
LSAP: A Machine Learning Method for Leaf-Senescence-Associated Genes Prediction


Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Datasets and Preprocessing
2.2. Features Selection
2.3. Machine Learning Model Development
2.4. Performance Evaluation
2.5. Large-Scale Prediction of SAGs
2.6. Database Construction
3. Results
3.1. The Results of SVM Performances
3.2. The Results of XGBoost Performances
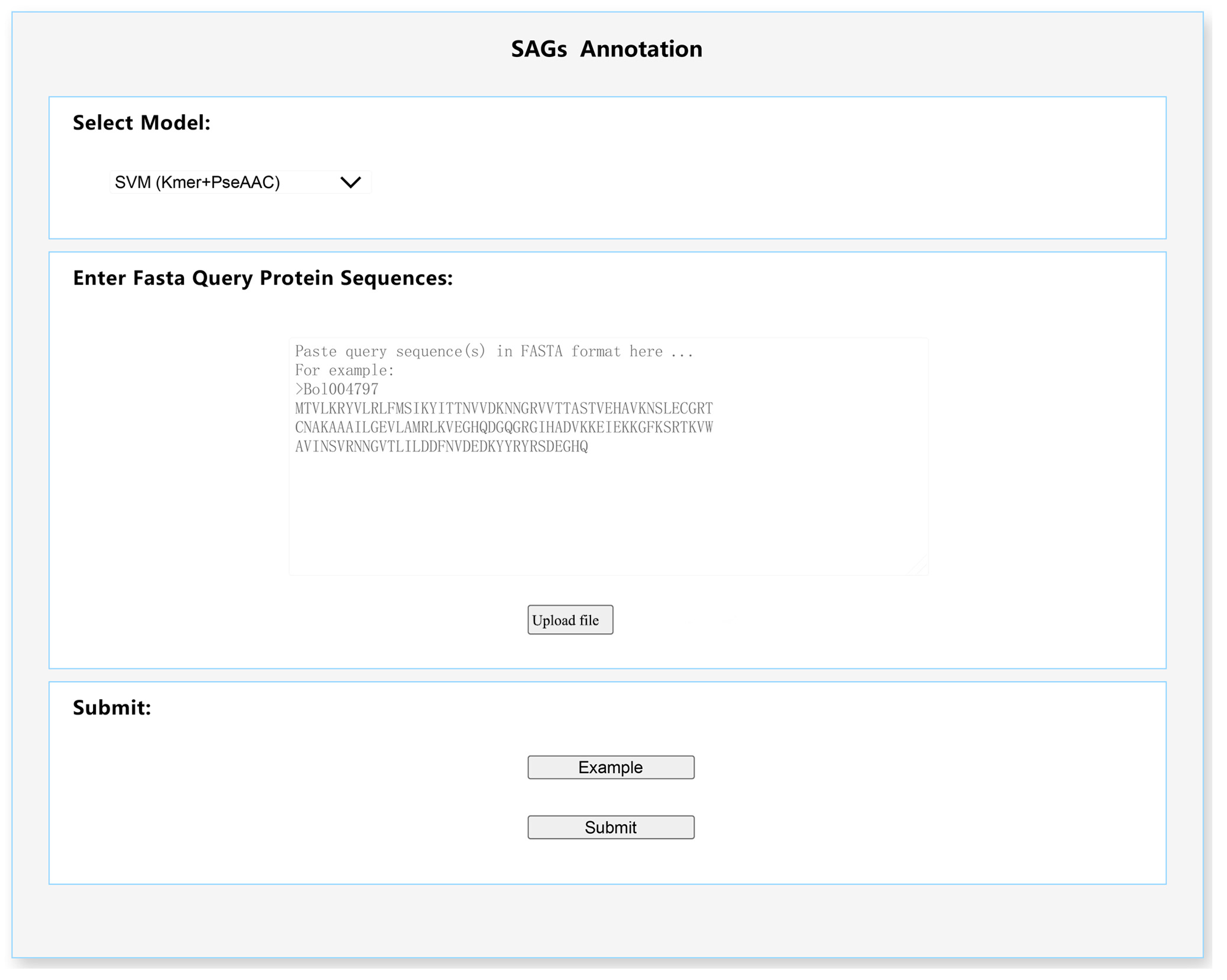
3.3. A Plant SAGs Predict Tool for Users
3.4. Large-Scale Prediction SAGs
3.5. Comparative Analysis of SAGs in Plants
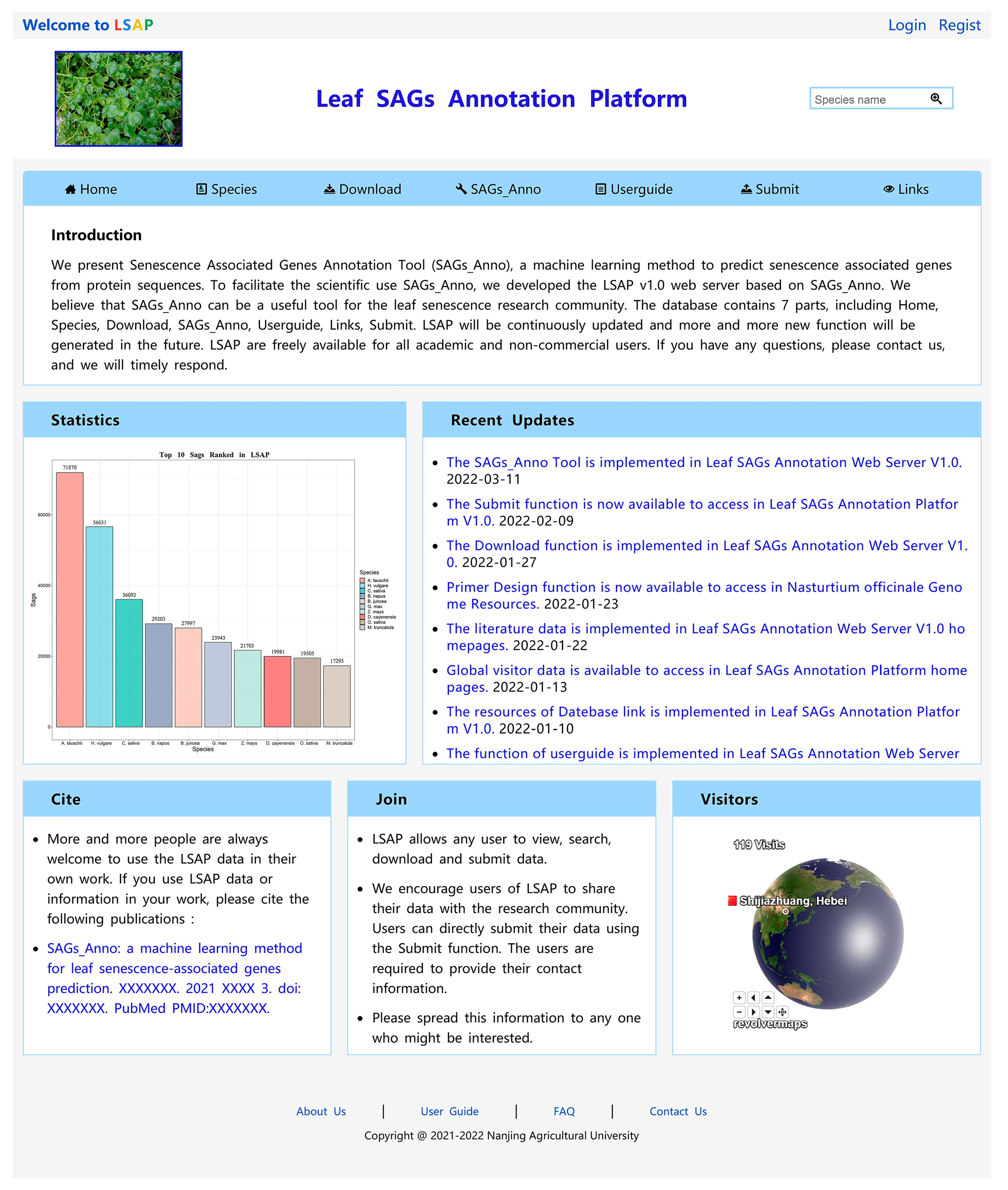
3.6. Plant Leaf SAGs Database Construction
3.7. SAGs_Anno
3.8. Browse Examined Species SAGs Dataset
3.9. Download
3.10. Userguide, Submit, Home, and Links
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Z.; Zhang, Y.; Zou, D.; Zhao, Y.; Wang, H.L.; Zhang, Y.; Xia, X.; Luo, J.; Guo, H.; Zhang, Z. LSD 3.0: A comprehensive resource for the leaf senescence research community. Nucleic Acids Res. 2020, 48, D1069–D1075. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Z.; Jiang, Z.; Zhao, Y.; Peng, J.; Jin, J.; Guo, H.; Luo, J. LSD: A leaf senescence database. Nucleic Acids Res. 2011, 39, D1103–D1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhao, Y.; Liu, X.; Peng, J.; Guo, H.; Luo, J. LSD 2.0: An update of the leaf senescence database. Nucleic Acids Res. 2014, 42, D1200–D1205. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Cai, N.; Pacheco, P.P.; Narrandes, S.; Wang, Y.; Xu, W. Applications of Support Vector Machine (SVM) Learning in Cancer Genomics. Cancer Genom. Proteom. 2018, 15, 41–51. [Google Scholar]
- Ghanat Bari, M.; Ung, C.Y.; Zhang, C.; Zhu, S.; Li, H. Machine Learning-Assisted Network Inference Approach to Identify a New Class of Genes that Coordinate the Functionality of Cancer Networks. Sci. Rep. 2017, 7, 6993. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef]
