
Pharmacological Targets in Chronic Heart Failure with Reduced Ejection Fraction


Abstract
:1. Introduction
2. Materials and Methods
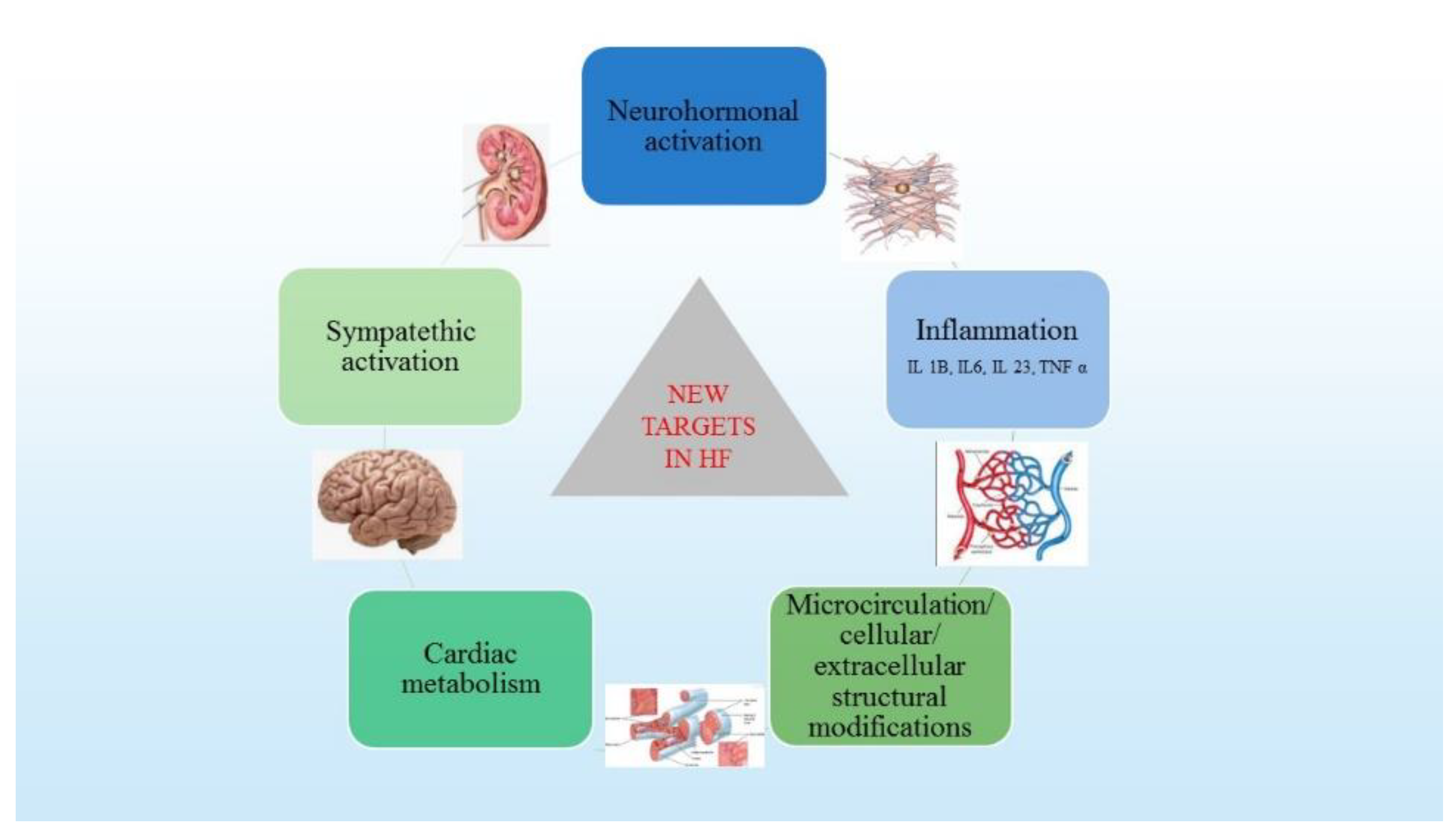
3. Pathophysiological Mechanisms in HF
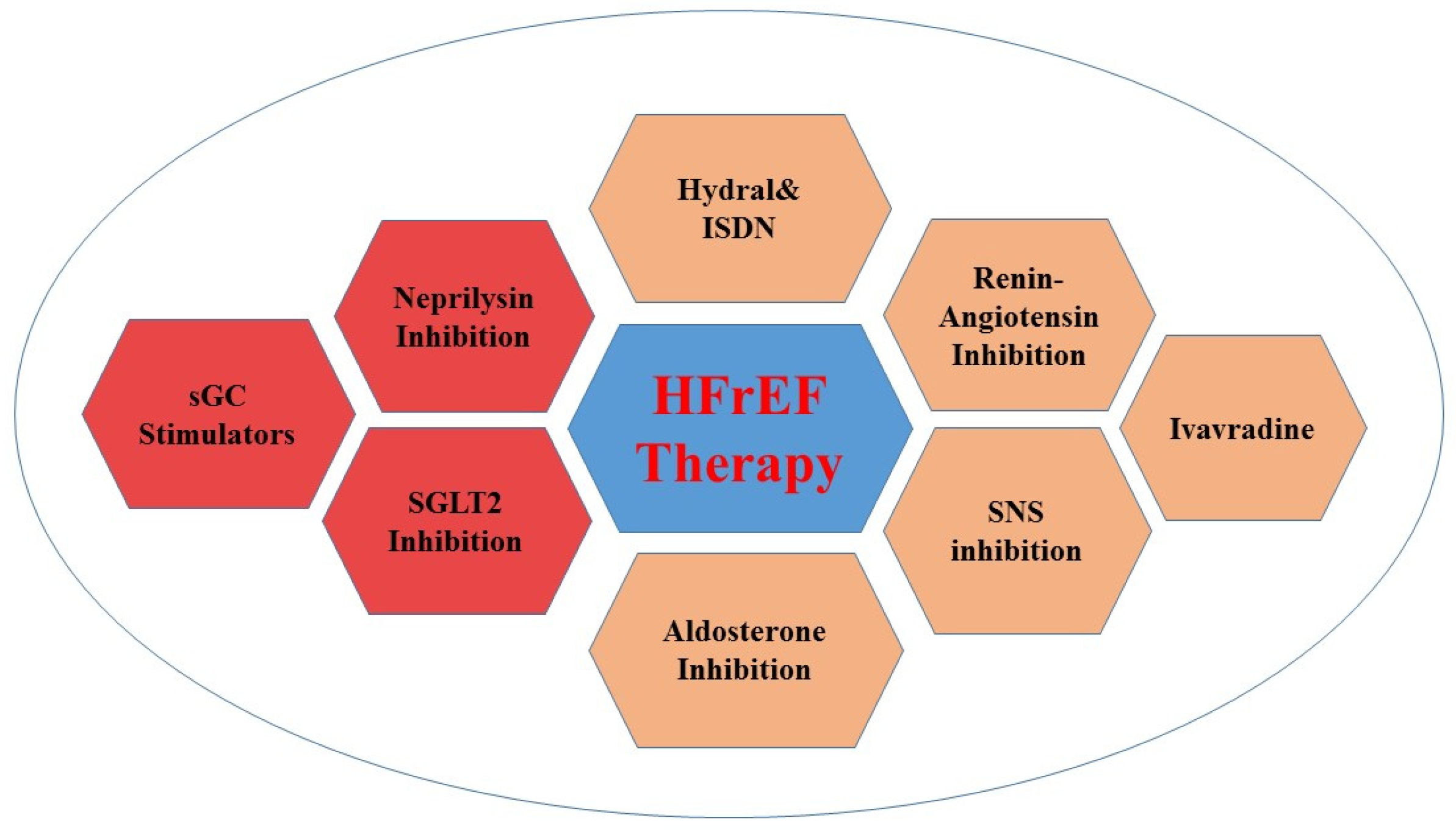
4. Guideline Recommendations for Therapeutic Targets
4.1. Novel Guideline Therapeutic Targets
4.2. New Approaches for Studying the Additional Benefits of Some Previous Therapeutic Targets
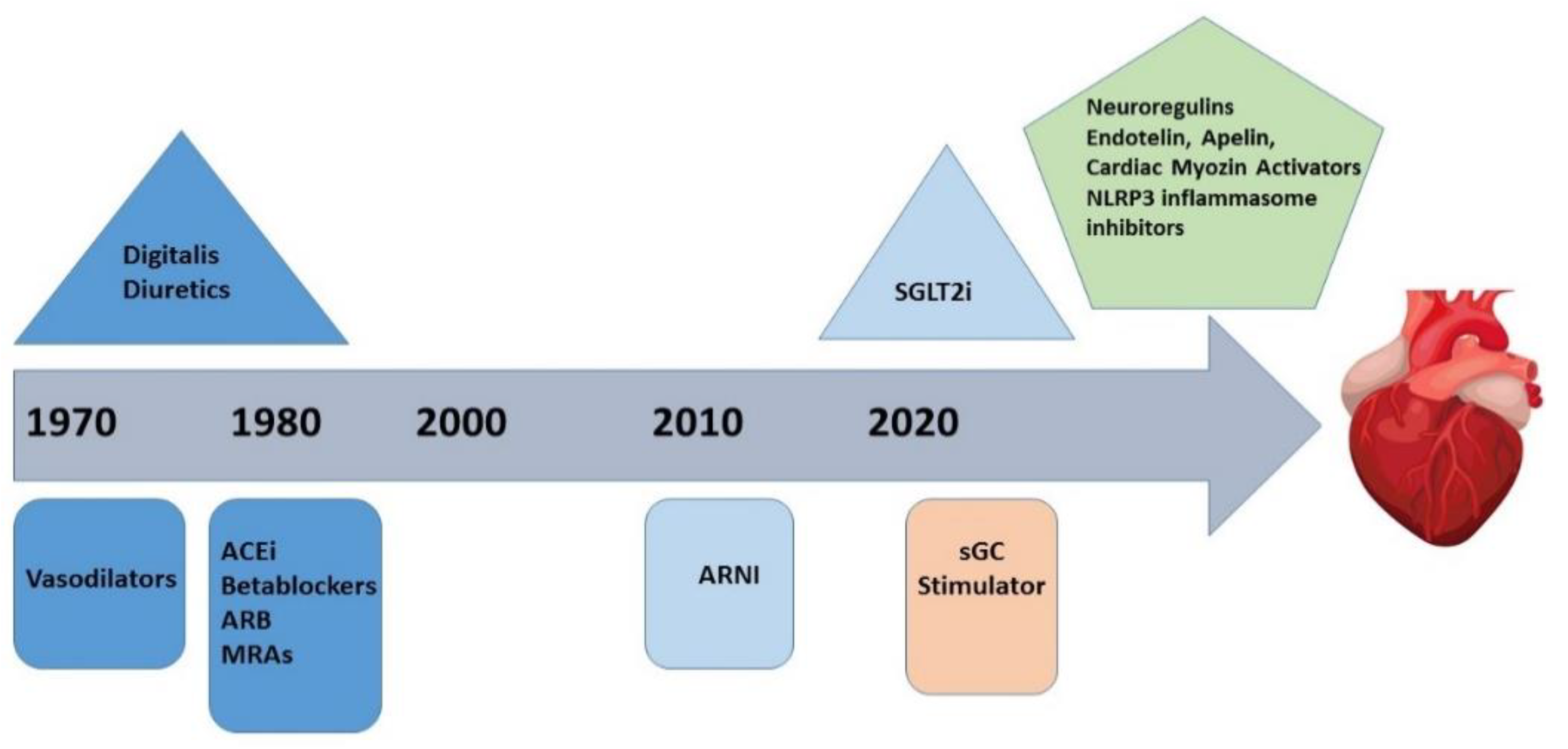
5. Evolving Therapeutic Targets
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pellicori, P.; Khan, M.J.I.; Graham, F.J.; Cleland, J.G.F. New perspectives and future directions in the treatment of heart failure. Heart Fail. Rev. 2020, 25, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correale, M.; Tricarico, L.; Fortunato, M.; Mazzeo, P.; Nodari, S.; Di Biase, M.; Brunetti, N.D. New Targets in Heart Failure Drug Therapy. Front. Cardiovasc. Med. 2021, 8, 665797. [Google Scholar] [CrossRef] [PubMed]
- Nabeebaccus, A.; Zheng, S.; Shah, A.M. Heart failure-potential new targets for therapy. Br. Med. Bull. 2016, 1, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Tkacs, N.C. Current concepts of neurohormonal activation in heart failure: Mediators and mechanisms. AACN Adv. Crit. Care 2008, 19, 364–385. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.V.; Bacal, F. Pathophysiology and Current Terrapeutic Implication. Int. J. Cardiovasc. Sci. 2020, 33, 439–446. [Google Scholar]
- Chaggar, P.S.; Malkin, C.J.; Shaw, S.M.; Williams, S.G.; Channer, K.S. Neuroendocrine effects on the heart and targets for therapeutic manipulation in heart failure. Cardiovasc. Ther. 2009, 27, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.E.; Dunlap, M.E. Renal hemodynamics in heart failure: Implications for treatment. Curr. Opin. Nephrol. Hypertens. 2008, 17, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Lopatin, Y.M.; Rosano, G.M.; Fragasso, G.; Lopaschuk, G.D.; Seferovic, P.M.; Gowdak, L.H.; Vinereanu, D.; Hamid, M.A.; Jourdain, P.; Ponikowski, P. Rationale and benefits of trimetazidine by acting on cardiac metabolism in heart failure. Int. J. Cardiol. 2016, 203, 909–915. [Google Scholar] [CrossRef]
- Giani, D.; Chan, J.; Gwathmey, J.K.; del Monte, F.; Hajjar, J.R. SERCA2a in heart failure: Role and therapeutic prospects. J. Bioenerg. Biomembr. 2005, 37, 375–380. [Google Scholar] [CrossRef]
- McDonagh, T.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Scientific Document Group, 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef] [PubMed]
