
Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors


Institute of Physiology, 1st Faculty of Medicine, Charles University, Albertov 5, 128 00 Prague, Czech Republic
Life 2022, 12(5), 606; https://doi.org/10.3390/life12050606
Submission received: 21 March 2022
/
Revised: 11 April 2022
/
Accepted: 17 April 2022
/
Published: 19 April 2022
(This article belongs to the Special Issue The Physiology and Pharmacology of Dopamine Receptors: From Normal Signaling to Pathology)
Abstract
:The dopaminergic system is one of the most important neurotransmitter systems in the central nervous system (CNS). It acts mainly by activation of the D1-like receptor family at the target cell. Additionally, fine-tuning of the signal is achieved via pre-synaptic modulation by the D2-like receptor family. Some dopamine drugs (both agonists and antagonists) bind in addition to DRs also to α2-ARs and 5-HT receptors. Unfortunately, these compounds are often considered subtype(s) specific. Thus, it is important to consider the presence of these receptor subtypes in specific CNS areas as the function virtually elicited by one receptor type could be an effect of other—or the co-effect of multiple receptors. However, there are enough molecules with adequate specificity. In this review, we want to give an overview of the most common off-targets for established dopamine receptor ligands. To give an overall picture, we included a discussion on subtype selectivity. Molecules used as antipsychotic drugs are reviewed too. Therefore, we will summarize reported affinities and give an outline of molecules sufficiently specific for one or more subtypes (i.e., for subfamily), the presence of DR, α2-ARs, and 5-HT receptors in CNS areas, which could help avoid ambiguous results.
1. Introduction
The dopaminergic system is one of the most important neurotransmitter systems in the CNS. Dopamine receptors (DRs, see Abbreviations for abbreviation list) belong to G protein-coupled receptor (GPCR) family. According to their structural similarities, DRs are divided into two groups (for a review, see [1]): D1-like (D1 and D5 subtypes) and D2-like (D2, D3, and D4 subtypes). The families of DRs differ in the coupling to G proteins and subsequent steps of intracellular signalization. While D1-like DRs activate adenylyl cyclase via Gs protein, the D2-like family (mainly pre-synaptic D2 DRs) inhibits adenylyl cyclase via Gi protein activation. However, in detail, D1-like DRs activate not only adenylyl cyclase but also increase phosphoinositide metabolism [2]. Similarly, coupling with Gq protein allows D2 DRs to activate phospholipase C (see note about receptor variants below). D1-like receptors are characterized by non-simple interactions with various other mediators and receptor systems, which can be activity-dependent, comprise heterological oligomerization, dynamic compartmentalization of signaling components, and system integration for exquisite functional regulation (see [2] for detail). The adenylyl cyclase response is associated with the D1 subtype, while the phosphoinositide responses may be preferentially mediated through stimulation of the D5 receptor [2].
The genes for D1-like and D2-like families differ in the presence of introns in their coding sequence. While the D1-like family does not contain introns [3,4], the D2-like family does [5,6,7,8]. This fact allows the generation of receptor variants, “long” and “short” D2 receptor isoforms. These two isoforms exhibit largely similar pharmacological characteristics, but their differences in G protein coupling [9] suggest different functions [10].
1.1. D1-like Family
D1-like family is the main element of the dopamine post-synaptic action (despite its pre-synaptic localization). Its members, D1 and D5 DRs, are pharmacologically indistinguishable. However, the affinities of D5 DR to the agonists are up to 10 times higher than that of D1 ones [11]. This fact could be of importance when one transmitter is supposed to have two effects—one through the high-affinity sites and the second one through the low-affinity sites in tissue expressing both subtypes. This could explain the different functions of striatal D1 and D5 DRs in synaptic plasticity [12]. Another difference between these two subtypes that is interesting to mention is that the D5 dopamine receptor, unlike the D1 subtype, is constitutively (agonist-independently) active [13]. Moreover, D1 DRs couple preferentially to G protein heterotrimers that contain γ7 subunits [14]. D1 DRs can also couple to another G protein, Golf (which also stimulates adenylyl cyclase) that is highly expressed in some brain areas, such as the caudate nucleus, nucleus accumbens, and olfactory tubercle. Some coupling of D1 DR with Golf was even suggested to be preferential [15]. The generation of D5 DR knockout mouse uncovered possible involvement of this subtype in the pathology of hypertension, as the mutant mice were hypertensive [16].
1.2. D2-like Family
D2 DRs are as D1 DRs [17] localized both pre- and postsynaptically. D2 DR has a relatively low (nanomolar) affinity for dopamine, which supports its importance as a modulatory (pre-synaptic) receptor. D2 DR isoforms (long and short) are differently distributed and thus may possess distinct functions. The short isoform seems to serve as an autoreceptor, whereas the long isoform is primarily a post-synaptic receptor [18]. Using genetically targeted deletion of the D2 dopamine receptor gene in mice revealed that other members of the receptor family were not affected [19] and these mutants had reduced locomotion and less coordinated movement [19].
D3 subtype of DR appears to have similar distribution as the D2 dopamine receptor [1]. Similar to D2 DR, alternative splicing variants of D3 DR were observed. These variants were hypothesized to contribute to the availability of active D3 DRs in some psychiatric conditions [20]. This hypothesis suggests that inactive D3 DRs affect ligand binding to the active D3 DRs and thus influence their function.
1.3. DR Ligand Targets
We have described above that signaling through DRs is far from to be simple. What is more, some DR ligands bind not only to DRs, but the spectrum of targets is much wider. Surprisingly, this is valid for dopamine itself. This natural neurotransmitter binds not only to DRs (D1-D5 pKis [see Abbreviations for abbreviation list and the elucidation of differences between pKi and pEC50 in the next paragraph] vary between 4.3–7.6 [7,8,23]), and dopamine transporter (DAT, pKi = 5.3 [24]) but also to other transporters (norepinephrine transporter—NET (pKi = 4.55 [25]), serotonin transporter—SERT, pKi = 4.53 [25]), to other receptors (α1-ARs (pKi-5.6, [26]), α2-ARs (pKi = 6.01, [26]), β1-, β2-ARs (pKi = 5.0, pKi = 4.3, respectively [27]) and to melatonin receptors MT1A, 1B, pKi = 5.15, pKi = 5.04, respectively). Looking at these numbers, it is possible to conclude that dopamine is bound with a similar affinity to D1 and D2 DRs (pKi = 4.3–5.6, pKi = 5.3–6.4, respectively) and DAT, NET, SERT, α1-, and α2-ARs, β1-, β2-ARs, and to melatonin receptors MT1A, 1B (see the pKis above). Other DRs have to dopamine a higher affinity (pKi = 6.3–7.4, 7.6, 6.6, respectively, for D3, D4, and D5 DRs).
It is necessary to mention (please see the values in this review) that binding assessed parameters (i.e., pKis) differ from the values determined using functional studies (i.e., dose-response determined constants, pEC50s [28]). This is because in studies based on dose-response determined parameters; the ligand usually discards the presence of other receptors on the studied effect by a combination of pharmacological means to attribute properly the receptor involved. Another possibility is that in dose-response studies, the formation of a ligand-receptor complex with activation of G protein and further with target second messenger producer activation is more complicated than the binding of ligand to the receptor in binding studies. The interesting correlation between pKi and pEC50 has been demonstrated for neurokinin NK1 receptors [29]. Although this is a specific example for specific receptors and specific ligands, we can assume that a similar correlation can be found for DRs and their ligands too. As reported here, the pKis and pEC50s differ for D1-like DR to SKF 38393. With some methodological reservation, one could construct the correlation between these values reported in [13,23,30,31,32,33,34] in humans and rats.
A similar multitarget binding can be found for DR agonists and antagonists. This review will focus on such interactions that can broaden the physiological effects elicited by dopamine ligands in the central nervous system. Besides, these interactions could present the potential problem with results interpretation: the ligand activating more neurotransmitter receptors that have similar affinity to them can distort the conclusions made. With this point of view, this review could help with careful interpretation of the results obtained. We will focus on orthosteric binding sites only, although there are also described allosteric binding sites on D2 DR [35]. The allosteric binding sites [36,37] and their interaction with other molecules exceed the topics of this review. The inclusion criteria were the ability to bind to other targets with pKi ≥ 7.0, pKIC50 ≥ 7.0 if the pKi for DRs is between 8 and 9. Interestingly, some papers report a surprisingly high concentration of drugs used as proof of specific dopamine subtype involvement even though the selectivity of such ligand is limited (e.g., SKF 38393 in concentration 100 μmol/L affects all dopamine receptor subtypes and also α2C-AR). If the ligand is sufficiently specific to dopamine receptors (i.e., the affinity differs at least two orders of magnitude), then it is not reviewed here.
The interested researcher should search available databases carefully for the ligands with well-documented selectivity to specific DR subtypes and not rely on the information from the manufacturer. The specific ligand should be at least two orders of magnitude more specific for the respective DR subtype than to the others. In other words: ΔpKis(pKi1, pKi2) ≥ 2. The examples of such ligands are shown in Table 1. On the other hand, the new research can bring new knowledge, and the supposed selectivity of the specific ligand could be doubted. Thus, it is necessary, before the choice of ligand, carefully check the present knowledge to avoid the use of non-specific ligands.
When using radioligand for receptor detection, one should be aware that a better option is to use an antagonist than an agonist because of stronger binding and lower possibility of dissociation of such ligand from the receptors.
2. DR Agonists
2.1. So-Called Selective Dopamine Receptor Agonists
The typical problem with dopamine ligand lies in the fact that manufacturers usually declare the ligand as selective, which could be, in some cases, far from reality. This could be misleading, and it could distort the conclusions made with such a “selective” drug. In the following paragraphs, we will describe the DR agonist in which the selectivity is limited. Other ligands that are selective according to present knowledge will not be mentioned.
We can generalize that dopamine drugs (both agonists and antagonists) bind in addition to DRs also to α2-ARs and 5-HT receptors. Thus, it is important to consider the presence of these receptor subtypes in specific CNS areas as the function virtually elicited by one receptor type could be the effect of other—or the co-effect of multiple receptors. The presence of neurotransmitter receptors in the CNS is shown in Table 2. In addition to that, dopamine ligands often bind to H1 histamine receptors. These receptors are present in many CNS structures [39]: cerebral cortex, hippocampal dentate gyrus, amygdaloid complex, basal forebrain, nucleus accumbens, islands of Calleja, septal nuclei, thalamus, hypothalamus (medial preoptic area, dorsomedial, ventromedial, and most posterior nuclei, including the tuberomammillary complex), nuclei of origin of most cranial nerves, and in the dorsal horn of spinal cord.
As an example, we can use SKF 38393. One of the manufacturers claims that this is a prototypical D1-like DR selective partial agonist. The careful search for pKi values (pEC50 values, respectively, see the discounts in Section 1.3), however, can indicate pKi = 6.41–6.8 [13,23,30] in human, pKi = 7.19 in rat [31], pEC50 = 5.0–8.96 in human for D1 DR [32,34], pKi = 6.91–7.0 for D5 DR in human [23,33], and pKi = 5.16 for D2 DR in rat [31]. These values indicate selectivity to D1-like DRs, but still show some effect on D2 DR. More importantly, SKF 38393 is also bound by α2C-AR with pKi = 7.08 [45], i.e., in the rank in which D1 and D5 DRs are activated.
This is important in tissues in which are DRs and ARs co-expressed (see Table 2). D1-like DRs are present [40] together with α2C-ARs [41] in the following brain areas: the cerebral cortex and amygdala. In general, α2C-ARs presence is described in the basal ganglia, and D1 DRs are abundantly present in the subthalamic nucleus and caudate-putamen. The D2 DRs (although they have a lower affinity to SKF 38393) are simultaneously present in α2C-ARs in the substantia nigra pars compacta and the ventral tegmental area. In those brain areas, one should be careful when interpreting the results obtained with SKF 38393 as both effects on DRs and α2-ARs can be present. Ignoring the fact that SKF 38393 activates D1-like DRs and blocks α2C-ARs could lead to misinterpretation of the results.
