
Epigenetic Regulation of Chondrocytes and Subchondral Bone in Osteoarthritis

 , , , and
, , , and
Abstract
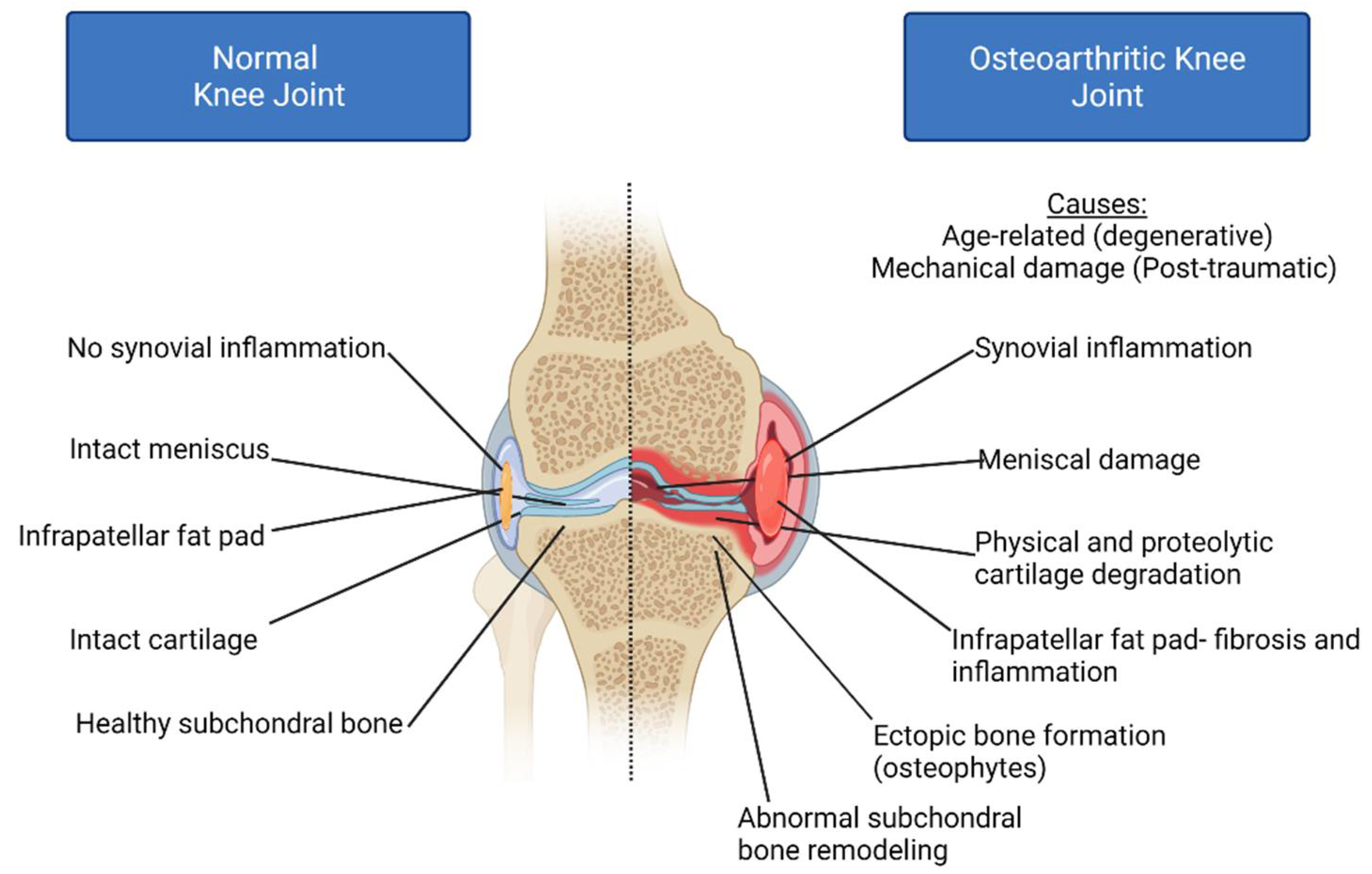
:1. Introduction
2. Methods
3. DNA Methylation
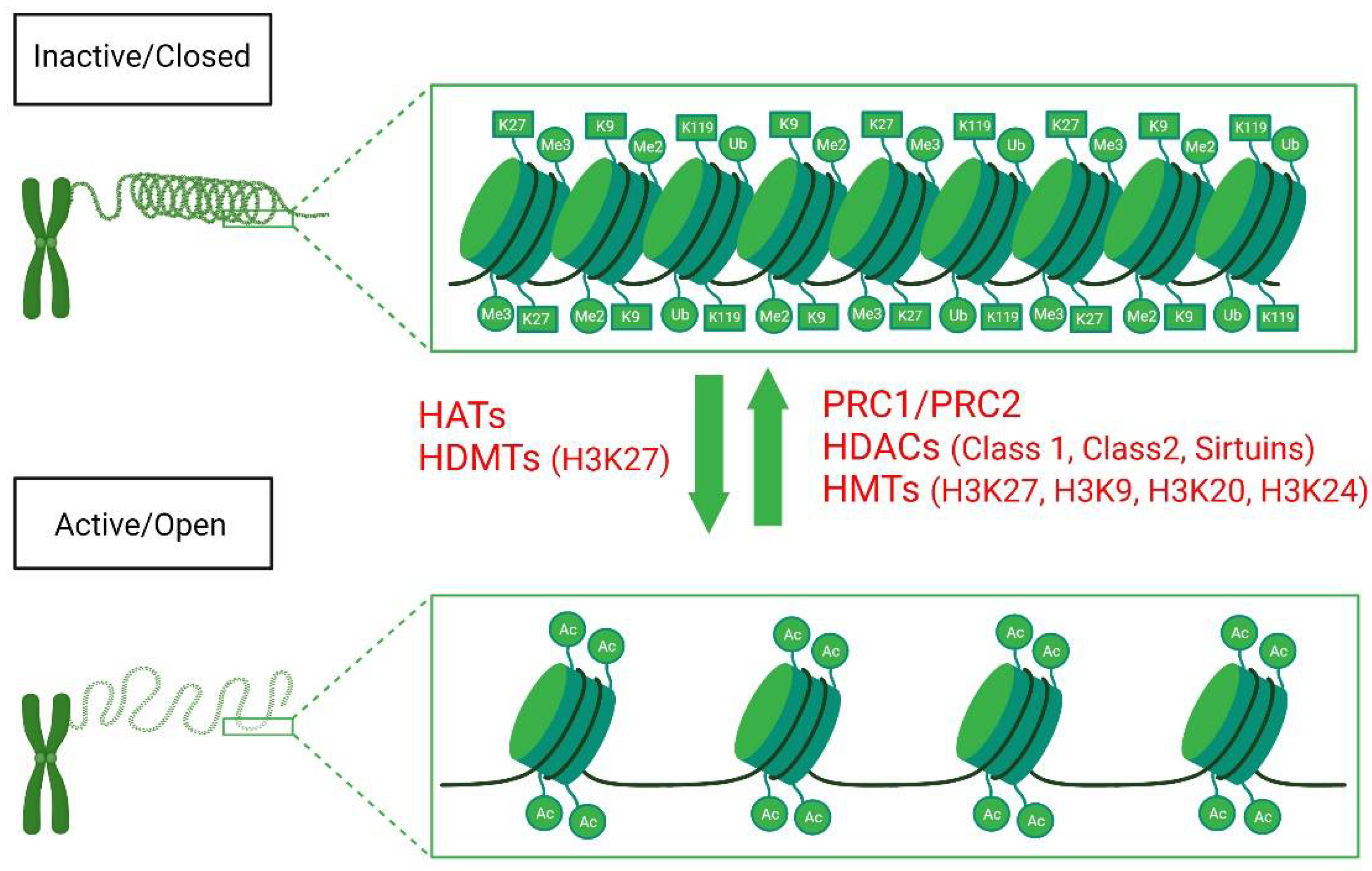
4. Histone Methylation
5. Histone Acetylation
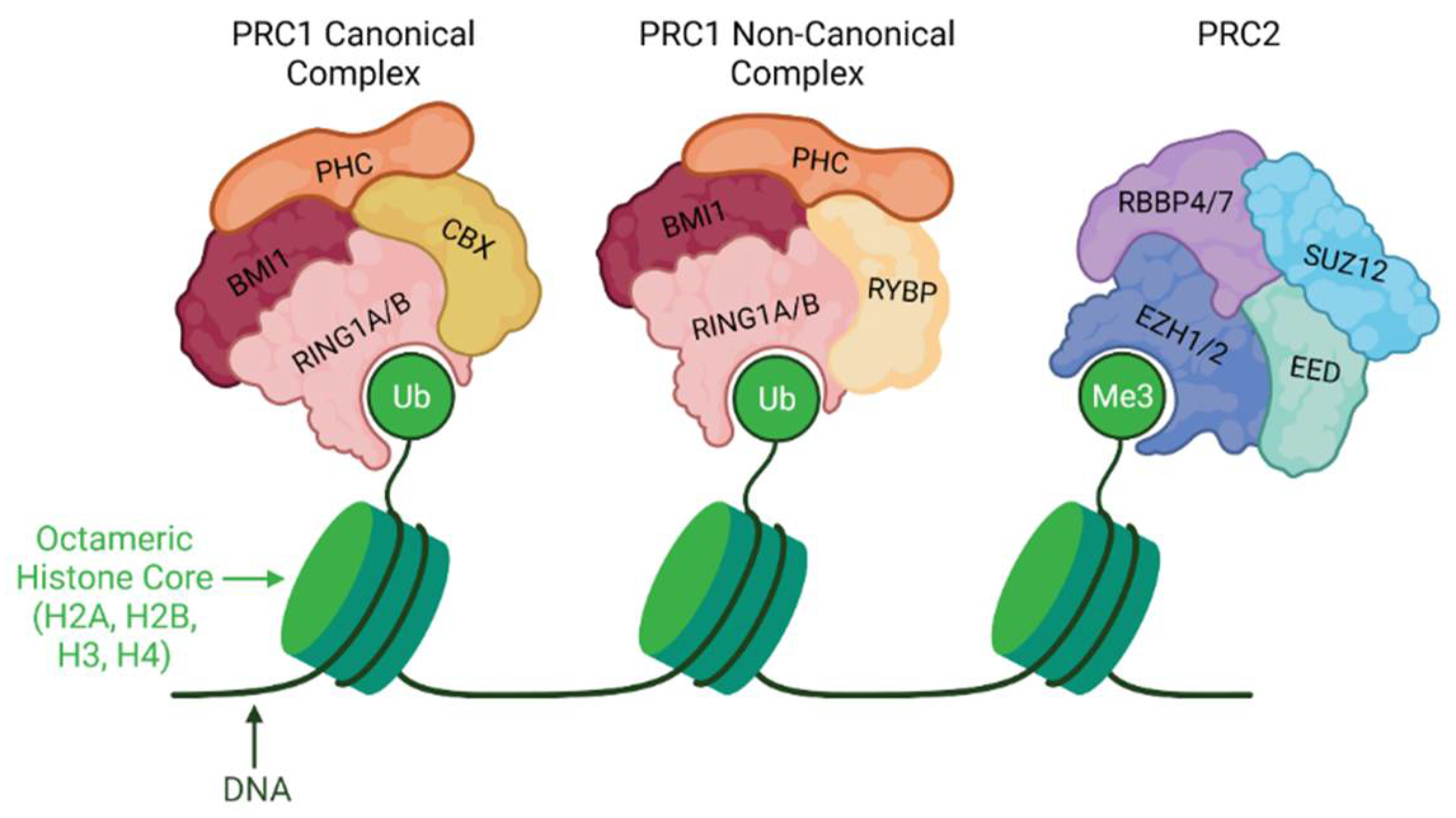
6. Polycomb Repressive Complexes (PRCs)
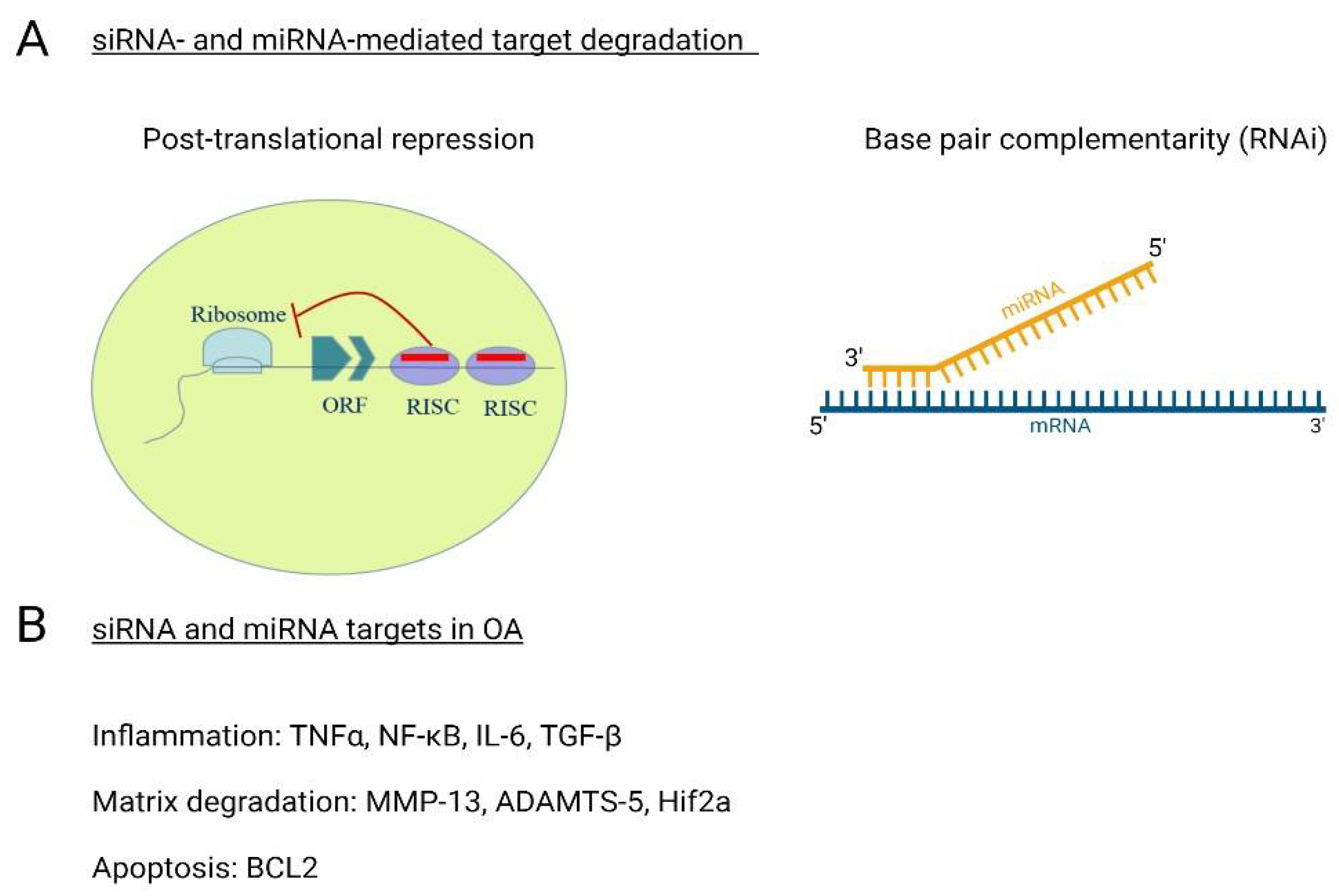
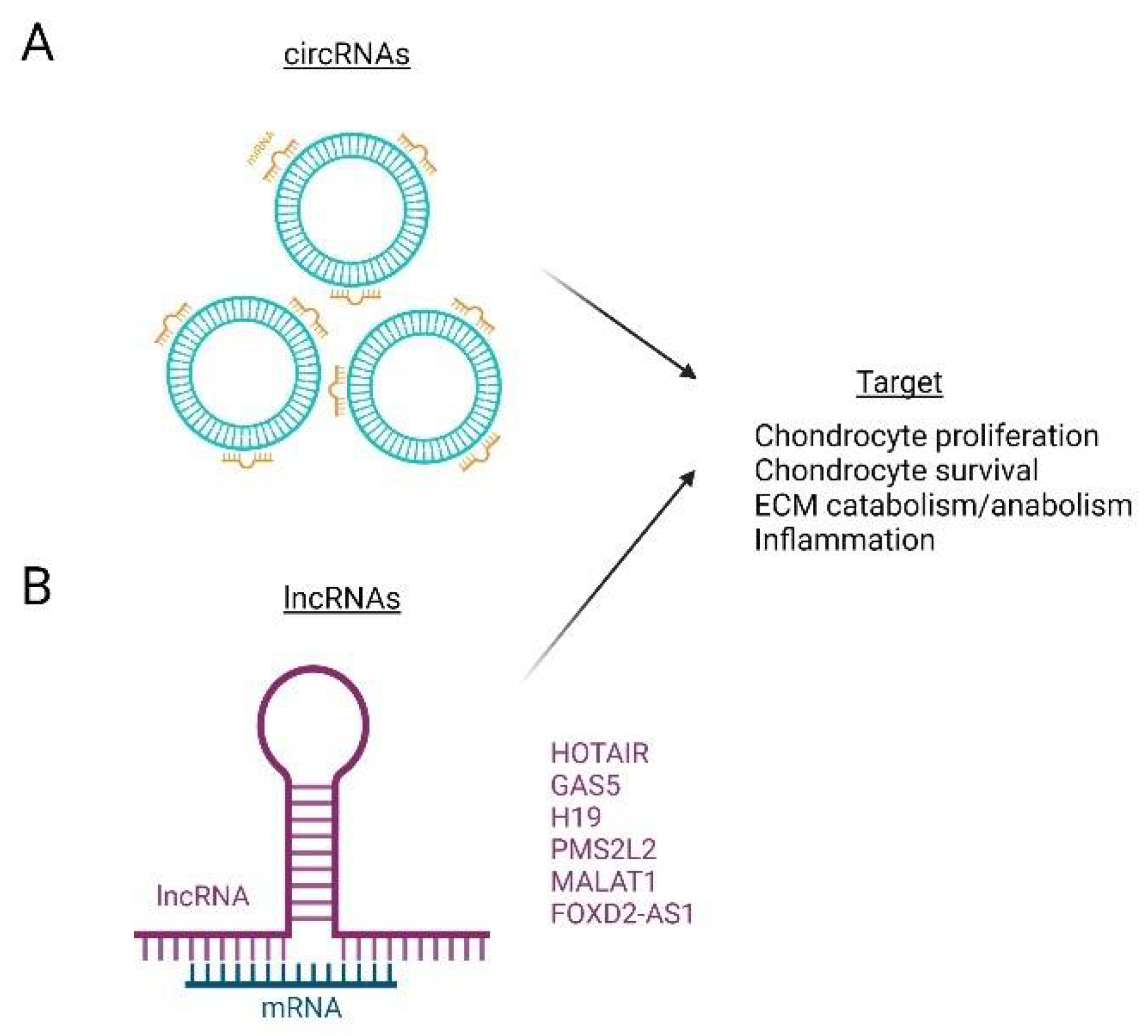
7. Non-Coding RNAs (ncRNAs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ncRNA | OA Function | References |
|---|---|---|
| miR-9 | Enhance ECM degradation | [171,172] |
| miR-98 | Enhance ECM degradation | [171,172] |
| miR-140 | Inhibit ECM degradation | [173,174,175,176,177] |
| miR-148a | Enhanced ECM anabolism Inhibit hypertrophy | [178] |
| miR-4784 | Enhances ECM anabolism Inhibit ECM degradation | [179,180] |
| miR-98 | Inhibit ECM degradation Inhibit inflammation | [171] |
| miR-181a | Inhibit ECM degradation Inhibit inflammation | [171] |
| miR-101 | Enhance ECM degradation | [183] |
| miR-145 | Enhance ECM degradation | [183] |
| miR-381a-3p | Proinflammatory | [185,186,187,188,189] |
| miR-34a | Proinflammatory | [185,186,187,188,189] |
| miR-146a | Proinflammatory | [185,186,187,188,189] |
| miR-181a | Proinflammatory | [185,186,187,188,189] |
| circCDK14 | Inhibit ECM degradation | [205] |
| miR-125 | Increased angiogenesis | [185,186,187,188,189] |
| HOTAIR | Enhance ECM degradation | [203] |
| GAS5 | Proinflammatory | [202] |
| H19 | Proinflammatory | [201] |
| MALAT1 | Inhibit apoptosis Increase proliferation | [204,205] |
| FOXD2-AS1 | Enhance ECM degradation | [206,207] |
8. Epigenetics of Obesity
9. Epigenetic Regulators as Therapeutic Options
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Waddington, C.H. An Introduction to Modern Genetics; The Macmillan Company: New York, NY, USA, 1939. [Google Scholar]
- Greally, J.M. A user’s guide to the ambiguous word ‘epigenetics’. Nat. Rev. Mol. Cell. Biol. 2018, 19, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, T.; Faivre, L.; Baurle, I.; Schubert, D. Chromatin-based mechanisms of temperature memory in plants. Plant Cell Environ. 