- Gupta, P.; Naithani, S.; Tello-Ruiz, M.K.; Chougule, K.; D’Eustachio, P.; Fabregat, A.; Jiao, Y.; Keays, M.; Lee, Y.K.; Kumari, S.; et al. Gramene Database: Navigating Plant Comparative Genomics Resources. Curr. Plant Biol. 2016, 7–8, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhao, M.; Wang, X.; Tong, C.; Huang, S.; Tehrim, S.; Liu, Y.; Hua, W.; Liu, S. Bolbase: A comprehensive genomics database for Brassica oleracea. BMC Genom. 2013, 14, 664. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, Y.; Liu, T.; Zhang, C.; Xiao, D.; Hou, X. Non-Heading Chinese Cabbage Database: An Open-Access Platform for the Genomics of Brassica campestris (syn. Brassica rapa) ssp. chinensis. Plants 2022, 11, 1005. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wu, S.; Bai, Y.; Sun, H.; Jiao, C.; Guo, S.; Zhao, K.; Blanca, J.; Zhang, Z.; Huang, S.; et al. Cucurbit Genomics Database (CuGenDB): A central portal for comparative and functional genomics of cucurbit crops. Nucleic Acids Res. 2019, 47, D1128–D1136. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.V.; Conners, S.I.; Huang, W.; Wilkey, A.P.; Grant, D.; Weeks, N.T.; Cannon, S.B.; Graham, M.A.; Nelson, R.T. A new decade and new data at SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 2021, 49, D1496–D1501. [Google Scholar] [CrossRef]
- Jayakodi, M.; Choi, B.S.; Lee, S.C.; Kim, N.H.; Park, J.Y.; Jang, W.; Lakshmanan, M.; Mohan, S.V.G.; Lee, D.Y.; Yang, T.J. Ginseng Genome Database: An open-access platform for genomics of Panax ginseng. BMC Plant Biol. 2018, 18, 62. [Google Scholar] [CrossRef]