- Maddox, T.M.; Januzzi, J.L.; Allen, L.A.; Breathett, K.; Butler, J.; Davis, L.L.; Fonarow, G.C.; Ibrahim, N.E.; Lindenfeld, J.; Masoudiet, F.A.; et al. 2021 Update to the 2017 ACC Expert Consensus Decision Pathway for Optimization of Heart Failure Treatment: Answers to 10 Pivotal Issues About Heart Failure With Reduced Ejection Fraction: A Report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2021, 6, 772–810. [Google Scholar]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.C.; Swedberg, K.; et al. For the PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez, E.; Morrow, D.A.; DeVore, A.; Duffy, C.I.; Ambrosy, A.P.; McCague, K.; Rocha, R.; Braunwald, E.; For the PIONEER-HF Investigators. Angiotensin–Neprilysin Inhibition in Acute Decompensated Heart Failure. N. Engl. J. Med. 2019, 380, 539–548. [Google Scholar]
- Gaziano, T.A.; Fonarow, G.C.; Velazquez, E.J.; Morrow, D.A.; Eugene Braunwald, E.; Solomon, S.D. Cost-effectiveness of sacubitril-valsartan in hospitalized patients who have heart failure with reduced ejection fraction. JAMA Cardiol. 2020, 5, 1236–1244. [Google Scholar] [CrossRef]
- Kuchulakanti, P.K. ARNI in cardiovascular disease: Current evidence and future perspectives. Future Cardiolog. 2020, 16, 505–515. [Google Scholar] [CrossRef]
- McMurray, J.J.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.A.; Bělohlávek, J.; et al. For the DAPA-HF Trial Committees and Investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Fitchett, D.; Zinman, B.; Wanner, C.; Lachin, J.M.; Hantel, S.; Salsali, A.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Inzucchi, S.E.; et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascularrisk: Results of the EMPA-REG OUTCOME® trial. Eur. Heart J. 2016, 37, 1526–1534. [Google Scholar]
- Rosano, G.; Moura, B.; Metra, M.; Böhm, M.; Bauersachs, J.; Gal, T.B.; Adamopoulos, S.; Abdelhamid, M.; Bistola, V.; Čelutkienė, J.; et al. Patient profiling in heart failure for tailoring medical therapy. Eur. J. Heart Fail. 2021, 23, 872–881. [Google Scholar] [CrossRef]
- Deepak, L.; Bhatt, D.L.; Szarek, M.; Steg, G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; et al. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar]
- Armstrong, P.W.; Roessig, L.; Patel, M.J.; Anstrom, K.J.; Butler, J.; Voors, A.A.; Lam, C.S.P.; Ponikowski, P.; Temple, T.; Pieske, B.; et al. A Multicenter, randomized, double-blind, placebo-controlled trial of the efficacy and safety of the oral soluble guanylate cyclase stimulator. The VICTORIATrial. JACC Heart Fail. 2018, 6, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L.; SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 11, 875–885. [Google Scholar]