2019, 42, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Pagiatakis, C.; Musolino, E.; Gornati, R.; Bernardini, G.; Papait, R. Epigenetics of aging and disease: A brief overview. Aging Clin. Exp. Res. 2021, 33, 737–745. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Naylor, R.M.; Baker, D.J.; van Deursen, J.M. Senescent cells: A novel therapeutic target for aging and age-related diseases. Clin. Pharmacol. Ther. 2013, 93, 105–116. [Google Scholar] [CrossRef]
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef] [Green Version]
- Lind, L.; Ingelsson, E.; Sundstrom, J.; Siegbahn, A.; Lampa, E. Methylation-based estimated biological age and cardiovascular disease. Eur. J. Clin. Investig. 2018, 48, e12872. [Google Scholar] [CrossRef]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef]
- Qiu, C.; Huang, S.; Park, J.; Park, Y.; Ko, Y.A.; Seasock, M.J.; Bryer, J.S.; Xu, X.X.; Song, W.C.; Palmer, M.; et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med. 2018, 24, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrell, C.R.; Markovic, B.S.; Fellabaum, C.; Arsenijevic, A.; Volarevic, V. Mesenchymal stem cell-based therapy of osteoarthritis: Current knowledge and future perspectives. Biomed. Pharmacother. 2019, 109, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Aiello, F.C.; Szychlinska, M.A.; Di Rosa, M.; Castrogiovanni, P.; Mobasheri, A. Osteoarthritis in the XXIst century: Risk factors and behaviours that influence disease onset and progression. Int. J. Mol. Sci. 2015, 16, 6093–6112. [Google Scholar] [CrossRef]
- Hawker, G.A. Osteoarthritis is a serious disease. Clin. Exp. Rheumatol. 2019, 37, S3–S6. [Google Scholar]
- Aburto, J.M.; Villavicencio, F.; Basellini, U.; Kjaergaard, S.; Vaupel, J.W. Dynamics of life expectancy and life span equality. Proc. Natl. Acad. Sci. USA 2020, 117, 5250–5259. [Google Scholar] [CrossRef] [Green Version]
- Rice, S.J.; Beier, F.; Young, D.A.; Loughlin, J. Interplay between genetics and epigenetics in osteoarthritis. Nat. Rev. Rheumatol. 2020, 16, 268–281. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, W.; Yong, H.; He, M.; Yang, Y.; Deng, Z.; Li, Y. Macrophages in osteoarthritis: Pathophysiology and therapeutics. Am. J. Transl. Res. 2020, 12, 261–268. [Google Scholar]
- Jang, S.; Lee, K.; Ju, J.H. Recent Updates of Diagnosis, Pathophysiology, and Treatment on Osteoarthritis of the Knee. Int. J. Mol. Sci. 2021, 22, 2619. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [Green Version]
