
Phosphorylation and Dephosphorylation of Beta-Amyloid Peptide in Model Cell Cultures: The Role of Cellular Protein Kinases and Phosphatases
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Beta-Amyloid Peptides
- Synthetic beta-amyloid peptide Aβ42:
2.2. Cell Culture
2.3. The Expression and Purification of Casein Kinase 2α (CK2α)
2.4. Phosphorylation and Dephosphorylation of Aβ in Cell Lysates
2.5. Phosphorylation of Aβ by Cell Surface Kinases
2.6. Intracellular Phosphorylation of Exogenous Aβ
2.7. Immunoprecipitation of Samples
2.8. Analysis of Phosphorylation and Dephosphorylation of Aβ Using Monoclonal Antibodies
2.9. Analysis of CK2α and β-Actin Expression in bEnd.3 and HEK293 Cell Lines
2.10. Analysis of the Degree of Aβ Phosphorylation by Autoradiography
2.11. Synthesis of 125I-Labeled Protein Standards
2.11.1. Synthesis of 125I-Aβ
2.11.2. Synthesis of 125I-Bovine Serum Albumin
2.12. Statistical Data Processing
3. Results
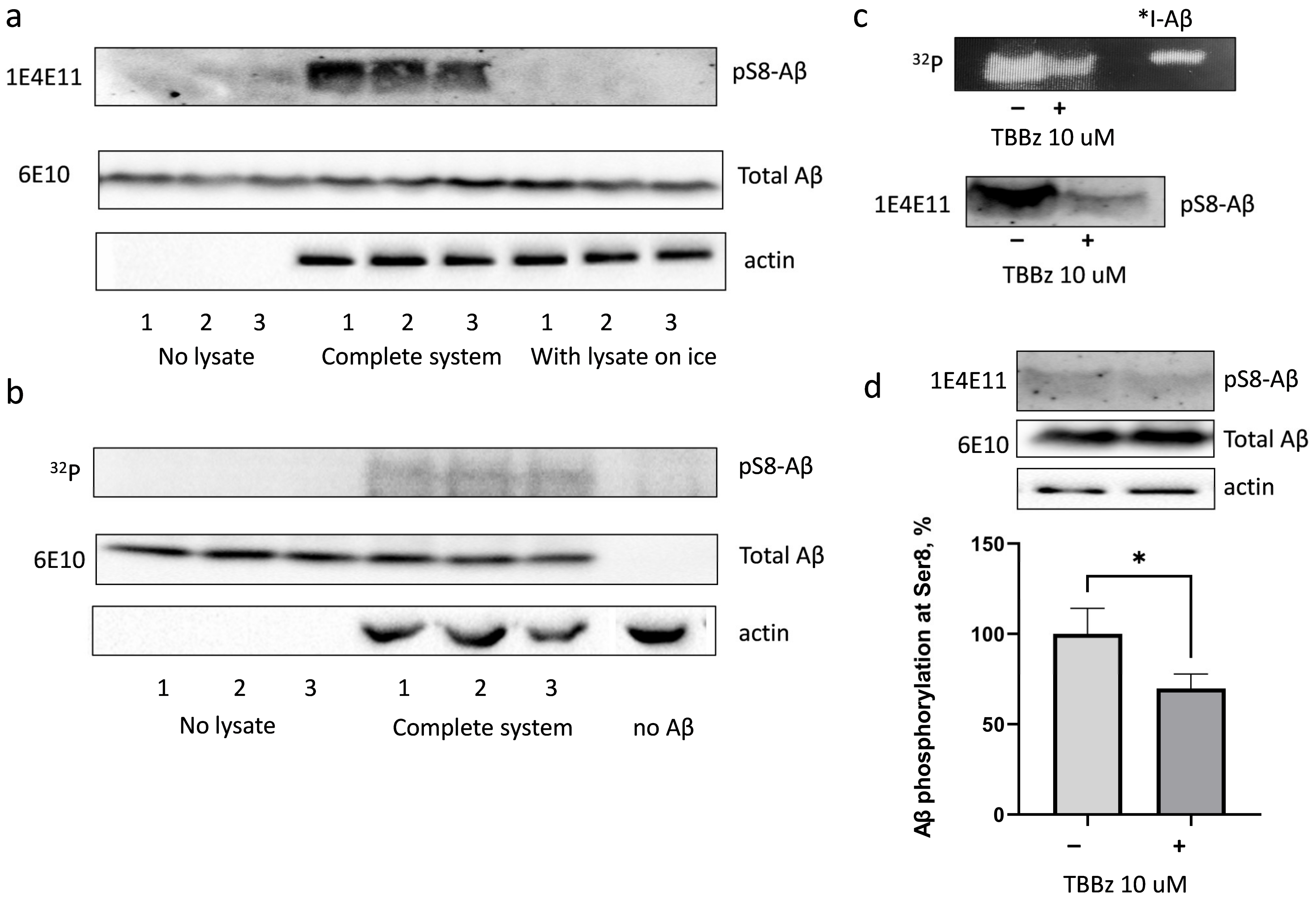
3.1. Amyloid Beta Is Phosphorylated in HEK293 Cell Lysate at Ser8 with the Involvement of Casein Kinase 2
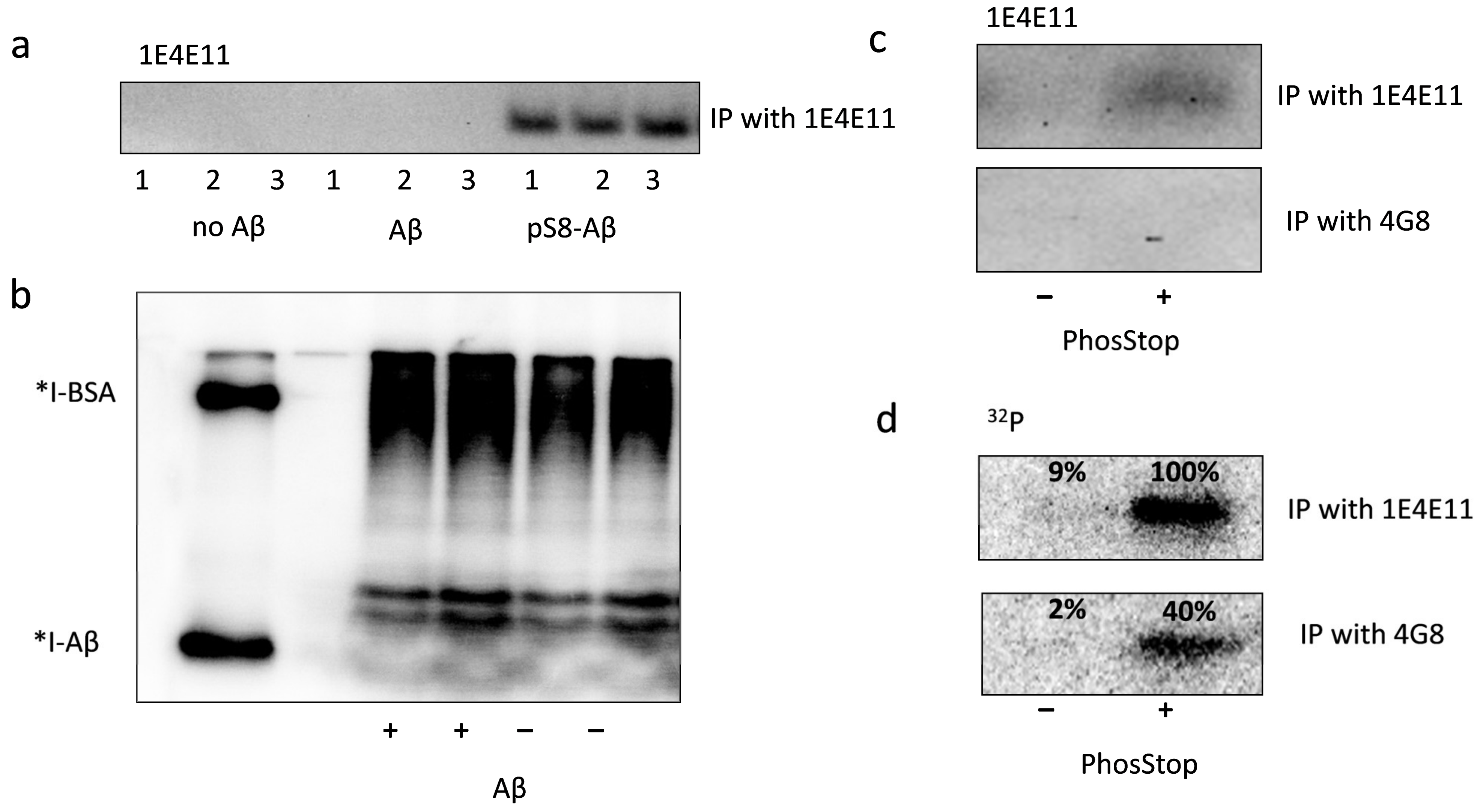
3.2. Exogenous Beta-Amyloid Is Not Phosphorylated by HEK293 Cells after Internalisation but Is Phosphorylated by Surface Kinases of These Cells
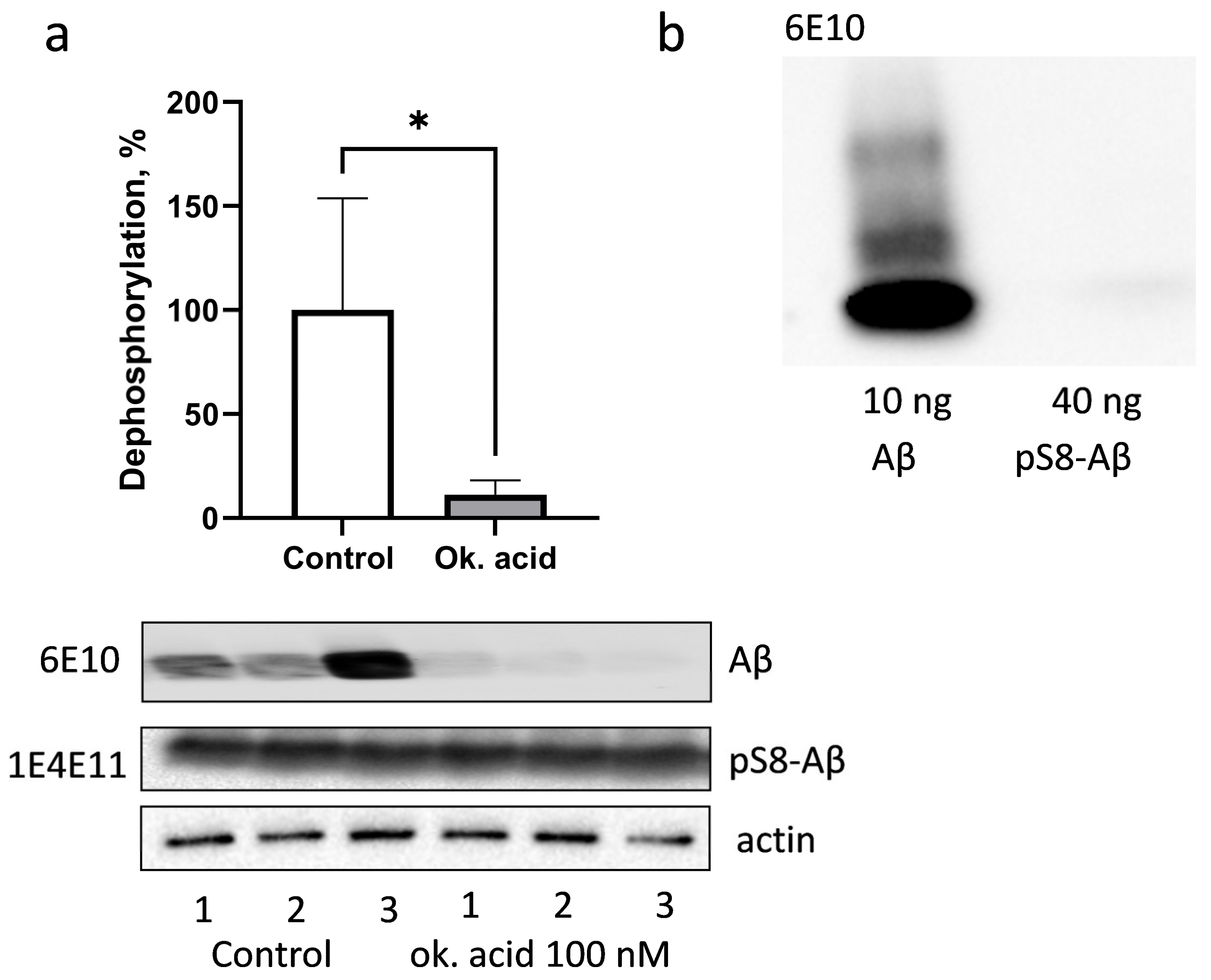
3.3. PP1 and PP2A Phosphatases Participate in Dephosphorylation of pS8-Aβ by HEK293 Cells
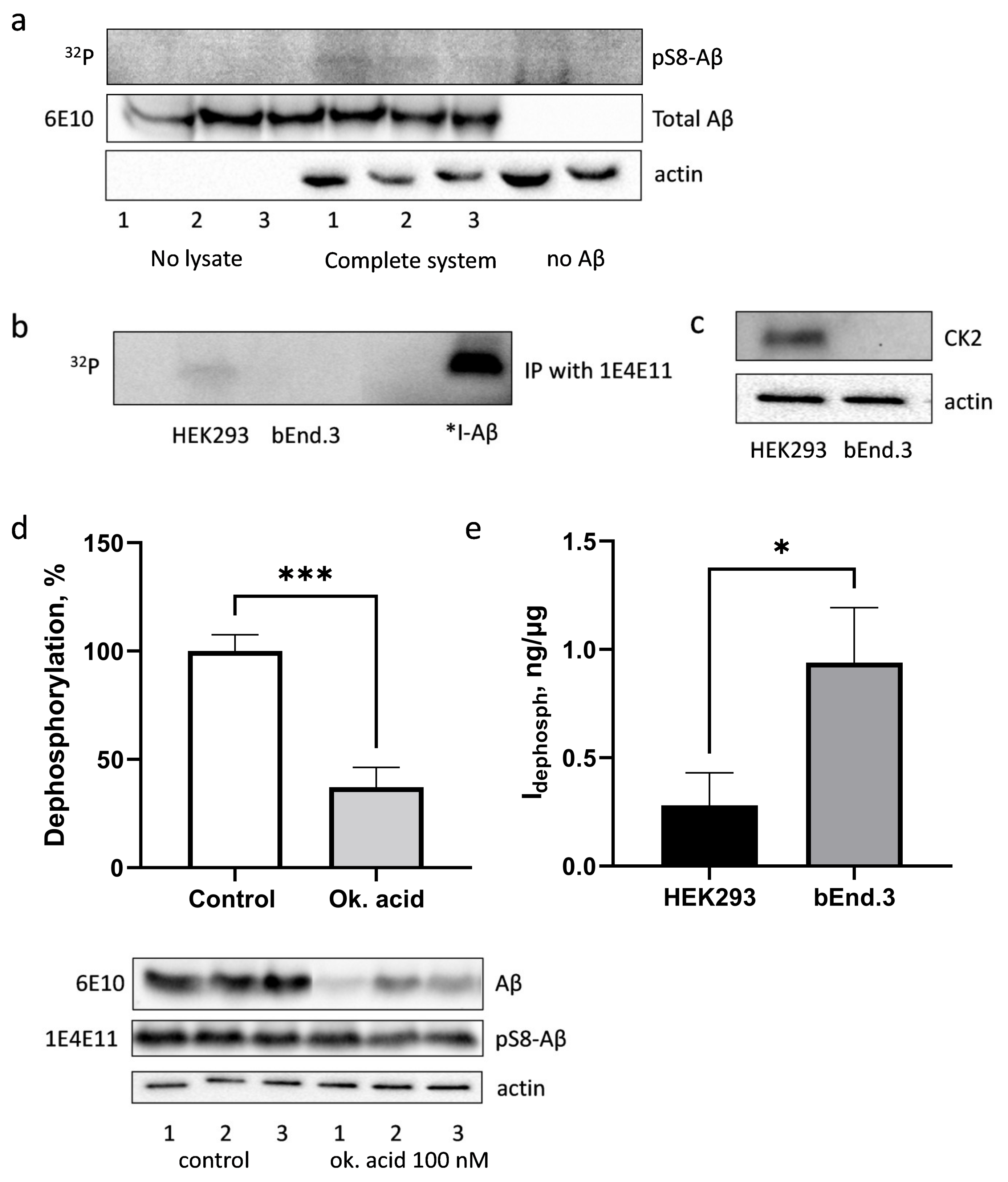