- Ahmed, A.; Rich, M.W.; Love, T.E.; Lloyd-Jones, D.M.; Aban, I.B.; Colucci, W.S.; Adams, K.F.; Gheorghiade, M. Digoxin and reduction in mortality and hospitalization in heart failure: A comprehensive post hoc analysis of the DIG trial. Eur. Heart J. 2006, 27, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.; Pitt, B.; Rahimtoola, S.H.; Waagstein, F.; White, M.; Love, T.E.; Braunwald, E. Effects of digoxin at low serum concentrationson mortality and hospitalization in heart failure: A propensity matched study of the DIG trial. Int. J. Cardiol. 2008, 123, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Abraham, B.; Megaly, M.; Sous, M.; Fransawyalkomos, M.; Saad, M.; Fraser, R.; Topf, J.; Goldsmith, S.; Simegn, M.; Bart, B.; et al. Meta-Analysis Comparing Torsemide Versus Furosemide in Patients with Heart Failure. Am. J. Cardiol. 2020, 1, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Greene, S.J.; Velazquez, E.J.; Anstrom, K.J.; Eisenstein, E.L.; Sapp, S.; Morgan, S.; Harding, T.; Sachdev, V.; Ketema, F.; Kim, D.Y.; et al. Pragmatic Design of Randomized Clinical Trials for Heart Failure: Rationale and Design of the TRANSFORM-HF Trial. JACC Heart Fail. 2021, 5, 325–335. [Google Scholar] [CrossRef]
- Mullens, W.; Verbrugge, F.H.; Nijst, P.; Martens, P.; Tartaglia, K.; Theunissen, E.; Bruckers, L.; Droogne, W.; Troisfontaines, P.; Damman, K.; et al. Rationale and design of the ADVOR (Acetazolamide in Decompensated Heart Failure with Volume Overload) trial. Eur. Heart J. 2018, 20, 1591–1600. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.J.; Yang, J.; Yang, J.; Fan, Z.X. Arginine vasopressin antagonist tolvaptan in the treatment of heart failure: A meta-analysis of randomized controlled trials. Int. J. Clin. Exp. Med. 2015, 8, 22117–22128. [Google Scholar]
- Van Tassell, B.; Mihalick, V.; Thomas, G.; Marawan, A.; Talasaz, A.H.; Lu, J.; Le Kang, L.; Ladd, A.; Damonte, J.I.; Dixon, D.L.; et al. Rationale and design of interleukin-1 blockade in recently decompensated heart failure (REDHART2): A randomized, double blind, placebo controlled, single center, phase 2 study. J. Transl. Med. 2022, 20, 270. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin, C.J.; Filippatos, G.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; Kirwan, B.A.; et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef] [Green Version]
- Bavry, A.A.; Bhatt, D.L. Patiromer for the Management of Hyperkalemia in Subjects Receiving RAASi for HFrEF—DIAMOND. Available online: https://www.acc.org/latest-in-cardiology/clinical-trials/2022/04/02/15/56/diamond (accessed on 3 July 2022).