Another “selective” D1 DR ligand is the partial agonist A68930, although also designated as sub-family selective. This compound was reported to have a similar effect on rat D1 and D5 DRs (pEC50 = 6.82 and 6.6, respectively, [46]). The other data showed higher pEC50 at D1 DRs in the rat (pEC50 = 8.71, when pKi = 8.8 [47]). This study also determined pKi = 6.09, and pEC50 = 4.99 at D2 DRs in the rat. This drug also binds to 5-HT1A, 5-HT2C serotonin receptors, and β1-ARs with pKi = 5.59, 5.0, and 5.0, respectively [47]. Although the affinity of 5-HT1A, 5-HT2C serotonin receptors, and β1-ARs is lower than D1-like DRs (when considering the data from [47]), the data from [46] are quite similar, and one should be cautious with the interpretation of the results obtained with this drug.
Quinpirole is very often declared by manufacturers as a selective dopamine D2 DR (or D2-like) agonist. As an example, quinpirole sensitization was used as a model of obsessive-compulsive disorder [48], targeting the D2 and D3 DRs. However, the pKi values for D2, D3, D4, and D1 DRs, respectively (pKi = 4.9–7.7 [49], pKi = 7.3–7.7 [49], pKi = 7.5 [50], pKi = 4.06–7.2 [51,52], respectively) do not reveal the full selectivity. The spectrum of quinpirole action is much wider: 5-HT2B, 5-HT2A, and 5-HT2C receptors reveal pKi = 5.0–6.5 [50], and 5-HT1A receptor reveal pKi = 5.8 [53]. These values are apparently in the rank of DR action. Quinpirole also produces significant THC-like effects when metabolic degradation of anandamide is inhibited, supporting the hypothesis that these effects of quinpirole are mediated by cannabinoid CB1 receptors [54].
Sumanirole (PNU-95,666) is assumed as a highly selective D2 DR full agonist, the first of its kind to be discovered [55] with D2 DR pKi = 8.1 [56]. 5-HT1A receptor reveals pKi = 7.14 [57] to sumanirole, which is too close to the pKi for D2 DR and co-effect should exist. There is also agonist activity of sumanirole at human D3 DR transfected in HEK293T cells, revealing pKi = 6.73 [58], suggesting slightly limited selectivity of sumanirole on D2 DR. It means that 50% of D2 DRs are occupied by approximately 8 nmol/L sumanirole and 50% of D3 DRs are occupied by approximately 189 nmol/L sumanirole. 20 nmol/L should completely block D2 DRs, but also 10% of D3 DRs.
2.2. Drugs–Dopamine Receptor Agonists with Multiple Targets of Action
Usually, the drugs used in the treatment have multiple targets of action, which can be an advantage as multiple targets are affected by one drug. In the following paragraphs, we will mention the drugs that: (1) also have DRs action, (2) are declared as a drug with multiple targets. This could help in the interpretation of the effects obtained with this drug that could be erroneously attributed to one target only.
An example of such a drug is fenoldopam, which causes arterial/arteriolar vasodilation decreasing blood pressure. Fenoldopam is used for the in-hospital, short-term (up to 48 h) management of severe hypertension, including malignant hypertension. It is declared as an agonist for D1 DRs with moderate affinity to α2-ARs and no significant affinity for D2 DRs, α1 and β-ARs, 5-HT1 and 5-HT2 receptors, or muscarinic receptors.
However, fenoldopam is also bound with similar affinity to D5 DR (pKi = 9.1 for D1 DR, pKi = 9.2 for D5 DR, respectively) and D2 DR (pKi = 8.5), and with lower affinity to D4 DR (pKi = 6.8) [59]. Some data indicate pKi to D2 DR is lower (4.89–5.89, [60]). Early evidence showed that fenoldopam had no effect on β-ARs, but had antagonistic activity on α1-ARs [61] (pA2 = 8.36 ± 0.21), although in some papers characterized as weak (pKi = 5.41, [62], or modest pKi = 6.82 [26]) and α2-ARs [63] (pKi = 7.60–7.78, [62]). Fenoldopam thus represents the typical multiple targets drug. This is a disadvantage with respect to the specific effect of receptors when aiming to determine the subtype involved in the function but could be an advantage when targeting to specific therapeutic aim (e.g., acute severe hypertension treatment).
Another example of a drug with multitarget action is atypical antipsychotic aripiprazole. This drug acts as an atypical agonist on D2 DRs (pKi = 9.7 [64]) with expressed selectivity over D4 DRs (pKi = 7.3 [64]). However, on D4 DRs its action is antagonistic. The multitargeting of this ligand comprises partial agonism on 5-HT1A and 5-HT2A serotonin receptors with pKi = 8.2 [65], and pKi = 7.5–8.1 [65], respectively. On 5-HT1D aripiprazole reveals full agonism with pKi = 7.2 [65]. Other serotonin receptors affected by aripiprazole are 5-HT7 (partial agonism, pKi = 7.8 [66]) and 5-HT2C (partial agonism, pKi = 7.6 [67]). H1 histamine receptors are antagonized by this ligand with pKi = 7.5 [67].
A wide spectrum of action also reveals cabergoline which is an ergot-derived, long-acting D2 DR agonist and prolactin inhibitor. However, the D2 DR selectivity is rather declared than it corresponds to the reality. This drug binds, besides to DRs, to other receptor proteins [50]: D2 DRs and D3 DRs bind this drug with similar affinity as a partial agonist (pKi = 9.0–9.2, and pKi = 9.1 for D2 DR and D3 DR, respectively), similar affinity reveal 5-HT2B receptors (pKi = 8.9, full agonist) and very close affinity show 5-HT2A and 5-HT1D (pKi = 8.2 and pKi = 8.1, respectively for 5-HT2A (full agonist) and 5-HT1D receptors [partial agonist]). On the other D2-like DRs (D4 DR) it also behaves as a partial agonist, but the affinity is lower (pKi = 7.3). Besides these effects cabergoline acts also as an antagonist on α2A-AR, α2C-AR, α2B-AR, and α1A-AR (with pKi = 7.9, pKi = 7.7, pKi = 7.1, and pKi = 7.1, respectively on α2A-AR, α2C-AR, α2B-AR, and α1A-AR) and as a full agonist on 5-HT1A receptor (pKi = 7.7) [50]. One should be cautious when thinking about the D2 DR or D2-like selectivity. Although about 1.5 order of magnitude difference (pKi about 9.0 for D2 DRs), the affinity of D1-like receptors could still play a role in the action of cabergoline: on D5 DR it behaves like a full agonist with pKi = 7.7, on the D1 DR it reveals a similar type of action (full agonism), but the pKi = 6.7 is significantly lower [50]. The affinity (full agonism) of 5-HT1B and 5-HT2C is much lower than the affinity of other receptors (pKi = 6.3 and pKi = 6.2, respectively) [50].
One of the typical drugs that has been used for almost 50 years for the treatment of pituitary tumors, Parkinson’s disease, hyperprolactinemia, neuroleptic malignant syndrome, and, as an adjunct, type 2 diabetes is an ergoline derivative and dopamine agonist bromocriptine. Typically, this drug has many targets of actions: 5-HT1D receptor (acts as partial agonist) with pKi = 8.0 [50], α2A-AR (acts as antagonist) with pKi = 8.0 [50], 5-HT1A receptor (acts as partial agonist) with pKi = 7.9 [50], D2 DR (acts as full agonist [50]; however, in rats it is a partial agonist [7]) with pKi = 7.3–8.3, 5-HT7 receptor (acts as full agonist) with pKi = 7.3–8.0 [68], D3 DR (acts as partial agonist [50]; however, in rats it is a full agonist [7]) with pKi = 7.1–8.2 [50], α2C-AR (acts as antagonist) with pKi = 7.6, 5-HT6 receptor (act as full agonist [69]; however, in rats it is a partial agonist [70]) with pKi = 7.5, α2B-AR (acts as antagonist) with pKi = 7.5 [50], 5-HT2B receptor (act as antagonist) with pKi = 7.3 [50], 5-HT2A receptor (act as partial agonist [50]) with pKi = 7.0, Other receptors (5-HT1B receptor, D4 DR, D5 DR, D1 DR, and 5-HT2C receptor reveal lower affinity with pKis < 7.0 [50]). When applied to experimental animals one should count all effects listed above.
The drug with declared multiple effects is apomorphine, historically used to relieve anxiety and craving in alcoholics, as an emetic, or in treating erectile dysfunction. Currently, apomorphine is used in the treatment of Parkinson’s disease but should be used together with antiemetics. Contrary to its name, apomorphine does not contain morphine or its skeleton, nor does it bind to opioid receptors. It is declared as a non-selective dopamine agonist which activates both D2-like and, to a much lesser extent, D1-like receptors, an antagonist of 5-HT2 and α-AR with high affinity. In detail, D4 DR binds this compound as a partial agonist with pKi = 8.4 [50], rat and human D3 DR binds this compound as a partial agonist with pKi = 7.7 [7], and with pKi = 6.1–7.6 [50], respectively. Rat and human D2 DRs bind this compound as a partial agonist with pKi = 7.6 [7], and pKi = 5.7–7.5 [50], respectively. α2C-AR binds this compound as an antagonist with pKi = 7.4 [50], α2B-AR binds this compound as an antagonist with pKi = 7.2 [50], D5 DR binds this compound as a partial agonist with pKi = 6.4–7.8 [50], 5-HT2C receptors bind this compound as an antagonist with pKi = 7.0 [50], 5-HT1A receptors bind this compound as a partial agonist with pKi = 6.9 [50], 5-HT2A receptor binds this compound as an antagonist with pKi = 6.9 [50], 5-HT2B receptor binds this compound as an antagonist with pKi = 6.9 [50], α2A-AR binds this compound as a partial agonist with pKi = 6.9 [50]. All these values, except stated otherwise, come from human receptors.
Benzquinamide is more potent inhibitor of cyclooxygenase COX-2 (pIC50 = 8.3) than agonist on D2 DR (pKi = 5.4) [71].
3. DR Antagonists
3.1. So-Called Selective Dopamine Receptor Antagonists
An example of a drug declared as D1 (or D1-like family, pKi = 8.4 for D1 DR) selective antagonist is flupentixol [13]. However, this antagonist also affects σ3-receptors [72] (pKi = 8.86). In addition to that, this ligand also antagonizes the D2-like family (pKi = 8.82 for D2 DR, and pKi = 8.96 for D3 DR, respectively) [73].
Another example of a drug, declared as specific, is L-741,626 which is usually marked as a potent D2 DR selective antagonist over D3 DR and D4 DR, respectively (D2 DR: pKi = 7.95–8.35 [74], D3 DR: pKi = 6.79–7.04 [74], D3 DR: pKi = 5.82 [74]). However, this compound also binds to the σ-1 receptor with pKi = 7.71 [75].
Domperidone, acting peripherally, as it is extensively metabolized in the liver, and has the low central nervous system penetration, is the next example of a declared specific D2 and D3 DR antagonist (pKi = 7.9–8.4, and pKi = 7.1–7.6, for D2 and D3 DRs, respectively [73]) is also able to bind to 5-HT3A/5-HT3B receptors with pKIC50 = 7.0 [76].
Nafadotride is usually considered a highly potent and competitive, centrally active D3 DR antagonist (pKi = 9.5 [77]) over D2 DR (pKi = 8.8 [77]) and mainly over D4 DR (pKi = 6.4 [64]). However, also 5-HT1A receptor can be activated (full agonisms exist here [78]) by this drug with pKi = 7.3.
PG01037 is considered as D3 DR selective antagonist (pKi = 9.2 [79]). Some other papers indicate different affinity (from pKi = 8.68 [80]to pKi = 9.5 [81]), and some indicate significant affinity to D2 DR (pKi = 7.13 [81]), to 5-HT2C (pKi = 7.33 [79]), to 5-HT2A (pKi = 7.2 [79]), and to 5-HT1A (pKi = 7.07 [79]).
