
Micellar Composition Affects Lipid Accretion Kinetics in Molecular Dynamics Simulations: Support for Lipid Network Reproduction


Abstract
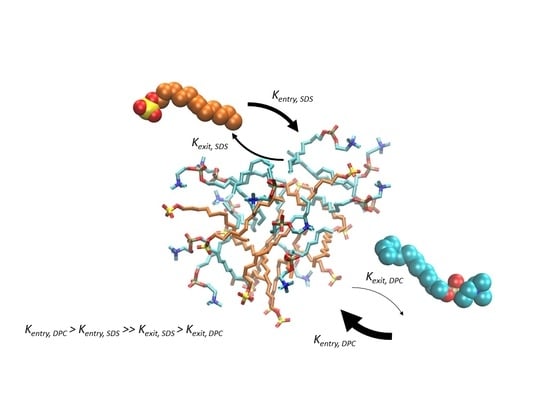
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Varied Lipid Combinations Elicit Discrete Dynamic Profiles of Self-Assembly
3.2. Exit Rates of Lipids from Pre-Micelles Are Significantly Affected by Compositional Variation
3.3. Entry Rates of Lipids into Pre-Micelles Show Sensitivity to Compositional Variation
3.4. Specific Headgroup Interactions Contribute to Observed Rate Modifications
3.5. Observed Accretion Kinetics Predicts Micelle Self-Reproduction at Non-Random Compositions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Dwars, T.; Paetzold, E.; Oehme, G. Reactions in micellar systems. Angew. Chem. Int. Ed. 2005, 44, 7174–7199. [Google Scholar] [CrossRef] [PubMed]
- La Sorella, G.; Strukul, G.; Scarso, A. Recent advances in catalysis in micellar media. Green Chem. 2015, 17, 644–683. [Google Scholar] [CrossRef]
- Serrano-Luginbühl, S.; Ruiz-Mirazo, K.; Ostaszewski, R.; Gallou, F.; Walde, P. Soft and dispersed interface-rich aqueous systems that promote and guide chemical reactions. Nat. Rev. Chem. 2018, 2, 306–327. [Google Scholar] [CrossRef]
- Otto, S.; Engberts, J.B.; Kwak, J.C. Million-fold acceleration of a Diels−Alder reaction due to combined Lewis acid and micellar catalysis in water. J. Am. Chem. Soc. 1998, 120, 9517–9525. [Google Scholar] [CrossRef] [Green Version]
- Szostak, J.W.; Bartel, D.P.; Luisi, P.L. Synthesizing life. Nature 2001, 409, 387–390. [Google Scholar] [CrossRef]
- Pressman, A.; Blanco, C.; Chen, I.A. The RNA world as a model system to study the origin of life. Curr. Biol. 2015, 25, R953–R963. [Google Scholar] [CrossRef] [Green Version]
- Deamer, D. The role of lipid membranes in life’s origin. Life 2017, 7, 5. [Google Scholar] [CrossRef]
- Sarkar, S.; Das, S.; Dagar, S.; Joshi, M.P.; Mungi, C.V.; Sawant, A.A.; Patki, G.M.; Rajamani, S. Prebiological membranes and their role in the emergence of early cellular life. J. Membr. Biol. 2020, 253, 589–608. [Google Scholar] [CrossRef]
- Sarkar, S.; Dagar, S.; Verma, A.; Rajamani, S. Compositional heterogeneity confers selective advantage to model protocellular membranes during the origins of cellular life. Sci. Rep. 2020, 10, 4483. [Google Scholar] [CrossRef] [Green Version]
- Lancet, D.; Zidovetzki, R.; Markovitch, O. Systems protobiology: Origin of life in lipid catalytic networks. J. R. Soc. Interface 2018, 15, 20180159. [Google Scholar] [CrossRef] [PubMed]
- Kahana, A.; Lancet, D. Self-reproducing catalytic micelles as nanoscopic protocell precursors. Nat. Rev. Chem. 2021, 5, 870–878. [Google Scholar] [CrossRef]
- Serra, R.; Villani, M. Sustainable growth and synchronization in protocell models. Life 2019, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahana, A.; Maslov, S.; Lancet, D. Dynamic lipid aptamers: Non-polymeric chemical path to early life. Chem. Soc. Rev. 2021, 50, 11741–11746. [Google Scholar] [CrossRef]
- Segré, D.; Shenhav, B.; Kafri, R.; Lancet, D. The molecular roots of compositional inheritance. J. Theor. Biol. 2001, 213, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Segré, D.; Ben-Eli, D.; Lancet, D. Compositional genomes: Prebiotic information transfer in mutually catalytic noncovalent assemblies. Proc. Natl. Acad. Sci. USA 2000, 97, 4112–4117. [Google Scholar] [CrossRef] [Green Version]