- Filippatos, G.; Anker, S.D.; Böhm, M.; Gheorghiade, M.; Køber, L.; Krum, H.; Maggioni, A.P.; Ponikowski, P.; Voors, A.A.; Zannad, F.; et al. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur. Heart J. 2016, 37, 2105–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargalovic, P.; Wong, P.; Onorato, J.; Finlay, H.; Wang, T.; Yan, M.; Crain, E.; St-Onge, S.; Héroux, M.; Bouvieret, M.; et al. In Vitro and In Vivo Evaluation of a Small-Molecule APJ (Apelin Receptor) Agonist, BMS-986224, as a Potential Treatment for Heart Failure. Circ. Heart Fail. 2021, 14, e007351. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.; Canada, J.; Carbone, S.; Trankle, C.; Buckley, L.; Erdle, C.O.; Abouzaki, N.A.; Dixon, D.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 blockade in recently decompensated systolic heart failure: Results from REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ. Heart Fail. 2017, 10, e004373. [Google Scholar] [CrossRef]
- Ponikowski, P.; Van Veldhuisen, D.J.; Comin, C.J.; Ertl, G.; Komajda, M.; Mareev, V.; McDonagh, T.; Parkhomenko, A.; Tavazzi, L.; Levesque, V.; et al. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur. Heart J. 2015, 36, 657–668. [Google Scholar] [CrossRef]
- Beadle, R.M.; Williams, L.K.; Kuehl, M.; Bowater, S.; Abozguia, K.; Leyva, F.; Yousef, Z.; Wagenmakers, A.J.M.; Thies, F.; Horowitz, J.; et al. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail. 2015, 3, 202–211. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.; Suen, C.; Jiang, B.; Deng, Y.; Weldrick, J.J.; Putinski, C.; Brunette, S.; Fernando, P.; Lee, T.T.; Flynn, P.; et al. Cardiotrophin 1 stimulates beneficial myogenic and vascular remodeling of the heart. Cell Res. 2017, 27, 1195–1215. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Aliaga, M.; Pérez-Echarri, N.; Marcos-Gómez, B.; Larequi, E.; Gil-Bea, F.J.; Viollet, B.; Gimenez, I.; Martínez, J.A.; Prieto, J.; Bustos, M. Cardiotrophin-1 is a key regulator of glucose and lipid metabolism. Cell Metab. 2011, 14, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Buckley, L.F.; Abbate, A. Interleukin-1 blockade in cardiovascular diseases: A clinical update. Eur. Heart J. 2018, 39, 2063–2069. [Google Scholar] [CrossRef] [Green Version]
- Wohlford, G.; VanTassell, B.; Billingsley, H.; Kadariya, D.; Canada, J.M.; Carbone, S.; Mihalick, V.L.; Bonaventura, A.; Vecchié, A.; Chiabrando, J.G.; et al. Phase 1B, randomized, double-blinded, dose escalation, single-center, repeat dose safety and pharmacodynamics study of the oral NLRP3 inhibitor dapansutrile in subjects with NYHA II-III systolic heart failure. J. Cardiovasc. Pharm. 2020, 77, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; McMurray, J.J.V.; Krum, H.; Kiowski, W.; Massie, B.M.; Caspi, A.; Pratt, C.M.; Petrie, M.C.; DeMets, D.; Kobrin, I.; et al. Long-Term Effect of Endothelin Receptor Antagonism with Bosentan on the Morbidity and Mortality of Patients with Severe Chronic Heart Failure: Primary Results of the ENABLE Trials. JACC Heart Fail. 2017, 5, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Zhang, J.; Cheng, L.; Kiowski, W.; Massie, B.M.; Caspi, A.; Pratt, C.M.; Petrie, M.C.; DeMets, D.; Kobrin, I.; et al. ENABLE Investigators and Committees. A Phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J. Am. Coll. Cardiol. 2010, 55, 1907–1914. [Google Scholar] [CrossRef] [Green Version]
- Beusekamp, J.C.; Tromp, J.; Van der Wal, H.H.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.; van der Harst, P.; Hillege, H.L.; Lang, C.C.; et al. Potassium and the use of renin-angiotensin-aldosterone system inhibitors in heart failure with reduced ejection fraction: Data from BIOSTAT-CHF. Eur. J. Heart Fail. 2018, 20, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Anker, S.D.; Kirwan, B.A.; Van Veldhuisen, D.J.; Filippatos, G.; Comin, C.J.; Ruschitzka, F.; Lüscher, T.F.; Arutyunov, G.P.; Motro, M.; Mori, C.; et al. Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron-deficient heart failure patients: An individual patient data meta-analysis. Eur. J. Heart Fail. 2018, 20, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Bomer, N.; Pavez-Giani, M.G.; Beverborg, N.G.; Cleland, J.G.F.; van Veldhuisen, D.J.; van der Meer, P. Micronutrient deficiencies in heart failure: Mitochondrial dysfunction as a common pathophysiological mechanism? J. Int. Med. 2022, 291, 713–731. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Iannuzzo, G.; Parlato, A.; Cuomo, G.; Crescenzo Testa, C.; Coppola, M.; D’Ambrosio, G.; Oliviero, D.A.; Sarullo, S.; Vitaleet, G.; et al. Clinical Evidence for Q10 Coenzyme Supplementation in Heart Failure: From Energetics to Functional Improvement. J. Clin. Med. 2020, 9, 1266. [Google Scholar] [CrossRef]
- Mortensen, A.L.; Rosenfeldt, F.; Filipiak, K.J. Effect of coenzyme Q10 in Europeans with chronic heart failure: A subgroup analysis of the Q-SYMBIO randomized double-blind trial. Cardiol. J. 2019, 26, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Pourmoghaddas, M.; Rabbani, M.; Shahabi, J.; Garakyaraghi, M.; Khanjani, R.; Hedayat, P. Combination of atorvastatin/coenzyme Q10 as adjunctive treatment in congestive heart failure: A double-blind randomized placebo-controlled clinical trial. ARYA Atheroscler. 2014, 10, 1–5. [Google Scholar]