The specific situation comes with spiperone. Spiperone is considered a D2-like dopamine receptor-specific ligand (pKi = 8.4–9.4 [82], 9.2 [83], and 9.3 [82] for D2, D3, and D4 DR, respectively) and is commercially available as a tritiated ligand. However, this ligand also exhibits similar affinities (pKi = 7.8–9.4) for 5-HT2A receptors [84], 5-HT1B receptors (pKi = 8.3) [85], and α1A, α1B and α1D-ARs (pKi = 8.3, 9.2, and 8.1, respectively) [86]. This is a very inconvenient feature as tritiated spiperone (3H-spiperone) is very often used as a specific ligand for binding of D2-like family: we found 1,156 results for 3H-spiperone in a Pubmed search (accessed on 21 March 2022). One should be cautious when interpreting the results obtained with 3H-spiperone in the cerebral cortex, striatum, olfactory tubercle, substantia nigra, globus pallidus, nucleus accumbens, CA1 region of hippocampus, hypothalamus, and cerebellum (see Table 2 for the presence of specific 5-HT subtypes). Moreover, the pKis of D1 and D5 DRs are 6.7, and 5.4, respectively [23].
On the other hand, another radiolabeled ligand, raclopride is specific for DR and has a similar affinity to D2 DR (pKi = 7.77 [87]) and D3 DR (pKi = 7.82 [87]) but do not bind significantly to D4 DR (pKi = 5.51 [87]) and also not to D1 DR (pKi = 4.43 [87]).
Another radiolabeled ligand used in DR assays, 7-OH-DPAT, binds to D3 DR with pKi = 5.85–9.6 [88,89]. It is necessary to say that the study with pKi = 5.85 [88] is exceptional, and usually, the pKi rank is between 8 and 9. The affinity to D2 DR is lower (pKi = 6.51 [90]-8.73 [91], as well as to D4 DR (pKi = 6.83 [92]). Besides these receptors, 7-OH-DPAT has also some affinity to 5-HT1A receptors (pKi = 7.33 [92]), and σ1-receptors (pKi = 7.63 [93]).
3.2. Drugs–Dopamine Receptor Antagonists with Multiple Targets of Action
Similar to agonists, there are some drugs used in the treatment of psychiatric/neurological disorders with multiple targets action. One of them is blonanserin, an atypical antipsychotic for the treatment of schizophrenia [94]. The spectrum of targets is relatively close, but in addition to D2 DRs (pKi = 9.9 [95]) it also antagonize the action on 5-HT2A receptors (pKi = 9.1 [95]) and on D3 DRs (pKi = 6.3 [96]). Blonanserin has a low affinity [97] for 5-HT2C, α1-ARs, histamine H1, and M1 muscarinic receptors but displays a relatively high affinity for 5-HT6 receptors (pKi = 7.93) [97].
Another atypical antipsychotic drug, risperidone, binds to 5-HT7 receptor in rat as an inverse agonist with pKd = 8.9–9.0 [98], to 5-HT2A receptor as an inverse agonist with pKi = 9.3–10.0 [67], to D2 DR as an antagonist with pKi = 9.4 [99], to 5-HT2A receptor in rat as an antagonist with pKi = 8.5 [100], to 5-HT7 receptor as an inverse agonist with pKi = 8.3–8.7 [101], to α1A-AR as an antagonist with pKi = 8.4 [86], to α1B-AR as an antagonist with pKi = 8.0 [86], to α2C-AR as an antagonist with pKi = 8.49 [102], to α2A-AR as an antagonist with pKi = 8.0 [102], to 5-HT1D receptor as an antagonist with pKi = 7.8–8.0 [103], to H1 histamine receptor as an antagonist with pKi = 7.6–7.8 [67,103], to 5-HT2C receptor as an inverse agonist with pKi = 7.5–7.6 [67], to 5-HT2B receptor as an antagonist with pKi = 7.7 [104], to 5-HT1A receptor as an antagonist with pKi = 7.68 [105], to α1D-adrenoceptor as an antagonist with pKi = 7.4 [86], to D3 DR as an antagonist with pKi = 7.0 [106], and to 5-HT1B receptor as antagonist with pKi = 6.6–7.3 [103]. Other targets (5-HT6, 5-HT1F receptors) have a lower affinity (pKi less 7.0).
Perphenazine, a typical antipsychotic, binds to a set of receptors: to D2 DR as an antagonist with pKi = 8.9–9.6 [67], to 5-HT2A receptor as an antagonist with pKi = 8.2 [67], to H1 histamine receptor as an antagonist with pKi =8.1 [67], to other 5-HT receptors (5-HT6, 5-HT7, 5-HT2C) the pKi vary between 7.8 and 6.9 [67,98,107].
Trifluoperazine, a typical antipsychotic drug, binds to D2 DR as an antagonist with pKi = 8.9–9.0 [67], to 5-HT2A receptor as an antagonist with pKi = 7.9 [67], to D4 DR as an antagonist with pKi = 7.4 [108], and to H1 histamine receptor as an antagonist with pKi = 7.2 [67].
Quetiapine, an anti-psychotic drug, is bound with the highest affinity by the H1 histamine receptor as an antagonist with pKi = 8.0–8.7 [67]. Lower affinity (antagonistic) is revealed by D2 DR (pKi = 7.2) [99]. Similar affinity as in D2 DR have 5-HT2A (pKi = 6.4–7.0, [67,103]) and 5-HT1A (pKi = 6.5–7.1, [103,104]) receptors. Interestingly, this drug can behave as an agonist [103] or as an antagonist [67] on 5-HT2A receptors. In addition to that, it also binds to α2C-AR as an antagonist with pKi = 7.0 [109], to α1A-AR, and α1B-AR as an antagonist with pKi = 7.0 [109], and M1 muscarinic receptors as an antagonist with pKi = 7.0–7.25 [105,110].
The typical antipsychotic drug, haloperidol, has a wide spectrum of actions, including antagonism on DRs (D4 DR pKi = 8.7–8.8, D2 DR pKi = 7.4–8.8, D3 DR pKi = 7.5–8.6, D1 DR pKi = 7.6–8.2) and antagonism on 5-HT receptors (5-HT2A receptor pKi = 6.7–7.3, other 5-HT receptors (5-HT1D, 5-HT7) have pKi < 7.0). Similarly, D5 DR and H1 histamine receptors reveal pKi < 7.0. Relatively high affinity to this drug also reveal α1A-AR (antagonist, pKi = 7.89–8.55 [111,112]), α1B-AR (antagonist, pKi = 8.00 [86]), α1D-AR (antagonist, pKi = 7.4 [86]), α2A-AR (antagonist, pKi = 7.6 [111]), and α2C-AR (antagonist, pKi = 7.6 [109]).
Sertindole is an atypical antipsychotic drug with high affinity to 5-HT2A receptor (antagonist, pKi = 9.2–9.4 [67]), to 5-HT2C receptor (inverse agonist, pKi = 9.0–9.2 [67]), to D2 DR (antagonist, pKi = 8.0–8.9 [67]), to α1A-AR (antagonist, pKi = 9.43 [113]), to α1B-AR (antagonist, pKi = 9.48 [113]), to α1D-AR (antagonist, pKi = 9.18 [113]), to H1 histamine receptor (antagonist, pKi = 9.29 [114]), and to D4 DR (antagonist, pKi = 7.8–9.1 [108]). Relatively high affinity reveal D3 DR (antagonist, pKi = 8.0–8.8 [103]), Kv11.1/HERG kalium channels (antagonist, pKIC50 = 8.57 [115]), 5-HT6 (antagonist, pKi = 8.3 [116]), and D1 DR (antagonist, pKi = 7.92 [117]). Possible targets are 5-HT1D receptor (antagonist, pKi = 7.2 [103]) and 5-HT1B receptor (antagonist, pKi = 7.0 [103]).
Loxapine is a typical antipsychotic drug that binds to a wide spectrum of targets: H1 histamine receptor, where it acts as an antagonist with pKi = 8.2 [67], D2 DR, where it acts as an antagonist with pKi = 7.9–8.3 [67], D4 DR, where it acts as an antagonist with pKi = 8.1, 5-HT2A receptor [108], where it acts as an inverse agonist with pKi = 8.1 [67], 5-HT2C receptor, where it acts as an inverse agonist with pKi = 7.8–8.0 [67], D3 DR, where it acts as an antagonist with pKi = 7.7 [118], 5-HT6 receptor, where it acts as an inverse agonist with pKi = 7.4–7.6 [107], 5-HT7 receptor, where it acts as an antagonist with pKi = 6.8–7.4 [98].
4. Discussion
The first thing that should be discussed is the similarity in the amino acid binding pocket of DRs with α2-ARs and 5-HT receptors. It is possible to deduce this statement from apparently similar affinities (pKis) for dopamine as given in the Introduction. This is given by the similarity of neurotransmitter structures: noradrenaline, adrenaline, dopamine, and serotonin (see Figure 1). However, as mentioned above, the main role plays in the relationship between specific G protein-coupled receptors, i.e., the sequence homology in the binding pocket between dopamine, serotonin receptors, and adrenoceptors. These homologies have been well documented for the second extracellular loop, as discussed in [121].
A second fact that implies the similarities in binding pocket/amino acid homology is that other ligands that bind to the similar amino acid residues in DRs as dopamine would also affect 5-HT receptors and α2-ARs. The examples of such ligands were listed above both for agonists and antagonists.
In general, the length, organization, and amino acid homology in the D1-like DR subfamily is quite high [122]. This is the reason for so far not synthesizing specific agonists to D5 DR (see below). The D1-like DRs have a shorter third intracellular loop and a longer carboxy-terminus compared to the D2-like DR subtypes [122]. The third intracellular loop and carboxy-terminus are not structures responsible for binding. The third intracellular loop and a carboxy-terminus play a role in the G protein binding. The receptor regions responsible for binding are transmembrane zones. More precisely, the predicted binding site of dopamine in D2 DR is located in the top third of the 7-TM barrel involving TM domains 3–6 [123]. These authors also divided dopamine ligands into two groups according to their binding properties: first, clozapine-like bulky antagonists; and second, ligands with two aromatic or ring moieties connected by a flexible linker with a protonated amine group as in haloperidol [123]. The first group occupies the region between TM3, TM4, TM5, and TM6 (the agonist binding pocket), and the second group occupies the region between TM2, TM3, TM6, and TM7, with minimal contact with TM4 and TM5 [123]. The binding pocket of D1 DR is slightly different comprising TM6, extracellular loop 2, TM5, and TM3 [121].
D3 DR and D2 DR subtypes have substantial amino acid sequence homology [122].
The main aim of this review is to show that drugs declared by manufacturers as specific could be, in some cases, able to bind to other targets than to DRs. This can produce ambiguous results. Importantly, there are enough ligands with sufficient specificity for DR subtypes (see Table 1). The interested researcher should search available databases carefully for the ligands with well-documented selectivity to specific DR subtypes and no to rely on the information from the manufacturer.
Nevertheless, one can experience different values for the same compound. As reported here, the affinities of 5-HT1A, 5-HT2C serotonin receptors, and β1-ARs to A68930 are similar to those of D1-like DRs (when considering the data from [47]), but the data from [46] are quite similar. Another example reported here is SKF 38393. The pKi values differ according to specific references in humans [13,23,30], which also vary from this value in rats. This can originate from different experimental conditions (temperature, incubation time, tissue, cell culture properties, and others). In such a case, one should be cautious with the selection of this compound for subtype determination or interpretation of results obtained with this drug in the literature. If possible, it is recommended to avoid such ligands.