- Man, G.; Mologhianu, G. Osteoarthritis pathogenesis–a complex process that involves the entire joint. J. Med. Life 2014, 7, 37. [Google Scholar] [PubMed]
- Di Laura Frattura, G.; Filardo, G.; Giunchi, D.; Fusco, A.; Zaffagnini, S.; Candrian, C. Risk of falls in patients with knee osteoarthritis undergoing total knee arthroplasty: A systematic review and best evidence synthesis. J. Orthop. 2018, 15, 903–908. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, Z.; Alexander, P.G.; Ocasio-Nieves, B.D.; Yocum, L.; Lin, H.; Tuan, R.S. Pathogenesis of Osteoarthritis: Risk Factors, Regulatory Pathways in Chondrocytes, and Experimental Models. Biology 2020, 9, 194. [Google Scholar] [CrossRef]
- Szilagyi, I.A.; Waarsing, J.H.; Schiphof, D.; van Meurs, J.B.J.; Bierma-Zeinstra, S.M.A. Towards sex-specific osteoarthritis risk models: Evaluation of risk factors for knee osteoarthritis in males and females. Rheumatology 2021, 61, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Ashinsky, B.G.; Bouhrara, M.; Dam, E.B.; Demehri, S.; Shifat, E.R.M.; Spencer, R.G.; Urish, K.L.; Rohde, G.K. Enabling early detection of osteoarthritis from presymptomatic cartilage texture maps via transport-based learning. Proc. Natl. Acad. Sci. USA 2020, 117, 24709–24719. [Google Scholar] [CrossRef] [PubMed]
- Bedson, J.; Croft, P.R. The discordance between clinical and radiographic knee osteoarthritis: A systematic search and summary of the literature. BMC Musculoskelet. Disord. 2008, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Katz, J.N.; Earp, B.E.; Gomoll, A.H. Surgical management of osteoarthritis. Arthritis Care Res. 2010, 62, 1220–1228. [Google Scholar] [CrossRef]
- Ronn, K.; Reischl, N.; Gautier, E.; Jacobi, M. Current surgical treatment of knee osteoarthritis. Arthritis 2011, 2011, 454873. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Marcu, K.B.; Goldring, M.B.; Otero, M. Phenotypic instability of chondrocytes in osteoarthritis: On a path to hypertrophy. Ann. N. Y. Acad. Sci. 2019, 1442, 17–34. [Google Scholar] [CrossRef]
