ subunit, which is upregulated. | Smits et al. (2012) [83]; Tantillo et al. (2023) [84] |
| GABA signaling decreases GBM proliferation and growth, but it may lead to epilepsy development. | Blanchart et al. (2017) [85]; Radin and Tsirka (2020) [86]; Tantillo et al. (2023) [84] | |
| GABAAR activity supports tumor cell quiescence. After tumor resection, its reduced activity may be responsible for GBM recurrence. | Smits et al. (2012) [83]; Blanchart et al. (2017) [85] | |
| Neurosteroids could modulate GBM cell line biology. | Zamora-Sánchez et al. (2017, 2022) [87,88]; Feng YH. et al. (2022) [89] | |
| nAchR | Few subtypes of nAchR are expressed in GBM cells. | Thompson et al. (2019) [90]; Pucci et al. (2022) [91] |
| nAchR controls the survival, proliferation, and infiltrative capacity of GBM. | Thompson et al. (2019) [90]; Pucci et al. (2021) [92] | |
| Activation of nAchR produces an increase in [Ca2+]int that leads to intracellular pathways activation (Akt, ERK). | Pucci et al. (2022) [91] | |
| nAchR antagonists decrease the viability, proliferation, and migration of GBM cells. | Spina et al. (2016) [93]; Pucci et al. (2022) [91]; Bavo et al. (2023) [94] | |
| iGluRs | Stimulation of iGluRs leads to GBM progression and contributes to seizures in the peritumoral area. | Venkataramani et al. (2019) [95]; Jung et al. (2020) [96] |
| NMDAR, AMPAR, and KAR expressed in GBM can be activated by glutamate released by neurons and by GBM itself. | Lyons et al. (2007) [97]; Nandakumar et al. (2019) [98]; Venkataramani et al. (2019) [95]; Venkatesh et al. (2019) [99] | |
| Ca2+ permeable AMPARs and NMDARs promote tumor progression in the glutamate-rich environment of GBM. | Nandakumar et al. (2019) [98] | |
| iGluR antagonists may slow GBM progression while limiting neurodegeneration and seizure onset. | Grossman et al. (2009) [100]; Cacciatore et al. (2017) [101]; Yamada et al. (2020) [102]; Albayrak et al. (2021) [103]; Blyufer et al. (2021) [104]; Venkataramani et al. (2021) [105] | |
| Cx43 | Cx43 expression is reduced in the tumor core and increased at the tumor–astrocyte interface. Due to its heterogeneous distribution, Cx43 acts both as a tumor suppressor and a promoter of cell migration. | Caltabiano et al. (2010) [106]; Sin et al. (2016) [107]; Uzu et al. (2018) [108]; McCutcheon and Spray (2022) [109] |
| Cx43 regulates apoptosis, impacts cell homeostasis, and promotes epileptic activity in the peritumoral zone. | Sin et al. (2016) [107]; Dong et al. (2017) [110]; Xing et al. (2019) [111] | |
| Cx43 expression in GBM cells is associated with the development of resistance to TMZ treatment. | Gielen et al. (2013) [112] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lia, A.; Di Spiezio, A.; Vitalini, L.; Tore, M.; Puja, G.; Losi, G. Ion Channels and Ionotropic Receptors in Astrocytes: Physiological Functions and Alterations in Alzheimer’s Disease and Glioblastoma. Life 2023, 13, 2038. https://doi.org/10.3390/life13102038
Lia A, Di Spiezio A, Vitalini L, Tore M, Puja G, Losi G. Ion Channels and Ionotropic Receptors in Astrocytes: Physiological Functions and Alterations in Alzheimer’s Disease and Glioblastoma. Life. 2023; 13(10):2038. https://doi.org/10.3390/life13102038
Chicago/Turabian StyleLia, Annamaria, Alessandro Di Spiezio, Lorenzo Vitalini, Manuela Tore, Giulia Puja, and Gabriele Losi. 2023. "Ion Channels and Ionotropic Receptors in Astrocytes: Physiological Functions and Alterations in Alzheimer’s Disease and Glioblastoma" Life 13, no. 10: 2038. https://doi.org/10.3390/life13102038