- Segré, D.; Lancet, D. Composing life. EMBO Rep. 2000, 1, 217–222. [Google Scholar] [CrossRef]
- Budin, I.; Prywes, N.; Zhang, N.; Szostak, J.W. Chain-length heterogeneity allows for the assembly of fatty acid vesicles in dilute solutions. Biophys. J. 2014, 107, 1582–1590. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, H.; Mizogami, M. Discrimination of stereoisomers by their enantioselective interactions with chiral cholesterol-containing membranes. Molecules 2018, 23, 49. [Google Scholar] [CrossRef] [Green Version]
- Bell, T.N.; Feng, K.; Calvin, G.; Van Winkle, D.H.; Lenhert, S. Organic composomes as supramolecular aptamers. ACS Omega 2020, 5, 27393–27400. [Google Scholar] [CrossRef]
- Lojewska, Z.; Loew, L.M. Insertion of amphiphilic molecules into membranes is catalyzed by a high molecular weight non-ionic surfactant. Biochim. Biophys. Acta BBA-Biomembr. 1987, 899, 104–112. [Google Scholar] [CrossRef]
- Adamala, K.P.; Engelhart, A.E.; Szostak, J.W. Collaboration between primitive cell membranes and soluble catalysts. Nat. Commun. 2016, 7, 11041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachakis, D.; Bencurova, E.; Papangelopoulos, N.; Kossida, S. Current state-of-the-art molecular dynamics methods and applications. Adv. Protein Chem. Struct. Biol. 2014, 94, 269–313. [Google Scholar] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwier, M.C.; Chong, L.T. Reaching biological timescales with all-atom molecular dynamics simulations. Curr. Opin. Pharmacol. 2010, 10, 745–752. [Google Scholar] [CrossRef]
- Kahana, A.; Lancet, D. Protobiotic systems chemistry analyzed by molecular dynamics. Life 2019, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Marrink, S.; Tieleman, D.; Mark, A. Molecular dynamics simulation of the kinetics of spontaneous micelle formation. J. Phys. Chem. B 2000, 104, 12165–12173. [Google Scholar] [CrossRef] [Green Version]
- Tieleman, D.; Van der Spoel, D.; Berendsen, H. Molecular dynamics simulations of dodecylphosphocholine micelles at three different aggregate sizes: Micellar structure and chain relaxation. J. Phys. Chem. B 2000, 104, 6380–6388. [Google Scholar] [CrossRef]
- Chen, J.; Hao, J. Molecular dynamics simulation of cetyltrimethylammonium bromide and sodium octyl sulfate mixtures: Aggregate shape and local surfactant distribution. Phys. Chem. Chem. Phys. 2013, 15, 5563–5571. [Google Scholar] [CrossRef]
- Jójárt, B.; Poša, M.; Fiser, B.; Szőri, M.; Farkaš, Z.; Viskolcz, B. Mixed micelles of sodium cholate and sodium dodecylsulphate 1:1 binary mixture at different temperatures–experimental and theoretical investigations. PLoS ONE 2014, 9, e102114. [Google Scholar] [CrossRef] [Green Version]
- Kampf, J.P.; Cupp, D.; Kleinfeld, A.M. Different mechanisms of free fatty acid flip-flop and dissociation revealed by temperature and molecular species dependence of transport across lipid vesicles. J. Biol. Chem. 2006, 281, 21566–21574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; Wang, S.; Larson, R.G. Potentials of mean force and escape times of surfactants from micelles and hydrophobic surfaces using molecular dynamics simulations. Langmuir 2015, 31, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Mohan, G.; Kopelevich, D.I. A multiscale model for kinetics of formation and disintegration of spherical micelles. J. Chem. Phys. 2008, 128, 044905. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.; Mark, A. Molecular dynamics simulations of mixed micelles modeling human bile. Biochemistry 2002, 41, 5375–5382. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Abel, S.; Dupradeau, F.-Y.; Marchi, M. Molecular dynamics simulations of a characteristic DPC micelle in water. J. Chem. Theory Comput. 2012, 8, 4610–4623. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Bondi, A.V. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Xu, W.; Wang, X.; Zhong, Z.; Song, A.; Hao, J. Influence of counterions on lauric acid vesicles and theoretical consideration of vesicle stability. J. Phys. Chem. B 2013, 117, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Rharbi, Y.; Winnik, M.A. Solute exchange between surfactant micelles by micelle fragmentation and fusion. Adv. Colloid Interface Sci. 2001, 89, 25–46. [Google Scholar] [CrossRef]
- Mysona, J.A.; McCormick, A.V.; Morse, D.C. Mechanism of micelle birth and death. Phys. Rev. Lett. 2019, 123, 038003. [Google Scholar] [CrossRef] [PubMed]
- Burov, S.V.; Vanin, A.A.; Brodskaya, E.N. Principal role of the stepwise aggregation mechanism in ionic surfactant solutions near the critical micelle concentration. Molecular dynamics study. J. Phys. Chem. B 2009, 113, 10715–10720. [Google Scholar] [CrossRef]
- Klevens, H. Critical micelle concentrations as determined by refraction. J. Phys. Chem. 1948, 52, 130–148. [Google Scholar] [CrossRef]
- Akhter, M.S. Effect of acetamide on the critical micelle concentration of aqueous solutions of some surfactants. Colloids Surf. A Physicochem. Eng. Asp. 1997, 121, 103–109. [Google Scholar] [CrossRef]
- Lazaridis, T.; Mallik, B.; Chen, Y. Implicit solvent simulations of DPC micelle formation. J. Phys. Chem. B 2005, 109, 15098–15106. [Google Scholar] [CrossRef]
- Manzo, G.; Carboni, M.; Rinaldi, A.C.; Casu, M.; Scorciapino, M.A. Characterization of sodium dodecylsulphate and dodecylphosphocholine mixed micelles through NMR and dynamic light scattering. Magn. Reson. Chem. 2013, 51, 176–183. [Google Scholar] [CrossRef]
- Menjoge, A.; James-Smith, M.A.; Shah, D.; Vasenkov, S. Influence of breakup and reformation of micelles on surfactant diffusion in pure and mixed micellar systems. Microporous Mesoporous Mater. 2009, 125, 85–89. [Google Scholar] [CrossRef]
- Thomas, A.S.; Elcock, A.H. Direct measurement of the kinetics and thermodynamics of association of hydrophobic molecules from molecular dynamics simulations. J. Phys. Chem. Lett. 2011, 2, 19–24. [Google Scholar] [CrossRef]
- Hossain, S.; Joyce, P.; Parrow, A.; Jõemetsa, S.; Höök, F.; Larsson, P.; Bergström, C.A. Influence of bile composition on membrane incorporation of transient permeability enhancers. Mol. Pharm. 2020, 17, 4226–4240. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.R.; Lee, C.T.; Amaro, R.E. Quantitative ranking of ligand binding kinetics with a multiscale milestoning simulation approach. J. Phys. Chem. Lett. 2018, 9, 4941–4948. [Google Scholar] [CrossRef] [PubMed]
- LeBard, D.N.; Levine, B.G.; DeVane, R.; Shinoda, W.; Klein, M.L. Premicelles and monomer exchange in aqueous surfactant solutions above and below the critical micelle concentration. Chem. Phys. Lett. 2012, 522, 38–42. [Google Scholar] [CrossRef]
- Buch, I.; Giorgino, T.; De Fabritiis, G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 10184–10189. [Google Scholar] [CrossRef] [Green Version]