- Van der Kraan, P.; Matta, C.; Mobasheri, A. Age-Related Alterations in Signaling Pathways in Articular Chondrocytes: Implications for the Pathogenesis and Progression of Osteoarthritis—A Mini-Review. Gerontology 2017, 63, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Zengini, E.; Hatzikotoulas, K.; Tachmazidou, I.; Steinberg, J.; Hartwig, F.P.; Southam, L.; Hackinger, S.; Boer, C.G.; Styrkarsdottir, U.; Gilly, A.; et al. Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat. Genet. 2018, 50, 549–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Kang, D.; Cho, Y.; Kim, J.-H. Epigenetic regulation of chondrocyte catabolism and anabolism in osteoarthritis. Mol. Cells 2015, 38, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The Roles of Human DNA Methyltransferases and Their Isoforms in Shaping the Epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebayashi, S.I.; Ryba, T.; Wimbish, K.; Hayakawa, T.; Sakaue, M.; Kuriya, K.; Takahashi, S.; Ogata, S.; Hiratani, I.; Okumura, K.; et al. The Temporal Order of DNA Replication Shaped by Mammalian DNA Methyltransferases. Cells 2021, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Fathollahi, A.; Aslani, S.; Jamshidi, A.; Mahmoudi, M. Epigenetics in osteoarthritis: Novel spotlight. J. Cell Physiol. 2019, 234, 12309–12324. [Google Scholar] [CrossRef]
- Izda, V.; Martin, J.; Sturdy, C.; Jeffries, M.A. DNA methylation and noncoding RNA in OA: Recent findings and methodological advances. Osteoarthr. Cartil. Open 2021, 3, 100208. [Google Scholar] [CrossRef]
- Reynard, L.N. Analysis of genetics and DNA methylation in osteoarthritis: What have we learnt about the disease? Semin. Cell Dev. Biol. 2017, 62, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.C.; Jeffries, M.A. The Epigenomic Landscape in Osteoarthritis. Curr. Rheumatol. Rep. 2017, 19, 30. [Google Scholar] [CrossRef]
- De Andres, M.C.; Imagawa, K.; Hashimoto, K.; Gonzalez, A.; Roach, H.I.; Goldring, M.B.; Oreffo, R.O. Loss of methylation in CpG sites in the NF-kappaB enhancer elements of inducible nitric oxide synthase is responsible for gene induction in human articular chondrocytes. Arthritis Rheum. 2013, 65, 732–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K.; Otero, M.; Imagawa, K.; De Andrés, M.C.; Coico, J.M.; Roach, H.I.; Oreffo, R.O.; Marcu, K.B.; Goldring, M.B. Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1β (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J. Biol. Chem. 2013, 288, 10061–10072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T. Transcriptional regulation of endochondral ossification by HIF-2α during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Roach, H.I.; Yamada, N.; Cheung, K.S.; Tilley, S.; Clarke, N.M.; Oreffo, R.O.; Kokubun, S.; Bronner, F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005, 52, 3110–3124. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.I.; Park, Y.S.; Im, G.I. Changes in the epigenetic status of the SOX-9 promoter in human osteoarthritic cartilage. J. Bone Miner. Res. 2013, 28, 1050–1060. [Google Scholar] [CrossRef]
- Imagawa, K.; De Andrés, M.C.; Hashimoto, K.; Itoi, E.; Otero, M.; Roach, H.I.; Goldring, M.B.; Oreffo, R.O. Association of reduced type IX collagen gene expression in human osteoarthritic chondrocytes with epigenetic silencing by DNA hypermethylation. Arthritis Rheumatol. 2014, 66, 3040–3051. [Google Scholar] [CrossRef] [Green Version]
- Papathanasiou, I.; Kostopoulou, F.; Malizos, K.N.; Tsezou, A. DNA methylation regulates sclerostin (SOST) expression in osteoarthritic chondrocytes by bone morphogenetic protein 2 (BMP-2) induced changes in Smads binding affinity to the CpG region of SOST promoter. Arthritis Res. Ther. 2015, 17, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Fontenla, C.; Calaza, M.; Evangelou, E.; Valdes, A.M.; Arden, N.; Blanco, F.J.; Carr, A.; Chapman, K.; Deloukas, P.; Doherty, M. Assessment of osteoarthritis candidate genes in a meta-analysis of nine genome-wide association studies. Arthritis Rheumatol. 2014, 66, 940–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdes, A.M.; Evangelou, E.; Kerkhof, H.J.; Tamm, A.; Doherty, S.A.; Kisand, K.; Tamm, A.; Kerna, I.; Uitterlinden, A.; Hofman, A. The GDF5 rs143383 polymorphism is associated with osteoarthritis of the knee with genome-wide statistical significance. Ann. Rheum. Dis. 2011, 70, 873–875. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Mabuchi, A.; Shi, D.; Kubo, T.; Takatori, Y.; Saito, S.; Fujioka, M.; Sudo, A.; Uchida, A.; Yamamoto, S. A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat. Genet. 2007, 39, 529–533. [Google Scholar] [CrossRef]