However, the nature of drug properties reviewed here could be more complex. One should also consider the anatomical relationship between the terminals that release dopamine and other receptors—this concerns both 5-HT receptors and α2-ARs. Dopamine terminals are frequently localized in tight contact with other axons configuring a triad—a configuration in which a neuron is connected to both the pre-synaptic element and post-synaptic (usually dendritic) target. Triads are common in the hippocampus, striatum, and medial frontal cortex (for a review, see [124]). These triads can contain both dopamine and serotonin or adrenergic terminals. The first point on how the interaction between DRs and 5-HT receptors can occur is the formation of the heteroreceptor complexes of D2 DR and 5-HT2A receptors [125]. The heterocomplexes could explain the effects of atypical antipsychotic drugs [125]. One of the possible mechanisms is based on blocking the allosteric enhancement of D2 DR protomer signaling by 5-HT2A receptor protomer activation. Another mechanism by which dopamine can interact with serotonin is the release of L-DOPA as a “false (or substitute)” neurotransmitter in the serotonin synapse [126]. “False neurotransmitter” is considered as an ectopic neurotransmitter in a neuron, which replaces the normal neurotransmitter in storage vesicles. When it is the case of L-DOPA it is then able to increase the dopamine levels as L-DOPA is a dopamine precursor. Moreover, dopamine can also act as a “false neurotransmitter” in noradrenergic neurons [126].
Another aspect is given by the presence (although sometimes doubted in dopaminergic synapse) of volume transmission [127,128,129]. This type of connection allows the spreading of the neurotransmitter to a higher distance (more than 10 μm in comparison to 30–40 nm in classical synapse), affecting 200 other dopamine synapses instead of only one post-synaptic membrane in the classical synapse. This can further be the factor of cross action of dopamine.
On the other hand, we cannot consider this a problem; this is most probably the physiological role of the transmitter.
It can be deduced from Table 1 that a D5 DR agonist does not exist to date and that the selectivity of the antagonist comprises the other member of D1-like family—D1 DR. However, specific agonists (A77636, SKF-81297, and SKF-83959) exist for D1 DR. Thus, it is possible to distinguish between D1 DR and D5 DR using the D1 DR agonists.
Specific subtypes in the D2-like family can be distinguished using specific antagonists for D2 DR (pipotiazine, ML321), D3 DR (S33084, SB 277011-A, (+)-S-14297), and D4 DR (sonepiprazole, L745870, A-381393, L741742, ML398). One should also consider the presence of off-targets (Table 2) when evaluating the role of specific dopamine receptors, as some receptors have a lower affinity to relatively selective ligand, but if the density of off-target receptors is much higher than DR that the proportion of the binding could be shifted.
Even though the attribution of a drug to be DR agonist/antagonist can also be the result of the side effect on another receptor. Thus, some drugs can primarily bind to other receptors and also reveal dopaminergic action. Examples of such drugs are some antipsychotics listed above (bromocriptine acting mainly at 5-HT receptors [50], risperidone acting mainly at 5-HT receptors [67,98], quetiapine which is H1 histamine receptor antagonist [67], sertindole which has a high affinity to 5-HT receptors [67], and loxapine acting on H1 histamine receptors [67]). Other drugs that could bind to DRs as to “second target” are muscarinic receptor agonists AC-260584, 77-LH-28-1, and LY-593039, which bind similarly to M1 muscarinic receptors and to D2 DRs [130]. Another group of drugs binds primarily to 5-HT receptors. An example of such a drug is 8-OH-DPAT (the binding of related 7-OH-DPAT is mentioned above), which is used in the tritiated form as a radioligand for 5-HT receptors. [3H]8-OH-DPAT binds to 5-HT1A receptors with high affinity (pKi = 9.33 [131]). The affinity of 5-HT1B receptors is lower (pKi = 6.25 [132]) and corresponds to the affinity to DR (pKi = 7.07 [133]). Another compound acting on 5-HT receptors and with similar binding to DRs is iloperidone, an atypical antipsychotic drug. This compound binds to 5-HT1A, 5-HT6, and 5-HT7 receptors with pKi = 6.8–7.7 [134,135] and to D2 DR with pKi = 7.0 [136]. Another atypical antipsychotic drug zotepine has antagonistic activity at 5-HT receptors (5-HT1D pKi = 9.3 [103], 5-HT2A pKi = 8.6 [103]) and on D2 DR (pKi = 8.0 [103]), D3 DR (pKi = 8.2 [103]), D4 DR (pKi = 7.4 [103]). Besides that, zotepine also binds to H1 histamine receptors (pKi = 9.2 [103]) and to 5-HT6 and to 5-HT7 with pKi = 8.9, and pKi = 8.8, respectively [98]. These examples just illustrate the complexity of the cross bunding between drugs suggested to be selective to specific receptors. The number of such interactions would increase with the increase in our knowledge on this topic.
This review can also help with the interpretation of results obtained with antipsychotic drugs as it critically reviews the real binding to different targets, and the reader can compare the affinities of specific target molecules to these ligands. In Table 2 it is possible to find the presence of other receptors (subtypes of α2-ARs and 5-HT receptors) that can help the interpretation of data obtained with antipsychotic drugs.
We can conclude that one should be very cautious when selecting the DR ligand with the aim to determine the role of a specific DR subtype in studied CNS function. This review can help in such selection. Not only the selectivity but also the presence of typical off-targets to dopamine ligands (subtypes of α2-ARs and 5-HT receptors) should be considered, and finally, the new research can bring new knowledge, and the supposed selectivity of the specific ligand could be doubted.
Funding
This research was funded by Charles University, Cooperation program in Pharmaceutical Sciences.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
List of abbreviations and a short explanation of the terms used.
| Abbreviation | Explanation |
| DR(s) | Dopamine receptor(s) |
| ARs | Adrenoceptors |
| 5-HT | Serotonin |
| TM | Transmembrane zone |
| pKi | The negative logarithm of the Ki value (the molar concentration of the competing ligand that would occupy 50% of the receptors) |
| pKD | The negative logarithm of KD value (the equilibrium dissociation constant represents the concentration of radioligand occupying half of the maximum receptor population) |
| pA2 | The measure of the potency of an antagonist, negative logarithm of the molar concentration of an antagonist that would produce a two-fold shift in the concentration-response curve for an agonist |
| pEC50 | The negative logarithm of EC50 value (the molar concentration of an agonist that produces 50% of the maximum possible response for that agonist). This value can vary when comparing different activation pathways |
References
- Emilien, G.; Maloteaux, J.-M.; Geurts, M.; Hoogenberg, K.; Cragg, S. Dopamine receptors—Physiological understanding to therapeutic intervention potential. Pharmacol. Ther. 1999, 84, 133–156. [Google Scholar] [CrossRef]
- Undieh, A.S. Pharmacology of signaling induced by dopamine D1-like receptor activation. Pharmacol. Ther. 2010, 128, 37–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civelli, O.; Bunzow, J.K.; Grandy, D.K. Molecular Diversity of the Dopamine Receptors. Annu. Rev. Pharmacol. Toxicol. 1993, 33, 281–307. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.F. Structures of Dopamine Receptors. J. Neurochem. 1993, 60, 804–816. [Google Scholar] [CrossRef]
- Giros, B.; Sokoloff, P.; Martres, M.P.; Riou, J.F.; Emorine, L.J.; Schwartz, J.C. Alternative splicing directs the expression of two D2 dopamine receptor isoforms. Nature 1989, 342, 923–926. [Google Scholar] [CrossRef]
- Grandy, D.K.; Litt, M.; Allen, L.; Bunzow, J.R.; Marchionni, M.; Makam, H.; Reed, L.; Magenis, R.E.; Civelli, O. The human dopamine D2 receptor gene is located on chromosome 11 at q22–q23 and identifies a TaqI RFLP. Am. J. Hum. Genet. 1989, 45, 778–785. [Google Scholar]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef]
- Van Tol, H.H.; Bunzow, J.R.; Guan, H.C.; Sunahara, R.K.; Seeman, P.; Niznik, H.B.; Civelli, O. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 1991, 350, 610–614. [Google Scholar] [CrossRef]
- Liu, L.X.; Monsma, F.J., Jr.; Sibley, D.R.; Chiodo, L.A. D2L, D2S, and D3 dopamine receptors stably transfected into NG108-15 cells couple to a voltage-dependent potassium current via distinct G protein mechanisms. Synapse 1996, 24, 156–164. [Google Scholar] [CrossRef]
- Picetti, R.; Saiardi, A.; Abdel Samad, T.; Bozzi, Y.; Baik, J.H.; Borrelli, E. Dopamine D2 receptors in signal transduction and behavior. Crit. Rev. Neurobiol. 1997, 11, 121–142. [Google Scholar] [CrossRef]
- Weinshank, R.L.; Adham, N.; Macchi, M.; Olsen, M.A.; Branchek, T.A.; Hartig, P.R. Molecular cloning and characterization of a high affinity dopamine receptor (D1 beta) and its pseudogene. J. Biol. Chem. 1991, 266, 22427–22435. [Google Scholar] [CrossRef]
- Centonze, D.; Grande, C.; Saulle, E.; Martin, A.B.; Gubellini, P.; Pavón, N.; Pisani, A.; Bernardi, G.; Moratalla, R.; Calabresi, P. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J. Neurosci. 2003, 23, 8506–8512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiberi, M.; Caron, M.G. High agonist-independent activity is a distinguishing feature of the dopamine D1B receptor subtype. J. Biol. Chem. 1994, 269, 27925–27931. [Google Scholar] [CrossRef]
- Wang, Q.; Jolly, J.P.; Surmeier, J.D.; Mullah, B.M.; Lidow, M.S.; Bergson, C.M.; Robishaw, J.D. Differential dependence of the D1 and D5 dopamine receptors on the G protein gamma 7 subunit for activation of adenylylcyclase. J. Biol. Chem. 2001, 276, 39386–39393. [Google Scholar] [CrossRef] [Green Version]
- Herve, D.; Levi-Strauss, M.; Marey-Semper, I.; Verney, C.; Tassin, J.P.; Glowinski, J.; Girault, J.A. G(olf) and Gs in rat basal ganglia: Possible involvement of G(olf) in the coupling of dopamine D1 receptor with adenylyl cyclase. J. Neurosci. 1993, 13, 2237–2248. [Google Scholar] [CrossRef] [Green Version]
- Hollon, T.R.; Bek, M.J.; Lachowicz, J.E.; Ariano, M.A.; Mezey, E.; Ramachandran, R.; Wersinger, S.R.; Soares-da-Silva, P.; Liu, Z.F.; Grinberg, A.; et al. Mice lacking D5 dopamine receptors have increased sympathetic tone and are hypertensive. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 10801–10810. [Google Scholar] [CrossRef] [Green Version]