- Sakai, H.; Naito, K.; Takahashi, Y.; Sato, T.; Yamamoto, T.; Muto, I.; Itoh, T.; Tomooka, N. The Vigna Genome Server, ‘VigGS’: A Genomic Knowledge Base of the Genus Vigna Based on High-Quality, Annotated Genome Sequence of the Azuki Bean, Vigna angularis (Willd.) Ohwi & Ohashi. Plant Cell Physiol. 2016, 57, e2. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.J.; Baek, S.; Lee, Y.J.; Cho, A.; Mun, J.H. The radish genome database (RadishGD): An integrated information resource for radish genomics. Database 2019, 2019, baz009. [Google Scholar] [CrossRef] [Green Version]
- Plomion, C.; Aury, J.M.; Amselem, J.; Leroy, T.; Murat, F.; Duplessis, S.; Faye, S.; Francillonne, N.; Labadie, K.; Le Provost, G.; et al. Oak genome reveals facets of long lifespan. Nat Plants. 2018, 4, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; van Treuren, R.; Liu, X.; Zhang, Z.; Chen, J.; Liu, Y.; Dong, S.; Sun, P.; Yang, T.; Lan, T.; et al. Whole-genome resequencing of 445 Lactuca accessions reveals the domestication history of cultivated lettuce. Nat. Genet. 2021, 53, 752–760. [Google Scholar] [CrossRef]
- Wang, X.; Wu, J.; Liang, J.; Cheng, F.; Wang, X. Brassica database (BRAD) version 2.0: Integrating and mining Brassicaceae species genomic resources. Database 2015, 2015, bav093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, S.L.; Erthmann, P.O.; Agerbirk, N.; Bak, S.; Hauser, T.P.; Nagy, I.; Paina, C.; Asp, T. The genome sequence of Barbarea vulgaris facilitates the study of ecological biochemistry. Sci. Rep. 2017, 7, 40728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droc, G.; Lariviere, D.; Guignon, V.; Yahiaoui, N.; This, D.; Garsmeur, O.; Dereeper, A.; Hamelin, C.; Argout, X.; Dufayard, J.F.; et al. The banana genome hub. Database 2013, 2013, bat035. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Methods | Number of Feature | F1score | ACC | AUC |
|---|---|---|---|---|
| SVM-ACC | 27 | 0.811 | 0.767 | 0.721 |
| SVM-Kmer | 400 | 0.858 | 0.857 | 0.912 |
| SVM-PC-PseAAC | 22 | 0.838 | 0.834 | 0.900 |