- Bomer, N.; den Hollander, W.; Ramos, Y.F.; Bos, S.D.; van der Breggen, R.; Lakenberg, N.; Pepers, B.A.; van Eeden, A.E.; Darvishan, A.; Tobi, E.W.; et al. Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Rushton, M.D.; Reynard, L.N.; Young, D.A.; Shepherd, C.; Aubourg, G.; Gee, F.; Darlay, R.; Deehan, D.; Cordell, H.J.; Loughlin, J. Methylation quantitative trait locus analysis of osteoarthritis links epigenetics with genetic risk. Hum. Mol. Genet. 2015, 24, 7432–7444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meulenbelt, I.; Chapman, K.; Dieguez-Gonzalez, R.; Shi, D.; Tsezou, A.; Dai, J.; Malizos, K.N.; Kloppenburg, M.; Carr, A.; Nakajima, M. Large replication study and meta-analyses of DVWA as an osteoarthritis susceptibility locus in European and Asian populations. Hum. Mol. Genet. 2009, 18, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Bos, S.D.; Slagboom, P.E.; Meulenbelt, I. New insights into osteoarthritis: Early developmental features of an ageing-related disease. Curr. Opin. Rheumatol. 2008, 20, 553–559. [Google Scholar] [CrossRef]
- Lindner, C.; Thiagarajah, S.; Wilkinson, J.M.; Panoutsopoulou, K.; Day-Williams, A.G.; Consortium, A.; Cootes, T.F.; Wallis, G.A.; Loughlin, J.; Arden, N. Investigation of association between hip osteoarthritis susceptibility loci and radiographic proximal femur shape. Arthritis Rheumatol. 2015, 67, 2076–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynard, L.N.; Loughlin, J. Insights from human genetic studies into the pathways involved in osteoarthritis. Nat. Rev. Rheumatol. 2013, 9, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Bartova, E.; Krejci, J.; Harnicarova, A.; Galiova, G.; Kozubek, S. Histone modifications and nuclear architecture: A review. J. Histochem. Cytochem. 2008, 56, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Ryu, H.Y.; Hochstrasser, M. Histone sumoylation and chromatin dynamics. Nucleic Acids Res. 2021, 49, 6043–6052. [Google Scholar] [CrossRef]
- Aquila, L.; Atanassov, B.S. Regulation of Histone Ubiquitination in Response to DNA Double Strand Breaks. Cells 2020, 9, 1699. [Google Scholar] [CrossRef] [PubMed]
- Hananya, N.; Daley, S.K.; Bagert, J.D.; Muir, T.W. Synthesis of ADP-Ribosylated Histones Reveals Site-Specific Impacts on Chromatin Structure and Function. J. Am. Chem. Soc. 2021, 143, 10847–10852. [Google Scholar] [CrossRef] [PubMed]