- Teerlink, J.R.; Diaz, R.; Felker, M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Biering-Sørensen, T.; Böhm, M.; Bonderman, D.; Fang, J.C.; et al. Effect of Ejection Fraction on Clinical Outcomes in Patients Treated with Omecamtiv Mecarbil in GALACTIC-HF. J. Am. Coll. Cardiol. 2021, 78, 97–108. [Google Scholar] [CrossRef]
- Zsebo, K.; Yaroshinsky, A.; Rudy, J.J.; Wagner, K.; Greenberg, B.; Jessup, M.; Hajjar, R.J. Long-Term Effects of AAV1/SERCA2a Gene Transfer in Patients with Severe Heart Failure: Analysis of recurrent cardiovascular events and mortality. Circ. Res. 2014, 114, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Jessup, M.; Greenberg, B.; Mancini, D.; Cappola, T.; Pauly, D.F.; Jaski, B.; Yaroshinsky, A.; Zsebo, K.M.; Dittrich, H.; Hajjar, R.J. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID). Circulation 2011, 124, 304–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe, G.W.; Garcia, J. Role of cell death in the progression of heart failure. Heart Fail. Rev. 2016, 21, 157–167. [Google Scholar] [CrossRef]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavandero, S.; Chiong, M.; Rothermel, B.A.; Hill, J.A. Autophagy in cardiovascular biology. J. Clin. Investig. 2015, 125, 55–64. [Google Scholar] [CrossRef]
- McGinnis, M.; Goolsby, W.A.; Olsen, L.A. Leadership Commitments to Improve Value in Health Care; National Academies Press: Washington, DC, USA, 2009; pp. 224–225. [Google Scholar]
- Mann, D.L.; Felke, M.G. Mechanisms and Models in Heart Failure. A Translational Approach. Circ. Res. 2021, 128, 1435–1450. [Google Scholar] [CrossRef]
- Lara-Pezzi, E.; Menasché, P.; Trouvin, J.H.; Ioannidis, J.P.; Wu, J.C.; Hill, J.A.; Koch, W.J.; De Felice, A.F.; de Waele, P.; Steenwinckel, V.; et al. AM Guidelines for translational research in heart failure. J. Cardiovasc. Transl. Res. 2015, 8, 3–22. [Google Scholar] [CrossRef]
- Lauer, M.S.; Skarlatos, S. Translational Research for Cardiovascular Diseases at the NHLBI: Moving from Bench to Bedside and From Bedside to Community. Circulation 2010, 121, 929–933. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classic Pathophysiological Changes | Modern Pathophysiological Mechanisms |
|---|---|
| Hemodynamic changes Neurohumoral activation:
| Inflammation: cytokine release (TNF-α, interleukins) Release of oxygen free radicals Endothelial dysfunction
|
| Trial | EPLERENONE | IVABRADINE | EPLERENONE | ARNI | SGLT2i | VERICIGUAT | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| EPHESUS | SHIFT | EMPHASIS-HF | PARADIGM-HF | EMPA-REG Outcome | CANVAS Program | DECLARE TIMI 58 | DAPA-HF | EMPEROR-Reduced | VICTORIA | |
| Year | 2003 | 2010 | 2011 | 2014 | 2015 | 2017 | 2018 | 2019 | 2020 | 2020 |
| Number of patients | 3313 | 6558 | 2737 | 8442 | 7020 | 10142 | 17160 | 2373 | 3730 | 5050 |
| Treatment regimen | Eplerenone vs. placebo | Ivabradine vs. placebo | Eplerenone/ placebo | Sacubitril/ valsartan vs. enalapril | Empagliflozin vs. standard treatment | Canagliflozin vs. standard treatment | Dapagliflozin vs. standard treatment | Dapagliflozin vs. standard treatment | Empa gliflozin vs. placebo | Vericiguat /placebo |
| Follow-up time | 16 months | 22.9 months | 21 months | 27 months | 37.2 months | 28.8 months | 74.4 months | 18.2 months | 16 months | 10.3 months |
| Inclusion criteria | NYHA class III, IV LVEF < 35%, 3–14 days after acute myo-cardial infarction | NYHA class II-IV, LVEF < 35% Sinus rhythm Heart rate ≥ 70/per minute, hospitalization for HF within the previous year, on stable back-ground treatment including a beta blocker if tolerated | NYHA class II, history of chronic systolic HF of at least 4 weeks duration Ischemic/ non-ischemic etiology Optimal dose /maximally tolerated dose of standard HF therapy | NYHA class II-IV symptoms, LVEF ≤ 40% until 2010 and ≤35% after If no HF hospitalizations in prior year: BNP ≥ 150 pg/mL or NT-proBNP ≥ 600 pg/mL If a HF hospitalization in prior year: BNP ≥ 100 pg/mL or NT-proBNP ≥ 400 pg/mL ACE-I/ARB, beta blocker therapy | DM 2 HbA1c of ≥7.0% background glucose-lowering therapy unchanged for ≥12 weeks prior to randomization or, in the case of insulin, unchanged by >10% from the dose at randomization in the previous 12 weeks Body mass index ≤45 kg/m2 eGFR > 30 mL/min/1.73m2 Established CV disease | HbA1c ≥ 7.0% to ≤10.5% eGFR ≥ 30 mL/min/1.73 m2 Age ≥ 30 years and history of prior CV event or age ≥ 50 years with ≥2 CV risk factors | Age ≥ 40 years, DM2 HbA1c of ≥6.5% and ≤12%, eGFR of >60 mL/min /1.73 m2 Established CV disease/multiple risk factors including men ≥ 55 years or women ≥ 60 years with HT, dyslipidemia or tobacco use | LVEF ≤ 40% NT-proBNP ≥ 600 pg/mL or ≥900 pg/mL if atrial fibrillation | NYHA class II, III LVEF ≤ 40% Elevated NT-proBNP eGFR > 20 mL/min/1.73 m2 Guideline recommended medical therapy stable 1 week prior to first visit | Chronic HF, NYHA class II-IV LVEF < 45% and guideline-directed HF therapy Recent HF hospitalization or intravenous diuretic use Elevated natriuretic peptides |