3.4. Phosphorylation and Dephosphorylation of Aβ by bEnd.3 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Musiek, E.S.; Holtzman, D.M. Three Dimensions of the Amyloid Hypothesis: Time, Space, and “Wingmen”. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barage, S.H.; Sonawane, K.D. Amyloid Cascade Hypothesis: Pathogenesis and Therapeutic Strategies in Alzheimer’s Disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.; Bieger, S.C.; Brühl, B.; Tienari, P.J.; Ida, N.; Allsop, D.; Roberts, G.W.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; et al. Distinct Sites of Intracellular Production for Alzheimer’s Disease Aβ40/42 Amyloid Peptides. Nat. Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.A.; Makarov, A.A. Amyloid β Modification: A Key to the Sporadic Alzheimer’s Disease? Front. Genet. 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummer, M.P.; Heneka, M.T. Truncated and Modified Amyloid-Beta Species. Alzheimer’s Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate Modified Amyloid-Beta in Ageing and Alzheimer’s Disease. Acta Neuropathol. Commun. 2018, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Wirths, O.; Theil, S.; Gerth, J.; Bayer, T.A.; Walter, J. Early Intraneuronal Accumulation and Increased Aggregation of Phosphorylated Abeta in a Mouse Model of Alzheimer’s Disease. Acta Neuropathol. 2013, 125, 699–709. [Google Scholar] [CrossRef]
- Kumar, S.; Frost, J.L.; Cotman, C.W.; Head, E.; Palmour, R.; Lemere, C.A.; Walter, J. Deposition of Phosphorylated Amyloid-β in Brains of Aged Nonhuman Primates and Canines. Brain Pathol. 2018, 28, 427–430. [Google Scholar] [CrossRef]
- Jamasbi, E.; Separovic, F.; Akhter Hossain, M.; Donato Ciccotosto, G. Phosphorylation of a Full Length Amyloid-β Peptide Modulates Its Amyloid Aggregation, Cell Binding and Neurotoxic Properties. Mol. Biosyst. 2017, 13, 1545–1551. [Google Scholar] [CrossRef] [Green Version]