- Weiner, D.M.; Levey, A.I.; Sunahara, R.K.; Niznik, H.B.; O´Dowd, B.F.; Seeman, P.; Brann, M.R. D1 and D2 dopamine receptor mRNA in rat brain. Proc. Natl. Acad. Sci. USA 1991, 88, 1859–1863. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.U.; Mrzljak, L.; Gutierrez, A.; de la Calle, A.; Goldman-Rakic, P.S. Prominence of the dopamine D2 short isoform in dopaminergic pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 7731–7736. [Google Scholar] [CrossRef] [Green Version]
- Saiardi, A.; Abdel Samad, T.; Picetti, R.; Bozzi, Y.; Baik, J.H.; Borrelli, E. The physiological role of dopamine D2 receptors. Adv. Pharmacol. 1998, 42, 521–524. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Levesque, D.; Martres, M.P.; Sokoloff, P. Dopamine D3 receptor: Basic and clinical aspects. Clin. Neuropharmacol. 1993, 16, 295–314. [Google Scholar] [CrossRef]
- Dziedzicka-Wasylewska, M. Brain dopamine receptors—Research perspectives and potential sites of regulation. Pol. J. Pharmacol. 2004, 56, 659–671. [Google Scholar] [PubMed]
- Patel, S.; Patel, S.; Marwood, R.; Emms, F.; Marston, D.; Leeson, P.D.; Curtis, N.R.; Kulagowski, J.J.; Freedman, S.B. Identification and pharmacological characterization of [125I]L-750,667, a novel radioligand for the dopamine D4 receptor. Mol. Pharmacol. 1996, 50, 1658–1664. [Google Scholar] [PubMed]
- Sunahara, R.K.; Guan, H.C.; O´Dowd, B.F.; Seeman, P.; Laurier, L.G.; Ng, G.; George, S.R.; Torchia, J.; Van Tol, H.H.; Niznik, H.B. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature 1991, 350, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.M.; Kung, M.-P.; Kabalka, G.W.; Kung, H.F.; Switzer, R. Synthesis and Characterization of Radioiodinated N-(3-Iodopropen-1-yl)-2 β-carbomethoxy-3 β-(4-chlorophenyl)tropanes: Potential Dopamine Reuptake Site Imaging Agents. J. Med. Chem. 1994, 37, 1535–1542. [Google Scholar] [CrossRef]
- Meltzer, P.C.; McPhee, M.; Madras, B.K. Synthesis and biological activity of 2-Carbomethoxy-3-catechol-8-azabicyclo[3.2.1]octanes. Bioorg. Med. Chem. Lett. 2003, 13, 4133–4137. [Google Scholar] [CrossRef]
- Fisher, L.E.; Rosenkranz, R.P.; Clark, R.D.; Muchowski, J.M.; McClelland, D.L.; Michel, A.; Caroon, J.M.; Galeazzi, E.; Eglen, R.; Whiting, R.L. N,N-6-bis-[2-(3,4-dihydroxybenzyl)pyrrolidinyl]hexane, a potent, selective, orally active dopamine analog with hypotensive and diuretic activity. Bioorg. Med. Chem. Lett. 1995, 5, 2371–2376. [Google Scholar] [CrossRef]
- Lu, S.-F.; Herbert, B.; Haufe, G.; Laue, K.W.; Padgett, W.L.; Oshunleti, O.; Daly, J.W.; Kirk, K.L. Syntheses of (R)-and (S)-2- and 6-Fluoronorepinephrine and (R)- and (S)-2- and 6-Fluoroepinephrine: Effect of Stereochemistry on Fluorine-Induced Adrenergic Selectivities. J. Med. Chem. 2000, 43, 1611–1619. [Google Scholar] [CrossRef]
- Kenakin, T. What is pharmacological ‘affinity’? Relevance to biased agonism and antagonism. Trends Pharmacol. Sci. 2014, 35, 434–441. [Google Scholar] [CrossRef]
- Rupniak, N.M.J.; Perdona, E.; Griffante, C.; Cavallini, P.; Sava, A.; Ricca, D.J.; Thor, K.B.; Burgard, E.C.; Corsi, M. Affinity, potency, efficacy, and selectivity of neurokinin A analogs at human recombinant NK2 and NK1 receptors. PLoS ONE 2018, 13, e0205894. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Cai, W.; Yu, L.; Zhen, X.; Zhang, A. ‘Click’ D1 receptor agonists with a 5-HT1A receptor pharmacophore producing D2 receptor activity. Bioorg. Med. Chem. 2009, 17, 4873–4880. [Google Scholar] [CrossRef]
- DeNinno, M.P.; Schoenleber, R.; Asin, K.E.; MacKenzie, R.; Kebabian, J.W. (1R,3S)-1-(Aminomethyl)-3,4-dihydro-5,6-dihydroxy-3-phenyl-1H-2-benzopyran: A potent and selective D1 agonist. J. Med. Chem. 1990, 33, 2948–2950. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Zhang, Y.; Yang, Z.; Qiang, K.; Chen, C.; Sun, L.; Chen, M.; Zhang, J. Chemical synthesis, microbial transformation and biological evaluation of tetrahydroprotoberberines as dopamine D1/D2 receptor ligands. Bioorg. Med. Chem. 2019, 27, 2100–2111. [Google Scholar] [CrossRef] [PubMed]
- Lebar, M.D.; Hahn, K.N.; Mutka, T.; Maignan, P.; McClintock, J.B.; Amsler, C.D.; van Olphen, A.; Kyle, D.E.; Baker, B.J. CNS and antimalarial activity of synthetic meridianin and psammopemmin analogs. Bioorg. Med. Chem. 2011, 19, 5756–5762. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yan, W.; McCorvy, J.D.; Cheng, J. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure-Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. J. Med. Chem. 2018, 61, 9841–9878. [Google Scholar] [CrossRef] [PubMed]
- Kopinathan, A.; Scammells, P.J.; Lane, J.R.; Capuano, B. Multivalent approaches and beyond: Novel tools for the investigation of dopamine D2 receptor pharmacology. Future Med. Chem. 2016, 8, 1349–1372. [Google Scholar] [CrossRef] [PubMed]
- Żuk, J.; Bartuzi, D.; Miszta, P.; Kaczor, A.A. The Role of Lipids in Allosteric Modulation of Dopamine D(2) Receptor-In Silico Study. Molecules 2022, 27, 1335. [Google Scholar] [CrossRef]
- Jones-Tabah, J.; Mohammad, H.; Paulus, E.G.; Clarke, P.B.S.; Hébert, T.E. The Signaling and Pharmacology of the Dopamine D1 Receptor. Front. Cell. Neurosci. 2021, 15, 806618. [Google Scholar] [CrossRef]
- Free, R.B.; Chun, L.S.; Moritz, A.E.; Miller, B.N.; Doyle, T.B.; Conroy, J.L.; Padron, A.; Meade, J.A.; Xiao, J.; Hu, X.; et al. Discovery and characterization of a G protein-biased agonist that inhibits β-arrestin recruitment to the D2 dopamine receptor. Mol. Pharmacol. 2014, 86, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Bouthenet, M.L.; Ruat, M.; Sales, N.; Garbarg, M.; Schwartz, J.C. A detailed mapping of hist amine H1-receptors in guinea-pig central nervous system established by autoradiography with [125I]iodobolpyramine. Neuroscience 1988, 26, 553–600. [Google Scholar] [CrossRef]
- Vallone, D.; Picetti, R.; Borrelli, E. Structure and function of dopamine receptors. Neurosci. Biobehav. Rev. 2000, 24, 125–132. [Google Scholar] [CrossRef]
- Saunders, C.; Limbird, L.E. Localization and trafficking of α2-adrenergic receptor subtypes in cells and tissues. Pharmacol. Ther. 1999, 84, 193–205. [Google Scholar] [CrossRef]
- Nichols, D.E.; Nichols, C.D. Serotonin Receptors. Chem. Rev. 2008, 108, 1614–1641. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.J.; Smith, C.T.; Petersen, K.J.; Trujillo, P.; van Wouwe, N.C.; Donahue, M.J.; Kessler, R.M.; Deutch, A.Y.; Zald, D.H.; Claassen, D.O. [(18)F]fallypride characterization of striatal and extrastriatal D(2/3) receptors in Parkinson’s disease. Neuroimage Clin. 2018, 18, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Amenta, F.; Mignini, F.; Ricci, A.; Sabbatini, M.; Tomassoni, D.; Tayebati, S.K. Age-related changes of dopamine receptors in the rat hippocampus: A light microscope autoradiography study. Mech. Ageing Dev. 2001, 122, 2071–2083. [Google Scholar] [CrossRef]
- Szőllősi, E.; Bobok, A.; Kiss, L.; Vass, M.; Kurkó, D.; Kolok, S.; Visegrády, A.; Keserű, G.M. Cell-based and virtual fragment screening for adrenergic α2C receptor agonists. Bioorg. Med. Chem. 2015, 23, 3991–3999. [Google Scholar] [CrossRef] [Green Version]
- Nergårdh, R.; Oerther, S.; Fredholm, B.B. Differences between A 68930 and SKF 82958 could suggest synergistic roles of D1 and D5 receptors. Pharmacol. Biochem. Behav. 2005, 82, 495–505. [Google Scholar] [CrossRef]
- DeNinno, M.P.; Schoenleber, R.; Perner, R.J.; Lijewski, L.; Asin, K.E.; Britton, D.R.; MacKenzie, R.; Kebabian, J.W. Synthesis and dopaminergic activity of 3-substituted 1-(aminomethyl)-3,4-dihydro-5,6-dihydroxy-1H-2-benzopyrans: Characterization of an auxiliary binding region in the D1 receptor. J. Med. Chem. 1991, 34, 2561–2569. [Google Scholar] [CrossRef]
- Stuchlik, A.; Radostová, D.; Hatalova, H.; Vales, K.; Nekovarova, T.; Koprivova, J.; Svoboda, J.; Horacek, J. Validity of Quinpirole Sensitization Rat Model of OCD: Linking Evidence from Animal and Clinical Studies. Front. Behav. Neurosci. 2016, 10, 209. [Google Scholar] [CrossRef] [Green Version]
- Burris, K.D.; Pacheco, M.A.; Filtz, T.M.; Kung, M.P.; Kung, H.F.; Molinoff, P.B. Lack of discrimination by agonists for D2 and D3 dopamine receptors. Neuropsychopharmacology 1995, 12, 335–345. [Google Scholar] [CrossRef]
- Millan, M.J.; Maiofiss, L.; Cussac, D.; Audinot, V.; Boutin, J.A.; Newman-Tancredi, A. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J. Pharmacol. Exp. Ther. 2002, 303, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Möller, D.; Kling, R.C.; Skultety, M.; Leuner, K.; Hübner, H.; Gmeiner, P. Functionally selective dopamine D2, D3 receptor partial agonists. J. Med. Chem. 2014, 57, 4861–4875. [Google Scholar] [CrossRef]
- Elsner, J.; Boeckler, F.; Heinemann, F.W.; Hübner, H.; Gmeiner, P. Pharmacophore-guided drug discovery investigations leading to bioactive 5-aminotetrahydropyrazolopyridines. Implications for the binding mode of heterocyclic dopamine D3 receptor agonists. J. Med. Chem. 2005, 48, 5771–5779. [Google Scholar] [CrossRef] [PubMed]
- Newman-Tancredi, A.; Cussac, D.; Audinot, V.; Millan, M.J. Actions of roxindole at recombinant human dopamine D2, D3 and D4 and serotonin 5-HT1A, 5-HT1B and 5-HT1D receptors. Naunyn Schmiedebergs Arch. Pharmacol. 1999, 359, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Solinas, M.; Tanda, G.; Wertheim, C.E.; Goldberg, S.R. Dopaminergic augmentation of delta-9-tetrahydrocannabinol (THC) discrimination: Possible involvement of D(2)-induced formation of anandamide. Psychopharmacology 2010, 209, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.G.; Darlington, W.H.; McMillan, M.W. Synthesis of the Selective D2 Receptor Agonist PNU-95666E from d-Phenylalanine Using a Sequential Oxidative Cyclization Strategy. J. Org. Chem. 1997, 62, 6582–6587. [Google Scholar] [CrossRef]
- McCall, R.B.; Lookingland, K.J.; Bédard, P.J.; Huff, R.M. Sumanirole, a highly dopamine D2-selective receptor agonist: In vitro and in vivo pharmacological characterization and efficacy in animal models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2005, 314, 1248–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heier, R.F.; Dolak, L.A.; Duncan, J.N.; Hyslop, D.K.; Lipton, M.F.; Martin, I.J.; Mauragis, M.A.; Piercey, M.F.; Nichols, N.F.; Schreur, P.J.; et al. Synthesis and biological activities of (R)-5,6-dihydro-N,N-dimethyl-4H-imidazo[4,5,1-ij]quinolin-5-amine and its metabolites. J. Med. Chem. 1997, 40, 639–646. [Google Scholar] [CrossRef]