| Exclusion criteria | Use of potassium-sparing diuretics A serum creatinine concentration > 2.5 mg/dL A serum potassium concentration > 5.0 mmol/L | Recent myocardial infarction Ventricular/ atrioventricular pacing that is operative for more than 40% of the day, atrial fibrillation hypotension | Severe chronic systolic HF symptomatic at rest despite optimal medical therapy eGFR < 30 mL/min/1.73 m2 | Symptomatic hypotension SBP < 100 mmHg at screening or <95 mmHg at randomization eGFR < 30 mL/min/1.73 m2 Reduction in eGFR > 25% serum potassium level >5.2 mmol/L History of angioedema Unacceptable side effects with ACE-I or ARB | Uncontrolled hyperglycemia, liver disease Planned cardiac surgery or angioplasty within 3 months, bariatric surgery within the past 2 years and other gastrointestinal surgeries that induce chronic malabsorption. Cancer treatment with anti-obesity drugs, alcohol/drug abuse within the last 3 months Acute coronary syndrome, stroke/ transient ischemic attack within 2 months prior to informed consent | History of diabetic ketoacidosis DM 1 Pancreas or beta cell transplantation, or diabetes secondary to pancreatitis or pancreatectomy, severe hypo-glycemic episode within 6 months before screening | DM 1 Bladder cancer Radiation therapy to the lower abdomen or pelvis at any time Chronic cystitis and/or recurrent urinary tract infections, pregnant or breast-feeding patients | eGFR < 30 mL/min/ 1.73 m2 and SBP < 95 mmHg | Myocardial infarction Coronary artery bypass graft surgery, stroke Heart trans-plantation Acute decompensated HF SBP ≥ 180 mm Hg at visit 2 Symptomatic hypotension and/or a SBP < 100 mmHg, liver disease Impaired renal function defined as eGFR < 20 mL/min/1.73 m2 Use/prior use of a SGLT2i, pregnancy | Use of long-acting nitrates, phosphodiesterase type 5 inhibitor, riociguat Heart transplantation Continuous intravenous diuretics eGRF 15 mL/min/1.73 m2 or dialysis Severe pulmonary disease requiring continuous oxygen Severe hepatic insufficiency |
| Primary endpoint | Death from any cause and death from CV causes or HF hospitalization | CV death or hospital admission for worsening HF | Death from CV causes or hospitalization for HF | CV mortality or HF hospitalization | MACEAll-cause mortality or CV mortality, myocardial infarction, stroke CV hospitalization Disease Progression or renal mortality | MACE All-cause mortality/ CV mortality Myocardial infarction, stroke CV hospitalization Disease progression or renal mortality | MACEAll-cause mortality/ CV mortality. Myocardial infarction Stroke CV hospitalization Disease progression or renal mortality | Hospitalizationor visit to the emergency room due to HF Hospitalization for HF Visit to the emergency room due to HF or CV death | CV death or hospitalization for worsening HF | CV death or HF hospitalization |
| p (superiority) | RR 0.87; 95 CI 0.79 to 0.95; p = 0.002 | HR 0.82, 95% CI 0.75–0.90, p < 0.0001 | HR 0.63; 95% CI 0.54 to 0.74; p < 0.001 | HR 0.80; 95% CI 0.73 to 0.87; p < 0.001 | HR 0.86; 95.02% CI, 0.74 to 0.99; p = 0.04 for superiority | HR 0.78; 95% CI 0.67–0.91, p = 0,02 | CI < 1.3; p < 0.01 for non-inferiority | HR 0.74; 95% CI 0.65 to 0.85; p < 0.01 | HR 0.75; 95% CI 0.65 to 0.86; p < 0.001 | HR 0.90; 95% CI 0.82 to 0.98; p = 0.02 |
| Secondary endpoint | Death from any cause or any hospitalization | Hospital admissions for worsening HF/ deaths due to HF | All-cause mortality or HF hospitalization | CV mortality HF hospitalization All-cause mortality | Hospitalization due to HF | Total hospitalizations Composite renal outcome | Death from CV causes Hospitalization for HF | |||
| p | RR 0.92; 95 CI 0.86 to 0.98; p = 0.02 | 21% placebo vs. 16% with ivabradine; HR 0.74, 0.66-0.83; p < 0.0001 | 19.8% vs. 27.4%, HR 0.65; 95% CI 0.55 to 0.76, p < 0.001 | HR for death from any cause, 0.84; 95% CI, 0.76 to 0.93; p < 0.001 | 2.7% and 4.1%, respectively; 35% RR reduction p = 0.08 | HR, 0.70; 95% CI, 0.55–0.89, p < 0.001 | 4.9% vs. 5.8%; HR, 0.83; 95% CI, 0.73 to 0.95; p = 0.005 | - | HR 0.70; 95% CI, 0.58 to 0.85; p < 0.001 | HR 0.90; 95% CI, 0.83 to 0.98; p = 0.02 |
| Objective/Results | Number of Patients | Year of Completion | |
|---|---|---|---|
| NCT03783429 (Digoxin Evaluation in Chronic Heart Failure: Investigational Study In Outpatients in the Netherlands (DECISION) https://clinicaltrials.gov/ct2/show/NCT03783429 (accessed on 3 July 2022) | To evaluate whether lower doses of digoxin, guided by serum concentrations, will reduce HF hospitalizations and cardiovascular death rate. | recruiting | 2025 |
| TRANSFORM-HF (ToRsemide compArisoN With furoSemide FORManagement of Heart Failure) [27] https://clinicaltrials.gov/ct2/show/NCT03296813 (accessed on 3 July 2022) | To compare Torsemide efficiency to Furosemide and its effects on mortality and morbidity. | 2859 | 2022 |
| ADVOR (Acetazolamide in Decompensated Heart Failure With Volume OveRload) [28] https://clinicaltrials.gov/ct2/show/NCT03505788 (accessed on 3 July 2022) | To test the efficiency of the association of Acetazolamide in HF. | 519 | 2022 |