- Barykin, E.P.; Petrushanko, I.Y.; Kozin, S.A.; Telegin, G.B.; Chernov, A.S.; Lopina, O.D.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Phosphorylation of the Amyloid-Beta Peptide Inhibits Zinc-Dependent Aggregation, Prevents Na, K-ATPase Inhibition, and Reduces Cerebral Plaque Deposition. Front. Mol. Neurosci. 2018, 11, 302. [Google Scholar] [CrossRef]
- Kumar, S.; Rezaei-Ghaleh, N.; Terwel, D.; Thal, D.R.; Richard, M.; Hoch, M.; Mc Donald, J.M.; Wullner, U.; Glebov, K.; Heneka, M.T.; et al. Extracellular Phosphorylation of the Amyloid Beta-Peptide Promotes Formation of Toxic Aggregates during the Pathogenesis of Alzheimer’s Disease. Embo J. 2011, 30, 2255–2265. [Google Scholar] [CrossRef] [Green Version]
- Tseng, B.P.; Green, K.N.; Chan, J.L.; Blurton-Jones, M.; LaFerla, F.M. Aβ Inhibits the Proteasome and Enhances Amyloid and Tau Accumulation. Neurobiol. Aging 2008, 29, 1607–1618. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Beal, M.F. Amyloid Beta, Mitochondrial Dysfunction and Synaptic Damage: Implications for Cognitive Decline in Aging and Alzheimer’s Disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Hunter, T. Protein Kinases and Phosphatases: The Yin and Yang of Protein Phosphorylation and Signaling. Cell 1995, 80, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, W.L. Abeta Toxicity in Alzheimer’s Disease: Globular Oligomers (ADDLs) as New Vaccine and Drug Targets. Neurochem. Int. 2002, 41, 345–352. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Mitkevich, V.A.; Anashkina, A.A.; Adzhubei, A.A.; Burnysheva, K.M.; Lakunina, V.A.; Kamanina, Y.V.; Dergousova, E.A.; Lopina, O.D.; Ogunshola, O.O.; et al. Direct Interaction of Beta-Amyloid with Na, K-ATPase as a Putative Regulator of the Enzyme Function. Sci. Rep. 2016, 6, 27738. [Google Scholar] [CrossRef] [Green Version]
- Zimina, E.P.; Fritsch, A.; Schermer, B.; Bakulina, A.Y.; Bashkurov, M.; Benzing, T.; Bruckner-Tuderman, L. Extracellular Phosphorylation of Collagen XVII by Ecto-Casein Kinase 2 Inhibits Ectodomain Shedding. J. Biol. Chem. 2007, 282, 22737–22746. [Google Scholar] [CrossRef] [Green Version]
- Marin, O.; Meggio, F.; Draetta, G.; Pinna, L.A. The Consensus Sequences for Cdc2 Kinase and for Casein Kinase-2 Are Mutually Incompatible. A Study with Peptides Derived from the Beta-Subunit of Casein Kinase-2. FEBS Lett 1992, 301, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Nagele, R.G.; D’Andrea, M.R.; Anderson, W.J.; Wang, H.-Y. Intracellular Accumulation of β-Amyloid1–42 in Neurons Is Facilitated by the A7 Nicotinic Acetylcholine Receptor in Alzheimer’s Disease. Neuroscience 2002, 110, 199–211. [Google Scholar] [CrossRef]
- Fonte, V.; Kapulkin, W.J.; Taft, A.; Fluet, A.; Friedman, D.; Link, C.D. Interaction of Intracellular β Amyloid Peptide with Chaperone Proteins. PNAS 2002, 99, 9439–9444. [Google Scholar] [CrossRef]
- Lee, H.-K.; Kumar, P.; Fu, Q.; Rosen, K.M.; Querfurth, H.W. The Insulin/Akt Signaling Pathway Is Targeted by Intracellular β-Amyloid. Mol. Biol. Cell 2009, 20, 1533–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P. The Role of Protein Phosphorylation in Human Health and Disease. Eur. J. Biochem. 