- Dickson, A.; Tiwary, P.; Vashisth, H. Kinetics of ligand binding through advanced computational approaches: A review. Curr. Top. Med. Chem. 2017, 17, 2626–2641. [Google Scholar] [CrossRef]
- Sykes, D.A.; Stoddart, L.A.; Kilpatrick, L.E.; Hill, S.J. Binding kinetics of ligands acting at GPCRs. Mol. Cell. Endocrinol. 2019, 485, 9–19. [Google Scholar] [CrossRef]
- Baz, J.; Gebhardt, J.; Kraus, H.; Markthaler, D.; Hansen, N. Insights into noncovalent binding obtained from molecular dynamics simulations. Chem. Ing. Tech. 2018, 90, 1864–1875. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Caflisch, A. Small molecule binding to proteins: Affinity and binding/unbinding dynamics from atomistic simulations. ChemMedChem 2011, 6, 1578–1580. [Google Scholar] [CrossRef]
- Tang, Z.; Chang, C.-E.A. Binding thermodynamics and kinetics calculations using chemical host and guest: A comprehensive picture of molecular recognition. J. Chem. Theory Comput. 2018, 14, 303–318. [Google Scholar] [CrossRef]
- Pan, A.C.; Xu, H.; Palpant, T.; Shaw, D.E. Quantitative characterization of the binding and unbinding of millimolar drug fragments with molecular dynamics simulations. J. Chem. Theory Comput. 2017, 13, 3372–3377. [Google Scholar] [CrossRef]
- Zhang, Y.; McCammon, J.A. Studying the affinity and kinetics of molecular association with molecular-dynamics simulation. J. Chem. Phys. 2003, 118, 1821–1827. [Google Scholar] [CrossRef]
- Yiv, S.; Zana, R.; Ulbricht, W.; Hoffmann, H. Effect of alcohol on the properties of micellar systems: II. Chemical relaxation studies of the dynamics of mixed alcohol+ surfactant micelles. J. Colloid Interface Sci. 1981, 80, 224–236. [Google Scholar] [CrossRef]
- Frindi, M.; Michels, B.; Zana, R. Ultrasonic absorption studies of surfactant exchange between micelles and bulk phase in aqueous micellar solutions of nonionic surfactants with short alkyl chains. 1. 1,2-Hexanediol and 1,2,3-octanetriol. J. Phys. Chem. 1991, 95, 4832–4837. [Google Scholar] [CrossRef]
- Aniansson, E.; Wall, S.; Almgren, M.; Hoffmann, H.; Kielmann, I.; Ulbricht, W.; Zana, R.; Lang, J.; Tondre, C. Theory of the kinetics of micellar equilibria and quantitative interpretation of chemical relaxation studies of micellar solutions of ionic surfactants. J. Phys. Chem. 1976, 80, 905–922. [Google Scholar] [CrossRef]
- Kato, S.; Nomura, H.; Zieliński, R.; Ikeda, S. Ultrasonic relaxation study of the micelle—Monomer exchange process in aqueous solutions of dodecyltrimethylammonium bromide in the presence of NaBr. J. Colloid Interface Sci. 1991, 146, 53–62. [Google Scholar] [CrossRef]
- Segré, D.; Ben-Eli, D.; Deamer, D.W.; Lancet, D. The lipid world. Orig. Life Evol. Biosph. 2001, 31, 119–145. [Google Scholar] [CrossRef]
- Vijaykumar, A.; Bolhuis, P.G.; Ten Wolde, P.R. The intrinsic rate constants in diffusion-influenced reactions. Faraday Discuss. 2017, 195, 421–441. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Markovitch, O.; Lancet, D. Excess mutual catalysis is required for effective evolvability. Artif. Life 2012, 18, 243–266. [Google Scholar] [CrossRef]
- Cardoso, R.M.; Martins, P.A.; Gomes, F.; Doktorovova, S.; Vaz, W.L.; Moreno, M.J. Chain-length dependence of insertion, desorption, and translocation of a homologous series of 7-nitrobenz-2-oxa-1,3-diazol-4-yl-labeled aliphatic amines in membranes. J. Phys. Chem. B 2011, 115, 10098–10108. [Google Scholar] [CrossRef]