- Zou, M.F.; Keck, T.M.; Kumar, V.; Donthamsetti, P.; Michino, M.; Burzynski, C.; Schweppe, C.; Bonifazi, A.; Free, R.B.; Sibley, D.R.; et al. Novel Analogues of (R)-5-(Methylamino)-5,6-dihydro-4H-imidazo[4,5,1-ij]quinolin-2(1H)-one (Sumanirole) Provide Clues to Dopamine D2/D3 Receptor Agonist Selectivity. J. Med. Chem. 2016, 59, 2973–2988. [Google Scholar] [CrossRef]
- Villalón, C.M.; Ramírez-San Juan, E.; Sánchez-López, A.; Bravo, G.; Willems, E.W.; Saxena, P.R.; Centurión, D. Pharmacological profile of the vascular responses to dopamine in the canine external carotid circulation. Pharmacol. Toxicol. 2003, 92, 165–172. [Google Scholar] [CrossRef]
- Wilcox, R.E.; Huang, W.-H.; Brusniak, M.-Y.K.; Wilcox, D.M.; Pearlman, R.S.; Teeter, M.M.; DuRand, C.J.; Wiens, B.L.; Neve, K.A. CoMFA-Based Prediction of Agonist Affinities at Recombinant Wild Type versus Serine to Alanine Point Mutated D2 Dopamine Receptors. J. Med. Chem. 2000, 43, 3005–3019. [Google Scholar] [CrossRef]
- Martin, S.W.; Broadley, K.J. Renal vasodilatation by dopexamine and fenoldopam due to α1-adrenoceptor blockade. Br. J. Pharmacol. 1995, 115, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, E.H.; Zabko-Potapovich, B.; Berkowitz, B.A. The DA1 receptor agonist fenoldopam (SK & F 82526) is also an α2-adrenoceptor antagonist. Eur. J. Pharmacol. 1985, 118, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Nichols, A.J.; Ruffolo, R.R., Jr.; Brooks, D.P. The pharmacology of fenoldopam. Am. J. Hypertens. 1990, 3, 116S–119S. [Google Scholar] [CrossRef] [PubMed]
- Schetz, J.A.; Benjamin, P.S.; Sibley, D.R. Nonconserved residues in the second transmembrane-spanning domain of the D(4) dopamine receptor are molecular determinants of D(4)-selective pharmacology. Mol. Pharmacol. 2000, 57, 144–152. [Google Scholar]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [Google Scholar] [CrossRef] [Green Version]
- Lawler, C.P.; Prioleau, C.; Lewis, M.M.; Mak, C.; Jiang, D.; Schetz, J.A.; Gonzalez, A.M.; Sibley, D.R.; Mailman, R.B. Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology 1999, 20, 612–627. [Google Scholar] [CrossRef]
- Kroeze, W.K.; Hufeisen, S.J.; Popadak, B.A.; Renock, S.M.; Steinberg, S.; Ernsberger, P.; Jayathilake, K.; Meltzer, H.Y.; Roth, B.L. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 2003, 28, 519–526. [Google Scholar] [CrossRef]
- Shen, Y.; Monsma, F.J., Jr.; Metcalf, M.A.; Jose, P.A.; Hamblin, M.W.; Sibley, D.R. Molecular cloning and expression of a 5-hydroxytryptamine7 serotonin receptor subtype. J. Biol. Chem. 1993, 268, 18200–18204. [Google Scholar] [CrossRef]
- Kohen, R.; Metcalf, M.A.; Khan, N.; Druck, T.; Huebner, K.; Lachowicz, J.E.; Meltzer, H.Y.; Sibley, D.R.; Roth, B.L.; Hamblin, M.W. Cloning, characterization, and chromosomal localization of a human 5-HT6 serotonin receptor. J. Neurochem. 1996, 66, 47–56. [Google Scholar] [CrossRef]
- Boess, F.G.; Monsma, F.J., Jr.; Sleight, A.J. Identification of residues in transmembrane regions III and VI that contribute to the ligand binding site of the serotonin 5-HT6 receptor. J. Neurochem. 1998, 71, 2169–2177. [Google Scholar] [CrossRef]
- Gregori-Puigjané, E.; Setola, V.; Hert, J.; Crews, B.A.; Irwin, J.J.; Lounkine, E.; Marnett, L.; Roth, B.L.; Shoichet, B.K. Identifying mechanism-of-action targets for drugs and probes. Proc. Natl. Acad. Sci. USA 2012, 109, 11178–11183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, A.M.; Charifson, P.S.; Owens, C.E.; Kula, N.S.; McPhail, A.T.; Baldessarini, R.J.; Booth, R.G.; Wyrick, S.D. Conformational Analysis, Pharmacophore Identification, and Comparative Molecular Field Analysis of Ligands for the Neuromodulatory ς 3 Receptor. J. Med. Chem. 1994, 37, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.B.; Patel, S.; Marwood, R.; Emms, F.; Seabrook, G.R.; Knowles, M.R.; McAllister, G. Expression and pharmacological characterization of the human D3 dopamine receptor. J. Pharmacol. Exp. Ther. 1994, 268, 417–426. [Google Scholar]
- Grundt, P.; Husband, S.L.; Luedtke, R.R.; Taylor, M.; Newman, A.H. Analogues of the dopamine D2 receptor antagonist L741, 626: Binding, function, and SAR. Bioorg. Med. Chem. Lett. 2007, 17, 745–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangveravong, S.; Taylor, M.; Xu, J.; Cui, J.; Calvin, W.; Babic, S.; Luedtke, R.R.; Mach, R.H. Synthesis and characterization of selective dopamine D2 receptor antagonists. 2. Azaindole, benzofuran, and benzothiophene analogs of L-741, 626. Bioorg. Med. Chem. 2010, 18, 5291–5300. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, Y.; Morie, T.; Yamazaki, H.; Yoshida, N.; Kato, S. A novel series of N-(hexahydro-1,4-diazepin-6-yl) and N-(hexahydroazepin-3-yl)benzamides with high affinity for 5-HT3 and dopamine D2 receptors. Bioorg. Med. Chem. Lett. 1998, 8, 619–624. [Google Scholar] [CrossRef]
- Sautel, F.; Griffon, N.; Sokoloff, P.; Schwartz, J.C.; Launay, C.; Simon, P.; Costentin, J.; Schoenfelder, A.; Garrido, F.; Mann, A.; et al. Nafadotride, a potent preferential dopamine D3 receptor antagonist, activates locomotion in rodents. J. Pharmacol. Exp. Ther. 1995, 275, 1239–1246. [Google Scholar]
- Newman-Tancredi, A.; Gavaudan, S.; Conte, C.; Chaput, C.; Touzard, M.; Verrièle, L.; Audinot, V.; Millan, M.J. Agonist and antagonist actions of antipsychotic agents at 5-HT1A receptors: A [35S]GTPgammaS binding study. Eur. J. Pharmacol. 1998, 355, 245–256. [Google Scholar] [CrossRef]
- Grundt, P.; Prevatt, K.M.; Cao, J.; Taylor, M.; Floresca, C.Z.; Choi, J.K.; Jenkins, B.G.; Luedtke, R.R.; Newman, A.H. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: Potential substance abuse therapeutic agents. J. Med. Chem. 2007, 50, 4135–4146. [Google Scholar] [CrossRef]
- Newman, A.H.; Grundt, P.; Nader, M.A. Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J. Med. Chem. 2005, 48, 3663–3679. [Google Scholar] [CrossRef]
- Keck, T.M.; John, W.S.; Czoty, P.W.; Nader, M.A.; Newman, A.H. Identifying Medication Targets for Psychostimulant Addiction: Unraveling the Dopamine D3 Receptor Hypothesis. J. Med. Chem. 2015, 58, 5361–5380. [Google Scholar] [CrossRef]
- Tang, L.; Todd, R.D.; Heller, A.; O´Malley, K.L. Pharmacological and functional characterization of D2, D3 and D4 dopamine receptors in fibroblast and dopaminergic cell lines. J. Pharmacol. Exp. Ther. 1994, 268, 495–502. [Google Scholar] [PubMed]
- MacKenzie, R.G.; VanLeeuwen, D.; Pugsley, T.A.; Shih, Y.H.; Demattos, S.; Tang, L.; Todd, R.D.; O´Malley, K.L. Characterization of the human dopamine D3 receptor expressed in transfected cell lines. Eur. J. Pharmacol. 1994, 266, 79–85. [Google Scholar] [CrossRef]
- Sleight, A.J.; Stam, N.J.; Mutel, V.; Vanderheyden, P.M. Radiolabelling of the human 5-HT2A receptor with an agonist, a partial agonist and an antagonist: Effects on apparent agonist affinities. Biochem. Pharmacol. 1996, 51, 71–76. [Google Scholar] [CrossRef]
- Maroteaux, L.; Saudou, F.; Amlaiky, N.; Boschert, U.; Plassat, J.L.; Hen, R. Mouse 5HT1B serotonin receptor: Cloning, functional expression, and localization in motor control centers. Proc. Natl. Acad. Sci. USA 1992, 89, 3020–3024. [Google Scholar] [CrossRef] [Green Version]
- Yoshio, R.; Taniguchi, T.; Itoh, H.; Muramatsu, I. Affinity of serotonin receptor antagonists and agonists to recombinant and native alpha1-adrenoceptor subtypes. Jpn. J. Pharmacol. 2001, 86, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Stark, D.; Piel, M.; Hübner, H.; Gmeiner, P.; Gründer, G.; Rösch, F. In vitro affinities of various halogenated benzamide derivatives as potential radioligands for non-invasive quantification of D(2)-like dopamine receptors. Bioorg. Med. Chem. 2007, 15, 6819–6829. [Google Scholar] [CrossRef]
- Dörfler, M.; Tschammer, N.; Hamperl, K.; Hübner, H.; Gmeiner, P. Novel D3 selective dopaminergics incorporating enyne units as nonaromatic catechol bioisosteres: Synthesis, bioactivity, and mutagenesis studies. J. Med. Chem. 2008, 51, 6829–6838. [Google Scholar] [CrossRef]
- Ricci, A.; Veglio, F.; Amenta, F. Radioligand binding characterization of putative dopamine D3 receptor in human peripheral blood lymphocytes with [3H]7-OH-DPAT. J. Neuroimmunol. 1995, 58, 139–144. [Google Scholar] [CrossRef]
- Brown, D.A.; Mishra, M.; Zhang, S.; Biswas, S.; Parrington, I.; Antonio, T.; Reith, M.E.; Dutta, A.K. Investigation of various N-heterocyclic substituted piperazine versions of 5/7-{[2-(4-aryl-piperazin-1-yl)-ethyl]-propyl-amino}-5,6,7,8-tetrahydro-naphthalen-2-ol: Effect on affinity and selectivity for dopamine D3 receptor. Bioorg. Med. Chem. 2009, 17, 3923–3933. [Google Scholar] [CrossRef] [Green Version]
- Maggio, R.; Scarselli, M.; Novi, F.; Corsini, G.U. Heterodimerization of G-Protein-Coupled Receptors Reveals an Unexpected Level of Pharmacological Diversity. Med. Chem. Res. 2004, 13, 25–33. [Google Scholar] [CrossRef]
- Stjernlöf, P.; Lin, C.-H.; Sonesson, C.; Svensson, K.; Smith, M.W. (Dipropylamino)-tetrahydronaphthofurans: Centrally acting serotonin agonists and dopamine agonists-antagonists. Bioorg. Med. Chem. Lett. 1997, 7, 2759–2764. [Google Scholar] [CrossRef]
- Chumpradit, S.; Kung, M.P.; Vessotskie, J.; Foulon, C.; Mu, M.; Kung, H.F. Iodinated 2-aminotetralins and 3-amino-1-benzopyrans: Ligands for dopamine D2 and D3 receptors. J. Med. Chem. 1994, 37, 4245–4250. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D.; Keating, G.M. Blonanserin: A review of its use in the management of schizophrenia. CNS Drugs 2010, 24, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Sakamoto, M.; Minamida, A.; Suzuki, K.; Ueda, T.; Une, T.; Toda, H.; Matsumoto, K.; Terauchi, Y. Syntheses and properties of the major hydroxy metabolites in humans of blonanserin AD-5423, a novel antipsychotic agent. Bioorg. Med. Chem. Lett. 2005, 15, 1055–1059. [Google Scholar] [CrossRef]
- Hida, H.; Mouri, A.; Mori, K.; Matsumoto, Y.; Seki, T.; Taniguchi, M.; Yamada, K.; Iwamoto, K.; Ozaki, N.; Nabeshima, T.; et al. Blonanserin ameliorates phencyclidine-induced visual-recognition memory deficits: The complex mechanism of blonanserin action involving D3-5-HT2A and D1-NMDA receptors in the mPFC. Neuropsychopharmacology 2015, 40, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Tenjin, T.; Miyamoto, S.; Ninomiya, Y.; Kitajima, R.; Ogino, S.; Miyake, N.; Yamaguchi, N. Profile of blonanserin for the treatment of schizophrenia. Neuropsychiatr. Dis. Treat. 2013, 9, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L.; Craigo, S.C.; Choudhary, M.S.; Uluer, A.; Monsma, F.J., Jr.; Shen, Y.; Meltzer, H.Y.; Sibley, D.R. Binding of typical and atypical antipsychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J. Pharmacol. Exp. Ther. 1994, 268, 1403–1410. [Google Scholar] [PubMed]