2001, 268, 5001–5010. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau Phosphorylation: The Therapeutic Challenge for Neurodegenerative Disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and Tau: Two Convergence Points in Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 33, S141–S144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, D.S.; Van Etten, R.A. Tyrosine Kinases as Targets for Cancer Therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Kristjánsdóttir, K.; Rudolph, J. Cdc25 Phosphatases and Cancer. Chem. Biol. 2004, 11, 1043–1051. [Google Scholar] [CrossRef] [Green Version]
- Tonks, N.K. Protein Tyrosine Phosphatases: From Genes, to Function, to Disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-Related Changes in AMPK Activation: Role for AMPK Phosphatases and Inhibitory Phosphorylation by Upstream Signaling Pathways. Ageing Res. Rev. 2016, 28, 15–26. [Google Scholar] [CrossRef]
- Abidi, P.; Leers-Sucheta, S.; Cortez, Y.; Han, J.; Azhar, S. Evidence That Age-Related Changes in P38 MAP Kinase Contribute to the Decreased Steroid Production by the Adrenocortical Cells from Old Rats. Aging Cell 2008, 7, 168–178. [Google Scholar] [CrossRef]
- Pahlavani, M.A.; Vargas, D.M. Age-Related Decline in Activation of Calcium/Calmodulin-Dependent Phosphatase Calcineurin and Kinase CaMK-IV in Rat T Cells. Mech. Ageing Dev. 1999, 112, 59–74. [Google Scholar] [CrossRef]
- Aslantas, E.E.; Buzoglu, H.D.; Karapinar, S.P.; Cehreli, Z.C.; Muftuoglu, S.; Atilla, P.; Aksoy, Y. Age-Related Changes in the Alkaline Phosphatase Activity of Healthy and Inflamed Human Dental Pulp. J. Endod. 2016, 42, 131–134. [Google Scholar] [CrossRef]