- Dieker, J.; Muller, S. Epigenetic histone code and autoimmunity. Clin. Rev. Allergy Immunol. 2010, 39, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Theleman, J.L.; Lygrisse, K.A.; Wang, J. Epigenetic Mechanisms Underlying the Aging of Articular Cartilage and Osteoarthritis. Gerontology 2019, 65, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; FitzGerald, U.; Murphy, J.M. Interplay of Inflammatory Mediators with Epigenetics and Cartilage Modifications in Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Cao, W.; Azeem, I.; Shao, Z. Epigenetics of osteoarthritis: Histones and TGF-beta1. Clin. Chim. Acta 2020, 510, 593–598. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- El Mansouri, F.E.; Chabane, N.; Zayed, N.; Kapoor, M.; Benderdour, M.; Martel-Pelletier, J.; Pelletier, J.P.; Duval, N.; Fahmi, H. Contribution of H3K4 methylation by SET-1A to interleukin-1-induced cyclooxygenase 2 and inducible nitric oxide synthase expression in human osteoarthritis chondrocytes. Arthritis Rheum. 2011, 63, 168–179. [Google Scholar] [CrossRef]
- Ukita, M.M.; Kenji, M.; Tamura, M.; Yamaguchi, T. Histone H3K9 methylation is involved in temporomandicular joint osteoarthritis. Int. J. Mol. Med. 2020, 45, 607–614. [Google Scholar] [PubMed]
- Lefebvre, V.; Angelozzi, M.; Haseeb, A. SOX9 in cartilage development and disease. Curr. Opin. Cell Biol. 2019, 61, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Dvir-Ginzberg, M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res. 2017, 58, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Im, G.I. SOX trio decrease in the articular cartilage with the advancement of osteoarthritis. Connect. Tissue Res. 2011, 52, 496–502. [Google Scholar] [CrossRef]
- Rodova, M.; Lu, Q.H.; Li, Y.; Woodbury, B.G.; Crist, J.D.; Gardner, B.M.; Yost, J.G.; Zhong, X.B.; Anderson, H.C.; Wang, J.X. Nfat1 Regulates Adult Articular Chondrocyte Function Through Its Age-Dependent Expression Mediated by Epigenetic Histone Methylation. J. Bone Miner. Res. 2011, 26, 1974–1986. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Rodova, M.; Lu, Q.; Woodbury, B.; Zhong, X.B.; Anderson, H. Nfat1 regulates adult articular chondrocyte function through its age-dependent expression mediated by epigenetic histone methylation. Bone 2011, 48, S14–S142. [Google Scholar] [CrossRef]