| SVM-Kmer-ACC | 427 | 0.781 | 0.787 | 0.863 |
| SVM-Kmer-PC-PseAAC | 422 | 0.852 | 0.854 | 0.925 |
| SVM-ACC-PC-PseAAC | 49 | 0.782 | 0.789 | 0.852 |
| SVM-ACC-Kmer-PC-PseAAC | 449 | 0.802 | 0.807 | 0.883 |
| Methods | Number of Feature | F1score | ACC | AUC |
|---|---|---|---|---|
| SVM-PCA-Kmer-ACC | 401 | 0.816 | 0.810 | 0.857 |
| SVM-PCA-Kmer-PC-PseAAC | 410 | 0.866 | 0.862 | 0.922 |
| SVM-PCA-ACC-PC-PseAAC | 46 | 0.799 | 0.797 | 0.847 |
| SVM-PCA-ACC-Kmer-PC-PseAAC | 161 | 0.822 | 0.822 | 0.869 |
| Methods | Number of Feature | F1score | ACC | AUC |
|---|---|---|---|---|
| XGBoost-ACC | 27 | 0.790 | 0.754 | 0.728 |
| XGBoost-Kmer | 400 | 0.860 | 0.852 | 0.916 |
| XGBoost-PC-PseAAC | 22 | 0.840 | 0.835 | 0.901 |
| XGBoost-Kmer-ACC | 427 | 0.863 | 0.854 | 0.923 |
| XGBoost-Kmer-PC-PseAAC | 422 | 0.860 | 0.853 | 0.928 |
| XGBoost-ACC-PC-PseAAC | 49 | 0.850 | 0.844 | 0.909 |
| XGBoost-ACC-Kmer-PC-PseAAC | 449 | 0.865 | 0.860 | 0.925 |
| Methods | Number of Feature | F1score | ACC | AUC |
|---|---|---|---|---|
| XGBoost-PCA-Kmer-ACC | 411 | 0. 842 | 0.832 | 0.900 |
| XGBoost-PCA-Kmer-PC-PseAAC | 212 | 0.855 | 0.846 | 0.919 |
| XGBoost-PCA-ACC-PC-PseAAC | 46 | 0.839 | 0.829 | 0.894 |
| XGBoost-PCA-ACC-Kmer-PC-PseAAC | 425 | 0.844 | 0.832 | 0.900 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Tang, W.; You, X.; Hou, X. LSAP: A Machine Learning Method for Leaf-Senescence-Associated Genes Prediction. Life 2022, 12, 1095. https://doi.org/10.3390/life12071095
Li Z, Tang W, You X, Hou X. LSAP: A Machine Learning Method for Leaf-Senescence-Associated Genes Prediction. Life. 2022; 12(7):1095. https://doi.org/10.3390/life12071095
Chicago/Turabian StyleLi, Zhidong, Wei Tang, Xiong You, and Xilin Hou. 2022. "LSAP: A Machine Learning Method for Leaf-Senescence-Associated Genes Prediction" Life 12, no. 7: 1095. https://doi.org/10.3390/life12071095