- Liu, F.; Benashski, S.E.; Persky, R.; Xu, Y.; Li, J.; McCullough, L.D. Age-Related Changes in AMP-Activated Protein Kinase after Stroke. AGE 2012, 34, 157–168. [Google Scholar] [CrossRef]
- Kudo, K.; Wati, H.; Qiao, C.; Arita, J.; Kanba, S. Age-Related Disturbance of Memory and CREB Phosphorylation in CA1 Area of Hippocampus of Rats. Brain Res. 2005, 1054, 30–37. [Google Scholar] [CrossRef]
- Lee, S.J.; Chung, Y.H.; Joo, K.M.; Lim, H.C.; Jeon, G.S.; Kim, D.; Lee, W.B.; Kim, Y.S.; Cha, C.I. Age-Related Changes in Glycogen Synthase Kinase 3β (GSK3β) Immunoreactivity in the Central Nervous System of Rats. Neurosci. Lett. 2006, 409, 134–139. [Google Scholar] [CrossRef]
- Perluigi, M.; Barone, E.; Di Domenico, F.; Butterfield, D.A. Aberrant Protein Phosphorylation in Alzheimer Disease Brain Disturbs Pro-Survival and Cell Death Pathways. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 1871–1882. [Google Scholar] [CrossRef]
- Bugrova, A.E.; Strelnikova, P.A.; Indeykina, M.I.; Kononikhin, A.S.; Zakharova, N.V.; Brzhozovskiy, A.G.; Barykin, E.P.; Pekov, S.I.; Gavrish, M.S.; Babaev, A.A.; et al. The Dynamics of β-Amyloid Proteoforms Accumulation in the Brain of a 5xFAD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 27. [Google Scholar] [CrossRef]
- Kumar, S.; Kapadia, A.; Theil, S.; Joshi, P.; Riffel, F.; Heneka, M.T.; Walter, J. Novel Phosphorylation-State Specific Antibodies Reveal Differential Deposition of Ser26 Phosphorylated Aβ Species in a Mouse Model of Alzheimer’s Disease. Front. Mol. Neurosci. 2021, 13. [Google Scholar] [CrossRef]