- Bachmann, P.A.; Luisi, P.L.; Lang, J. Autocatalytic self-replicating micelles as models for prebiotic structures. Nature 1992, 357, 57–59. [Google Scholar] [CrossRef]
- Bissette, A.J.; Odell, B.; Fletcher, S.P. Physical autocatalysis driven by a bond-forming thiol–ene reaction. Nat. Commun. 2014, 5, 4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Early, J.T.; Yager, K.G.; Lodge, T.P. Direct observation of micelle fragmentation via in situ liquid-phase transmission electron microscopy. ACS Macro Lett. 2020, 9, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Colomer, I.; Borissov, A.; Fletcher, S.P. Selection from a pool of self-assembling lipid replicators. Nat. Commun. 2020, 11, 176. [Google Scholar] [CrossRef] [PubMed]
- Bukhryakov, K.V.; Almahdali, S.; Rodionov, V.O. Amplification of chirality through self-replication of micellar aggregates in water. Langmuir 2015, 31, 2931–2935. [Google Scholar] [CrossRef]
- Sharma, V.K.; Mitra, S.; Mukhopadhyay, R. Dynamic landscape in self-assembled surfactant aggregates. Langmuir 2019, 35, 14151–14172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Kamp, F.; Hamilton, J.A. Dissociation of long and very long chain fatty acids from phospholipid bilayers. Biochemistry 1996, 35, 16055–16060. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.R.; Pillai, B.K.; Hamilton, J.A. Fatty acid flip-flop in a model membrane is faster than desorption into the aqueous phase. Biochemistry 2008, 47, 9081–9089. [Google Scholar] [CrossRef] [PubMed]
- Brigati, G.; Franchi, P.; Lucarini, M.; Pedulli, G.F.; Valgimigli, L. The EPR study of dialkyl nitroxides as probes to investigate the exchange of solutes between micellar and water phases. Res. Chem. Intermed. 2002, 28, 131–141. [Google Scholar] [CrossRef]
- Yao, S.; Ghosh, I.; Zutshi, R.; Chmielewski, J. Selective amplification by auto-and cross-catalysis in a replicating peptide system. Nature 1998, 396, 447–450. [Google Scholar] [CrossRef]
- Vaidya, N.; Manapat, M.L.; Chen, I.A.; Xulvi-Brunet, R.; Hayden, E.J.; Lehman, N. Spontaneous network formation among cooperative RNA replicators. Nature 2012, 491, 72–77. [Google Scholar] [CrossRef]
- Lancet, D.; Kedem, O.; Pilpel, Y. Emergence of order in small autocatalytic sets maintained far from equilibrium: Application of a probabilistic receptor affinity distribution (RAD) model. Ber. Der Bunsenges. Für Phys. Chem. 1994, 98, 1166–1169. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ratio | Monomer Count | |
|---|---|---|
| 1 | 10%/90% | 6/48 |
| 2 | 30%/70% | 16/38 |
| 3 | 50%/50% | 27/27 |
| 4 | 70%/30% | 38/16 |
| 5 | 90%/10% | 48/6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kahana, A.; Lancet, D.; Palmai, Z. Micellar Composition Affects Lipid Accretion Kinetics in Molecular Dynamics Simulations: Support for Lipid Network Reproduction. Life 2022, 12, 955. https://doi.org/10.3390/life12070955
Kahana A, Lancet D, Palmai Z. Micellar Composition Affects Lipid Accretion Kinetics in Molecular Dynamics Simulations: Support for Lipid Network Reproduction. Life. 2022; 12(7):955. https://doi.org/10.3390/life12070955
Chicago/Turabian StyleKahana, Amit, Doron Lancet, and Zoltan Palmai. 2022. "Micellar Composition Affects Lipid Accretion Kinetics in Molecular Dynamics Simulations: Support for Lipid Network Reproduction" Life 12, no. 7: 955. https://doi.org/10.3390/life12070955