| QUEST (Efficacy and Safety of Tolvaptan in Heart Failure Patients with Volume Overload Despite the Standard Treatment with Conventional Diuretics) [29] https://clinicaltrials.gov/ct2/show/NCT01651156 (accessed on 3 July 2022) | To evaluate the efficacy and safety of tolvaptan in HFrEF patients with cardiac edema after current diuretic treatment. Increase in total kidney volume: 2.8% per year (95% CI, 2.5 to 3.1) in the tolvaptan group vs. 5.5% per year in the placebo group (95% CI, 5.1 to 6.0; p < 0.001). The composite endpoint favored tolvaptan over placebo (44 vs. 50 events per 100 person-years, p = 0.01), with lower rates of worsening kidney function (2 vs. 5 events per 100 person-years, p < 0.001) and kidney pain (5 vs. 7 events per 100 person-years, p = 0.007). | 244 | 2013 |
| EVEREST (The Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan) [29] https://clinicaltrials.gov/ct2/show/NCT00071331 (accessed on 3 July 2022) | To compare the safety and efficacy of tolvaptan versus placebo in the treatment of patients with worsening congestive HF. The composite endpoint of cardiovascular death or hospitalization for HF: 871 tolvaptan group patients (42%) and 829 placebo group patients (40.2%) (hazard ratio, 1.04; 95% CI, 0.95–1.14; p = 0.55). | 3600 | 2006 |
| NCT03797001—Interleukin-1 Blockade in Recently Decompensated Heart Failure-2 (REDHART2) [30] https://clinicaltrials.gov/ct2/show/NCT03797001 (accessed on 3 July 2022) | To evaluate the effects of anakinra (100 mg subcutaneous injection, daily for 24 weeks) on peak aerobic exercise capacity measured with a cardiopulmonary test after 24 weeks in patients with recently decompensated HFrEF and increased systemic inflammation. | 102 | 2024 |
| FAIR-HF2 (Intravenous Iron in Patients With Systolic Heart Failure and Iron Deficiency to Improve Morbidity and Mortality) https://clinicaltrials.gov/ct2/show/NCT03036462 (accessed on 3 July 2022) | To investigate the effect of a long-term therapy with ferric carboxymaltosis vs. placebo on decreasing the rate of recurrent hospitalizations and CV death in HfrEF. | recruiting | 2024 |
| NCT03388593 (Survival Study of the Recombinant Human Neuregulin-1β in Subjects With Chronic Heart Failure) https://clinicaltrials.gov/ct2/show/NCT03388593 (accessed on 3 July 2022) | To test whether daily intravenous neuroregulin 1 perfusion, followed by weekly bolus, is feasible and safe in HFrEF. | 1600 | 2023 |
| NCT03875183-Study to Evaluate Effects of INL1 in Patients With Heart Failure and Reduced Ejection Fraction (TRACER-HF) [31] https://clinicaltrials.gov/ct2/show/NCT03875183 (accessed on 3 July 2022) | To evaluate the efficacy and safety of three PO INL1 doses in HFrEF. The primary outcome measure is NT-proBNP serum level decrease. The secondary outcome measures are echocardiographic parameters and functional status changes. | 200 | 2023 |
| HEART-FID (Randomized Placebo-controlled Trial of FCM as Treatment for Heart Failure With Iron Deficiency) https://clinicaltrials.gov/ct2/show/NCT03037931 (accessed on 16 July 2022) | To evaluate the effects of intravenous ferric carboxymaltose FCM vs. placebo on the 12-month rate of death, hospitalization for worsening HF and the 6MWT distance in HfrEF patients with iron deficiency. | active, not recruiting 3068 participants | 2023 |
| IRONMAN (Intravenous Iron Treatment in Patients With Heart Failure and Iron Deficiency) https://clinicaltrials.gov/ct2/show/NCT02642562 (accessed on 3 July 2022) | To evaluate the additional effect of intravenous iron (ferric derisomaltose) vs. placebo on top of standard care in HFrEF patients with iron deficiency. | active, not recruiting 1160 participants | 2022 |
| NCT03888066—DIAMOND (Patiromer for the Management of Hyperkalemia in Subjects Receiving RAASi Medications for the Treatment of Heart Failure) [32] https://www.clinicaltrials.gov/ct2/show/NCT03888066 (accessed on 3 July 2022) | To evaluate patiromer compared to control among patients with HFrEF and a history of hyperkalemia. The primary endpoint, adjusted mean change in serum potassium level, was 0.03 mEq/L in the patiromer group vs. 0.13 mEq/L in the control group (p < 0.001). | 878 | 2022 |
| ARTS-HF (MinerAlocorticoid Receptor antagonist Tolerability Study-Heart Failure) [33] https://clinicaltrials.gov/ct2/show/NCT04435626 (accessed on 3 July 2022) | To investigate the safety and potential efficacy of finerenone in patients with worsening chronic HFrEF and at high risk of hyperkalaemia and worsening renal dysfunction. Finerenone demonstrated a decrease of >30% in plasma N-terminal pro-B-type natriuretic peptide during 90 days in 37.2% of patients vs. eplerenone. The composite endpoint (CV hospitalizations, acute worsening HF or 90-day all-cause mortality) was statistically significant only in the 10 to 20 mg group (hazard ratio 0.56, 95% CI, 0.35; 0.90; p = 0.02). | 1066 | 2021 |