- Nunan, J.; Small, D.H. Regulation of APP Cleavage by α-, β- and γ-Secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Koo, E.H.; Squazzo, S.L. Evidence That Production and Release of Amyloid Beta-Protein Involves the Endocytic Pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar] [CrossRef]
- Zheng, L.; Calvo-Garrido, J.; Hallbeck, M.; Hultenby, K.; Marcusson, J.; Cedazo-Minguez, A.; Terman, A. Intracellular Localization of Amyloid-β Peptide in SH-SY5Y Neuroblastoma Cells. J. Alzheimer’s Dis. 2013, 37, 713–733. [Google Scholar] [CrossRef]
- Aksenova, M.V.; Burbaeva, G.S.; Kandror, K.V.; Kapkov, D.V.; Stepanov, A.S. The Decreased Level of Casein Kinase 2 in Brain Cortex of Schizophrenic and Alzheimer’s Disease Patients. FEBS Lett. 1991, 279, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iimoto, D.S.; Masliah, E.; DeTeresa, R.; Terry, R.D.; Saitoh, T. Aberrant Casein Kinase II in Alzheimer’s Disease. Brain Res. 1990, 507, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Klement, E.; Medzihradszky, K.F. Extracellular Protein Phosphorylation, the Neglected Side of the Modification. Mol. Cell. Proteom. 2017, 16, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, J.; Schindzielorz, A.; Hartung, B.; Haass, C. Phosphorylation of the β-Amyloid Precursor Protein at the Cell Surface by Ectocasein Kinases 1 and 2. J. Biol. Chem. 2000, 275, 23523–23529. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Holmes, C.F.B.; Tsukitani, Y. Okadaic Acid: A New Probe for the Study of Cellular Regulation. Trends Biochem. Sci. 1990, 15, 98–102. [Google Scholar] [CrossRef]