- Arnt, J.; Skarsfeldt, T. Do novel antipsychotics have similar pharmacological characteristics? A review of the evidence. Neuropsychopharmacology 1998, 18, 63–101. [Google Scholar] [CrossRef]
- Egan, C.T.; Herrick-Davis, K.; Teitler, M. Creation of a constitutively activated state of the 5-hydroxytryptamine2A receptor by site-directed mutagenesis: Inverse agonist activity of antipsychotic drugs. J. Pharmacol. Exp. Ther. 1998, 286, 85–90. [Google Scholar]
- Thomas, D.R.; Gittins, S.A.; Collin, L.L.; Middlemiss, D.N.; Riley, G.; Hagan, J.; Gloger, I.; Ellis, C.E.; Forbes, I.T.; Brown, A.M. Functional characterisation of the human cloned 5-HT7 receptor (long form); antagonist profile of SB-258719. Br. J. Pharmacol. 1998, 124, 1300–1306. [Google Scholar] [CrossRef] [Green Version]
- Fernández, J.; Alonso, J.M.; Andrés, J.I.; Cid, J.M.; Díaz, A.; Iturrino, L.; Gil, P.; Megens, A.; Sipido, V.K.; Trabanco, A.A. Discovery of new tetracyclic tetrahydrofuran derivatives as potential broad-spectrum psychotropic agents. J. Med. Chem. 2005, 48, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Schotte, A.; Janssen, P.F.; Gommeren, W.; Luyten, W.H.; Van Gompel, P.; Lesage, A.S.; De Loore, K.; Leysen, J.E. Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology 1996, 124, 57–73. [Google Scholar] [CrossRef]
- Lange, J.H.; Reinders, J.H.; Tolboom, J.T.; Glennon, J.C.; Coolen, H.K.; Kruse, C.G. Principal component analysis differentiates the receptor binding profiles of three antipsychotic drug candidates from current antipsychotic drugs. J. Med. Chem. 2007, 50, 5103–5108. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.; Bristow, L.J.; Hutson, P.H. Current and novel approaches to the drug treatment of schizophrenia. J. Med. Chem. 2001, 44, 477–501. [Google Scholar] [CrossRef]
- Millan, M.J.; Peglion, J.L.; Vian, J.; Rivet, J.M.; Brocco, M.; Gobert, A.; Newman-Tancredi, A.; Dacquet, C.; Bervoets, K.; Girardon, S.; et al. Functional correlates of dopamine D3 receptor activation in the rat in vivo and their modulation by the selective antagonist, (+)-S 14297: 1. Activation of postsynaptic D3 receptors mediates hypothermia, whereas blockade of D2 receptors elicits prolactin secretion and catalepsy. J. Pharmacol. Exp. Ther. 1995, 275, 885–898. [Google Scholar]
- Purohit, A.; Smith, C.; Herrick-Davis, K.; Teitler, M. Stable expression of constitutively activated mutant h5HT6 and h5HT7 serotonin receptors: Inverse agonist activity of antipsychotic drugs. Psychopharmacology 2005, 179, 461–469. [Google Scholar] [CrossRef]
- Seeman, P.; Corbett, R.; Van Tol, H.H. Atypical neuroleptics have low affinity for dopamine D2 receptors or are selective for D4 receptors. Neuropsychopharmacology 1997, 16, 93–110; discussion 111–135. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyaya, A.; Rajagopalan, D.R.; Rath, N.P.; Herrold, A.; Rajagopalan, R.; Napier, T.C.; Tedford, C.E.; Rajagopalan, P. The synthesis and receptor binding affinities of DDD-016, a novel, potential, atypical antipsychotic. MedChemComm 2012, 3, 580–583. [Google Scholar] [CrossRef]
- Ablordeppey, S.Y.; Altundas, R.; Bricker, B.; Zhu, X.Y.; Kumar, E.V.; Jackson, T.; Khan, A.; Roth, B.L. Identification of a butyrophenone analog as a potential atypical antipsychotic agent: 4-[4-(4-chlorophenyl)-1,4-diazepan-1-yl]-1-(4-fluorophenyl)butan-1-one. Bioorg. Med. Chem. 2008, 16, 7291–7301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolós, J.; Anglada, L.; Gubert, S.; Planas, J.M.; Agut, J.; Príncep, M.; De la Fuente, N.; Sacristán, A.; Ortiz, J.A. 7-[3-(1-piperidinyl)propoxy]chromenones as potential atypical antipsychotics. 2. Pharmacological profile of 7-[3-[4-(6-fluoro-1, 2-benzisoxazol-3-yl)-piperidin-1-yl]propoxy]-3-(hydroxymeth yl)chromen -4-one (abaperidone, FI-8602). J. Med. Chem. 1998, 41, 5402–5409. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Tsai, K.C.; Xia, L. Pharmacophore identification of alpha(1A)-adrenoceptor antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 657–664. [Google Scholar] [CrossRef]
- Jørgensen, M.; Jørgensen, P.N.; Christoffersen, C.T.; Jensen, K.G.; Balle, T.; Bang-Andersen, B. Discovery of novel α1-adrenoceptor ligands based on the antipsychotic sertindole suitable for labeling as PET ligands. Bioorg. Med. Chem. 2013, 21, 196–204. [Google Scholar] [CrossRef]
- Kristensen, J.L.; Püschl, A.; Jensen, M.; Risgaard, R.; Christoffersen, C.T.; Bang-Andersen, B.; Balle, T. Exploring the neuroleptic substituent in octoclothepin: Potential ligands for positron emission tomography with subnanomolar affinity for α(1)-adrenoceptors. J. Med. Chem. 2010, 53, 7021–7034. [Google Scholar] [CrossRef]
- Kołaczkowski, M.; Marcinkowska, M.; Bucki, A.; Pawłowski, M.; Mitka, K.; Jaśkowska, J.; Kowalski, P.; Kazek, G.; Siwek, A.; Wasik, A.; et al. Novel arylsulfonamide derivatives with 5-HT6/5-HT7 receptor antagonism targeting behavioral and psychological symptoms of dementia. J. Med. Chem. 2014, 57, 4543–4557. [Google Scholar] [CrossRef] [PubMed]
- Krogsgaard-Larsen, N.; Jensen, A.A.; Kehler, J. Novel 7-phenylsulfanyl-1,2,3,4,10,10a-hexahydro-pyrazino[1,2-a]indoles as dual serotonin 5-HT2C and 5-HT6 receptor ligands. Bioorg. Med. Chem. Lett. 2010, 20, 5431–5433. [Google Scholar] [CrossRef]
- Balle, T.; Perregaard, J.; Ramirez, M.T.; Larsen, A.K.; Søby, K.K.; Liljefors, T.; Andersen, K. Synthesis and structure-affinity relationship investigations of 5-heteroaryl-substituted analogues of the antipsychotic sertindole. A new class of highly selective alpha(1) adrenoceptor antagonists. J. Med. Chem. 2003, 46, 265–283. [Google Scholar] [CrossRef]
- Seeman, P. Antipsychotic drugs, dopamine receptors, and schizophrenia. Clin. Neurosci. Res. 2001, 1, 53–60. [Google Scholar] [CrossRef]
- Burstein, E.S.; Ma, J.; Wong, S.; Gao, Y.; Pham, E.; Knapp, A.E.; Nash, N.R.; Olsson, R.; Davis, R.E.; Hacksell, U.; et al. Intrinsic efficacy of antipsychotics at human D2, D3, and D4 dopamine receptors: Identification of the clozapine metabolite N-desmethylclozapine as a D2/D3 partial agonist. J. Pharmacol. Exp. Ther. 2005, 315, 1278–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasudevan, S.R.; Moore, J.B.; Schymura, Y.; Churchill, G.C. Shape-based reprofiling of FDA-approved drugs for the H1 histamine receptor. J. Med. Chem. 2012, 55, 7054–7060. [Google Scholar] [CrossRef]
- Sun, B.; Feng, D.; Chu, M.L.-H.; Fish, I.; Lovera, S.; Sands, Z.A.; Kelm, S.; Valade, A.; Wood, M.; Ceska, T.; et al. Crystal structure of dopamine D1 receptor in complex with G protein and a non-catechol agonist. Nat. Commun. 2021, 12, 3305. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Taylor, M.; Griffin, S.A.; McInnis, T.; Sumien, N.; Mach, R.H.; Luedtke, R.R. Evaluation of Substituted N-Phenylpiperazine Analogs as D3 vs. D2 Dopamine Receptor Subtype Selective Ligands. Molecules 2021, 26, 3182. [Google Scholar] [CrossRef] [PubMed]
- Kalani, M.Y.S.; Vaidehi, N.; Hall, S.E.; Trabanino, R.J.; Freddolino, P.L.; Kalani, M.A.; Floriano, W.B.; Kam, V.W.T.; Goddard, W.A., 3rd. The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc. Natl. Acad. Sci. USA 2004, 101, 3815–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cover, K.K.; Mathur, B.N. Axo-axonic synapses: Diversity in neural circuit function. J. Comp. Neurol. 2021, 529, 2391–2401. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Ambrogini, P.; Narvaez, M.; Di Liberto, V.; Beggiato, S.; Ferraro, L.; Fores-Pons, R.; Alvarez-Contino, J.E.; Lopez-Salas, A.; Mudò, G.; et al. Serotonin Heteroreceptor Complexes and Their Integration of Signals in Neurons and Astroglia—Relevance for Mental Diseases. Cells 2021, 10, 1902. [Google Scholar] [CrossRef]
- Chagraoui, A.; Boulain, M.; Juvin, L.; Anouar, Y.; Barrière, G.; Deurwaerdère, P.D. L-DOPA in Parkinson’s Disease: Looking at the “False” Neurotransmitters and Their Meaning. Int. J. Mol. Sci. 2020, 21, 294. [Google Scholar] [CrossRef] [Green Version]
- Cachope, R.; Cheer, J.F. Local control of striatal dopamine release. Front. Behav. Neurosci. 2014, 8, 188. [Google Scholar] [CrossRef]
- Fuxe, K.; Borroto-Escuela, D.; Romero-Fernandez, W.; Zhang, W.-B.; Agnati, L. Volume transmission and its different forms in the central nervous system. Chin. J. Integr. Med. 2013, 19, 323–329. [Google Scholar] [CrossRef]
- Zoli, M.; Torri, C.; Ferrari, R.; Jansson, A.; Zini, I.; Fuxe, K.; Agnati, L.F. The emergence of the volume transmission concept. Brain Res. Rev. 1998, 26, 136–147. [Google Scholar] [CrossRef]
- Heinrich, J.N.; Butera, J.A.; Carrick, T.; Kramer, A.; Kowal, D.; Lock, T.; Marquis, K.L.; Pausch, M.H.; Popiolek, M.; Sun, S.-C.; et al. Pharmacological comparison of muscarinic ligands: Historical versus more recent muscarinic M1-preferring receptor agonists. Eur. J. Pharmacol. 2009, 605, 53–56. [Google Scholar] [CrossRef]
- Fujio, M.; Togo, Y.; Tomozane, H.; Kuroita, T.; Morio, Y.; Katayama, J.; Matsumoto, Y. N-[[1-(2-phenylethyl)pyrrolidin-2-yl]methyl]cyclohexanecarboxamides as selective 5-HT1A receptor agonists. Bioorg. Med. Chem. Lett. 2000, 10, 509–512. [Google Scholar] [CrossRef]
- Perez, M.; Jorand-Lebrun, C.; Pauwels, P.J.; Pallard, I.; Halazy, S. Dimers of 5HT1 ligands preferentially bind to 5HT1B/1D receptor subtypes. Bioorg. Med. Chem. Lett. 1998, 8, 1407–1412. [Google Scholar] [CrossRef]
- Haadsma-Svensson, S.R.; Svensson, K.; Duncan, N.; Smith, M.W.; Lin, C.H. C-9 and N-substituted analogs of cis-(3aR)-(−)-2,3,3a,4,5,9b-hexahydro-3-propyl-1H-benz[e]indole-9-carboxamide: 5-HT1A receptor agonists with various degrees of metabolic stability. J. Med. Chem. 1995, 38, 725–734. [Google Scholar] [CrossRef]
- Kalkman, H.O.; Subramanian, N.; Hoyer, D. Extended radioligand binding profile of iloperidone: A broad spectrum dopamine/serotonin/norepinephrine receptor antagonist for the management of psychotic disorders. Neuropsychopharmacology 2001, 25, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Kongsamut, S.; Roehr, J.E.; Cai, J.; Hartman, H.B.; Weissensee, P.; Kerman, L.L.; Tang, L.; Sandrasagra, A. Iloperidone binding to human and rat dopamine and 5-HT receptors. Eur. J. Pharmacol. 1996, 317, 417–423. [Google Scholar] [CrossRef]
- Strupczewski, J.T.; Bordeau, K.J.; Chiang, Y.; Glamkowski, E.J.; Conway, P.G.; Corbett, R.; Hartman, H.B.; Szewczak, M.R.; Wilmot, C.A.; Helsley, G.C. 3-[[(Aryloxy)alkyl]piperidinyl]-1,2-benzisoxazoles as D2/5-HT2 antagonists with potential atypical antipsychotic activity: Antipsychotic profile of iloperidone (HP 873). J. Med. Chem. 1995, 38, 1119–1131. [Google Scholar] [CrossRef]
Figure 1.