| GALACTIC-HF (Global Approach to Lowering Adverse Cardiac Outcomes Through Improving Contractility in Heart Failure) [34] https://clinicaltrials.gov/ct2/show/NCT02929329 (accessed on 3 July 2022) | To evaluate the selective cardiac myosin activator omecamtiv mecarbil compared to placebo among patients with HFrEF. The primary composite endpoint: omecamtiv mecarbil reduced CV death or HF events compared to placebo (hazard ratio 0.92 [95% CI, 0.86–0.99]; p = 0.02). | 8256 | 2021 |
| CANTOS (Cardiovascular Risk Reduction Study (Reduction in Recurrent Major CV Disease Events) [35] https://clinicaltrials.gov/ct2/show/NCT01327846 (accessed on 3 July 2022) | To test if canakinumab would prevent hospitalization for HF and the composite of HHF or HF-related mortality. A dose of 150 mg every 3 months significantly reduced the composite endpoint of HF hospitalization or HF–related mortality in patients with a history of acute myocardial infarction (hazard ratio vs. placebo, 0.83; 95% CI, 0.73 to 0.95; p = 0.005). | 10,061 | 2020 |
| ISRCTN94506234 (Q-SYMBIO trial) https://www.isrctn.com/ISRCTN94506234 (accessed on 3 July 2022) | To evaluate coenzyme Q10 as an adjunctive treatment in chronic HFrEF. Improvement of composite risk assessed by MACE (HR: 0.23; 95% CI = 0.11–0.51, p < 0.001). Improvement in NHYA class after 2 years of CoQ10 supplementation vs. placebo (48% vs. 25%, p = 0.003). Significant improvement in LVEF in Coq10 group of 6% from baseline (p = 0.021). | 420 | 2019 |
| REDHART (REcently Decompensated Heart failure Anakinra Response Trial) [36] https://clinicaltrials.gov/ct2/show/NCT01936909 (accessed on 3 July 2022) | To test inhibition of inflammatory response and improvement in peak aerobic exercise capacity in recently decompensated HFrEF after administration of IL-1 receptor antagonist (anakinra). Anakinra improved peak aerobic exercise capacity after 12 weeks of treatment in patients with LVEF < 50% (from 14.5 mL/kg/minute to 16.1 mL/kg/minute; p = 0.009). | 60 | 2017 |
| CONFIRM-HF (Ferric CarboxymaltOse evaluatioN on perFormance in patients with IRon deficiency in coMbination with chronic Heart Failure) [37] https://clinicaltrials.gov/ct2/show/NCT01453608 (accessed on 16 July 2022) | To determine, relative to placebo, the effect of intravenous ferric carboxymaltose (FCM) over a 1 year period on exercise capacity in patients with chronic heart failure and iron deficiency. FCM significantly prolonged 6MWT distance (difference FCM vs. placebo 36 ± 11 m, p < 0.001) A reduction in the risk of hospitalizations for worsening HF (hazard ratio-95% confidence interval: 0.39 (0.19–0.82), p = 0.009); a significant improvement in NYHA class symptoms and quality of life scores. | 304 | 2015 |
| NCT00454818—Efficacy and Safety Study of Genetically Targeted Enzyme Replacement Therapy for Advanced Heart Failure (CUPID) [34] https://clinicaltrials.gov/ct2/show/NCT00454818 (accessed on 3 July 2022) | To evaluate the effects of 3 doses of AAV1/SERCA2a versus placebo in patients with HF NYHA class III, IV, LVEF ≤ 35%, o2max ≤ 20 mL/kg per minute and ICD on optimal therapy. Primary endpoints: incidence of treatment adverse events at 12 months; length of CV-related hospitalizations at 6 months; change in NYHA class, MLWHFQ score, 6-min walk test, o2max, absolute levels of NT-proBNP, LVEF, LVESV at 6 months. Results: significant decrease in clinical events, hospitalization length and NT-proBNP levels, trending toward significant recovery of clinical evolution and functional capacity. | 51 | 2012 |
| NCT00841139 (Metabolic Manipulation in Chronic Heart Failure) [38] https://clinicaltrials.gov/ct2/show/NCT00841139 (accessed on 3 July 2022) | To test whether short-term treatment with perhexiline improves cardiac energetics, LVEF or symptoms of HF by altering substrate utilization. Perhexiline improves cardiac energetics (30% increase in the phosphocreatine/adenosine triphosphate ratio from 1.16 ± 0.39 to 1.51 ± 0.51; p < 0.001) and symptom status (p = 0.036) with no evidence of altered cardiac substrate utilization or changes in LVEF. | 50 | 2011 |
| FAIR-HF (Ferinject® Assessment in Patients With Iron Deficiency and Chronic Heart Failure) [31] https://clinicaltrials.gov/ct2/show/NCT00520780 (accessed on 16 July 2022) | To evaluate the efficacy of Ferinject® in improving symptoms of chronic HFrEF in patients with iron deficiency. Results: significant improvements in NYHA functional class at week 24 (odds ratio for improvement by one class, 2.40; 95% CI, 1.55 to 3.71; p < 0.001), in distance on the 6MWT and in quality of life at week 24 (p < 0.001 for all comparisons). | 456 | 2009 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moloce, M.-A.; Costache, I.-I.; Nicolae, A.; Onofrei Aursulesei, V. Pharmacological Targets in Chronic Heart Failure with Reduced Ejection Fraction. Life 2022, 12, 1112. https://doi.org/10.3390/life12081112
Moloce M-A, Costache I-I, Nicolae A, Onofrei Aursulesei V. Pharmacological Targets in Chronic Heart Failure with Reduced Ejection Fraction. Life. 2022; 12(8):1112. https://doi.org/10.3390/life12081112
Chicago/Turabian StyleMoloce, Maria-Angela, Irina-Iuliana Costache, Ana Nicolae, and Viviana Onofrei Aursulesei. 2022. "Pharmacological Targets in Chronic Heart Failure with Reduced Ejection Fraction" Life 12, no. 8: 1112. https://doi.org/10.3390/life12081112