- Gerth, J.; Kumar, S.; Rijal Upadhaya, A.; Ghebremedhin, E.; von Arnim, C.A.F.; Thal, D.R.; Walter, J. Modified Amyloid Variants in Pathological Subgroups of β-Amyloidosis. Ann. Clin. Transl. Neurol. 2018, 5, 815–831. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Sample Type | Sample, μL | IP Wash Buffer, μL | Magnetic Beads Suspension, μL | PhosStop, μL |
|---|---|---|---|---|---|
| Phosphorylation in cell lysates | Reaction mixture | 30 | 470 | 20 | 50 |
| Intracellular phosphorylation of exogenous Aβ | Cell lysate | 120 | 450 | 10 | 50 |
| Phosphorylation of Aβ by cell surface kinases | Cell medium | 500 | 500 | 15 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barykin, E.P.; Yanvarev, D.V.; Strelkova, M.A.; Valuev-Elliston, V.T.; Varshavskaya, K.B.; Mitkevich, V.A.; Makarov, A.A. Phosphorylation and Dephosphorylation of Beta-Amyloid Peptide in Model Cell Cultures: The Role of Cellular Protein Kinases and Phosphatases. Life 2023, 13, 147. https://doi.org/10.3390/life13010147
Barykin EP, Yanvarev DV, Strelkova MA, Valuev-Elliston VT, Varshavskaya KB, Mitkevich VA, Makarov AA. Phosphorylation and Dephosphorylation of Beta-Amyloid Peptide in Model Cell Cultures: The Role of Cellular Protein Kinases and Phosphatases. Life. 2023; 13(1):147. https://doi.org/10.3390/life13010147
Chicago/Turabian StyleBarykin, Evgeny P., Dmitry V. Yanvarev, Maria A. Strelkova, Vladimir T. Valuev-Elliston, Kseniya B. Varshavskaya, Vladimir A. Mitkevich, and Alexander A. Makarov. 2023. "Phosphorylation and Dephosphorylation of Beta-Amyloid Peptide in Model Cell Cultures: The Role of Cellular Protein Kinases and Phosphatases" Life 13, no. 1: 147. https://doi.org/10.3390/life13010147