Schematic structure of DR and above, potential ligands. Transmembrane zones important to dopamine binding are shown in orange (see text for details).
Figure 1.
Schematic structure of DR and above, potential ligands. Transmembrane zones important to dopamine binding are shown in orange (see text for details).


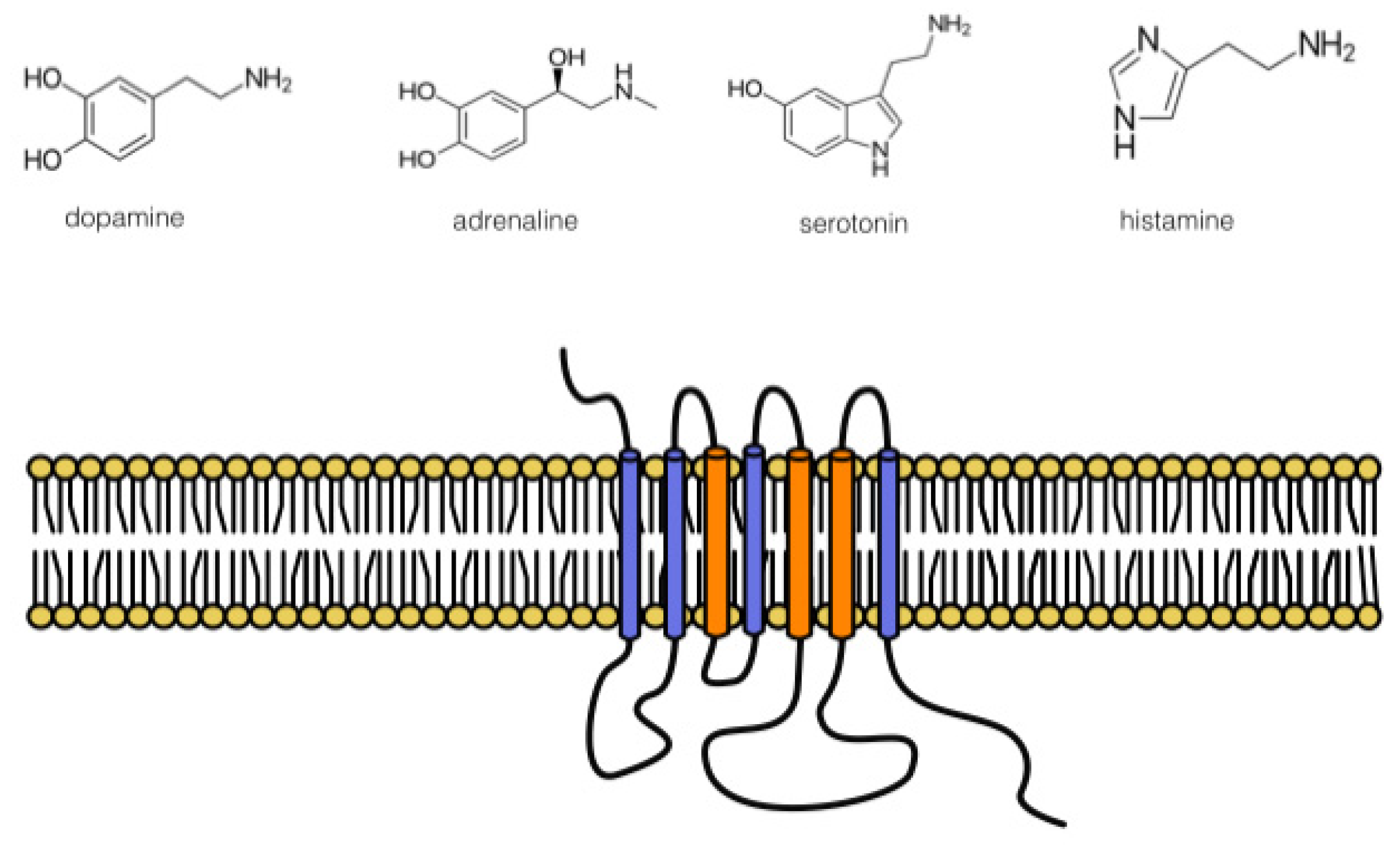{kind=link}
Table 1.
Selective ligands to dopamine receptor subtypes. Listed are both subtype(s) and family-specific compounds.
Table 1.
Selective ligands to dopamine receptor subtypes. Listed are both subtype(s) and family-specific compounds.
| D1 | D2 | D3 | D4 | D5 | |
|---|---|---|---|---|---|
| agonist | A77636 SKF-81297 SKF-83959 | MLS1547 2 Rotigotine 3 Ropinirole 4 Pramipexole 4 PD 128907 4 PD168,077 7 A412997 8 | Rotigotine 3 Ropinirole 4 Pramipexole 4 PD 128907 4 A412997 8 [3H]PD128907 9 | Rotigotine 3 PD168,077 7 A412997 8 | |
| antagonist | SKF-83566 1 SCH-23390 1 Ecopipam 1 [125I]SCH23982 1,9 | pipotiazine perospirone 5 raclopride 3 ML321 Prochlorperazine 4 Sulpiride 5 NGB 2904 6 | Perospirone 5 Raclopride 3 Prochlorperazine 4 Sulpiride 5 S33084 NGB 2904 6 SB 277011-A (+)-S-14297 | Perospirone 5 Sulpiride 5 sonepiprazole L745870 A-381393 L741742 ML398 [125I]L750667 9 [3H]NGD941 9 | SKF-83566 1 SCH-23390 1 Ecopipam 1 [125I]SCH23982 1,9 |
1 The selectivity is expressed to D1-like DRs. 2 Biased D2 DR agonist [38]: it antagonizes arrestin recruitment to D2 DR but behaves as an agonist in its capacity to induce D2 DR signaling. 3 D2 DR and D3 DR selective over D4 DR. 4 D2 DR and D3 DR selective. 5 The selectivity is expressed to D2-like DR. 6 D3 DR selective over D2 DR. 7 Slightly more selective to D4 DR than to D2 DR. 8 Selectivity D4 DR > D3 DR > D2 DR. 9 Please note that this is radioligand.
Table 2.
The co-presence of receptor types in specific brain areas.
| CNS Area | DR Presence | α2-AR Presence | 5-HT Presence | |
|---|---|---|---|---|
| Cerebral cortex | D1-like D2-like | α2C-AR | 5-HT2 5-HT4 5-HT6 5-HT1A 5-HT1B 5 5-HT1E 5-HT1F 5-HT5A | |
| Amygdala | D1-like | α2C-AR α2A-AR2 | 5-HT2C 5-HT6 5-HT1B 5 | |
| Substantia nigra | pars compacta | D2 DR | α2C-AR | 5-HT4 5-HT1B 6 5-HT1D 6 5-HT1F 6 |
| pars reticularis | 5-HT4 5-HT1B 6 5-HT1D 6 5-HT1F 6 | |||
| Striatum (Caudate-putamen) | D1 DR D2 DR D3 DR | α2C-AR 1 | 5-HT2A/2C 5-HT4 1 5-HT6 5-HT1B 2 5-HT1D 1 5-HT1F 7 | |
| Globus pallidus | D2-like | α2C-AR 1 | 5-HT4 1 5-HT1B 5-HT1D 1 5-HT1F | |
| Ncl. accumbens | D1 DR | 5-HT2A/2C 5-HT6 5-HT1B 2 | ||
| Hippocampus (without further specification) | D5 DR D4 DR | α2C-AR α2A-AR 2 α2B-AR 2 | 5-HT4 5-HT6 5-HT7 5-HT1A 5-HT1F 5-HT5A 2 | |
| CA1 | D1-like D2-like | 5-HT4 5-HT1A 5-HT1B 2 5-HT1E 5-HT5A 2 | ||
| CA3 | D1-like D2-like | 5-HT1E 5-HT5A 2 | ||
| Thalamus | D1 DR | α2B-AR α2C-AR2 | 5-HT2A 5-HT6 5-HT7 | |
| Ncl. subthalamicus | D1 DR | α2C-AR 1 | 5-HT1B 2 | |
| Hypothalamus | D5 DR D3 DR | α2A-AR 2 | 5-HT2C 3 5-HT6 5-HT7 4 5-HT1A 5-HT1B 5 5-HT5 4 | |
| Olfactory tubercle | D3 DR | α2C-AR | 5-HT2A/2C 5-HT6 | |
| Midbrain | D4 DR | α2A-AR 2 α2C-AR 2 | ||
| Ventral tegmental area | D2 DR | |||
| Cerebellum | D3 DR D4 DR | α2A-AR 2 α2B-AR 2 | 5-HT6 5-HT1B 2 5-HT5A 2 | |
The presence of specific receptor types was referred to by [1,40,41,42,43,44]. D1-like means the presence of D1 DRs and D5 DRs,.D2-like means the presence of D2 DRs, D3 DRs, and D4 DRs. The presence of receptors in the cerebral cortex can be more specific to layers, part of the cortex, etc. Please see [1,40,41,42,43,44] for detail. 1 Referenced as a presence of subtype in basal ganglia (no further specification). 2 mRNA expression only does not necessarily mean the presence of receptors binding sites. 3 Specifically in the dorsomedial hypothalamus and the paraventricular nucleus. 4 Generally in the hypothalamus, specifically in the suprachiasmatic nucleus. 5 Low autoradiography detected levels. 6 Referenced as a presence of subtype in substantia nigra (no further specification). 7 Specifically in the putamen.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Myslivecek, J. Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors. Life 2022, 12, 606. https://doi.org/10.3390/life12050606
AMA Style
Myslivecek J. Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors. Life. 2022; 12(5):606. https://doi.org/10.3390/life12050606
Chicago/Turabian StyleMyslivecek, Jaromir. 2022. "Dopamine and Dopamine-Related Ligands Can Bind Not Only to Dopamine Receptors" Life 12, no. 5: 606. https://doi.org/10.3390/life12050606
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.