- Castano Betancourt, M.C.; Cailotto, F.; Kerkhof, H.J.; Cornelis, F.M.; Doherty, S.A.; Hart, D.J.; Hofman, A.; Luyten, F.P.; Maciewicz, R.A.; Mangino, M.; et al. Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc. Natl. Acad. Sci. USA 2012, 109, 8218–8223. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Gene Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Hao, Z.; Liu, C.; Yuan, L.; Li, L.; Yin, M.; Li, Q.; Qi, Z.; Wang, Z. LEF1 mediates osteoarthritis progression through circRNF121/miR-665/MYD88 axis via NF-small ka, CyrillicB signaling pathway. Cell Death Dis. 2020, 11, 598. [Google Scholar] [CrossRef]
- Neefjes, M.; van Caam, A.P.M.; van der Kraan, P.M. Transcription Factors in Cartilage Homeostasis and Osteoarthritis. Biology 2020, 9, 290. [Google Scholar] [CrossRef]
- Lietman, C.; Wu, B.; Lechner, S.; Shinar, A.; Sehgal, M.; Rossomacha, E.; Datta, P.; Sharma, A.; Gandhi, R.; Kapoor, M.; et al. Inhibition of Wnt/beta-catenin signaling ameliorates osteoarthritis in a murine model of experimental osteoarthritis. JCI Insight 2018, 3, e96308. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, F.M.F.; de Roover, A.; Storms, L.; Hens, A.; Lories, R.J.; Monteagudo, S. Increased susceptibility to develop spontaneous and post-traumatic osteoarthritis in Dot1l-deficient mice. Osteoarthr. Cartil. 2019, 27, 513–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Yu, D.; Wang, Y.; Chen, Y.; Sun, H.; Zhang, X.; Zhu, S.; Pan, Z.; Heng, B.C.; Zhang, S. Kdm6b regulates cartilage development and homeostasis through anabolic metabolism. Ann. Rheum. Dis. 2017, 76, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xu, L.; Xu, L.; Xu, Q.; Li, D.; Yang, Y.; Karsenty, G.; Chen, C.D. JMJD3 promotes chondrocyte proliferation and hypertrophy during endochondral bone formation in mice. J. Mol. Cell Biol. 2015, 7, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetsunaga, T.; Nishida, K.; Furumatsu, T.; Naruse, K.; Hirohata, S.; Yoshida, A.; Saito, T.; Ozaki, T. Regulation of mechanical stress-induced MMP-13 and ADAMTS-5 expression by RUNX-2 transcriptional factor in SW1353 chondrocyte-like cells. Osteoarthr. Cartil. 2011, 19, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpio, L.R.; Westendorf, J.J. Histone Deacetylases in Cartilage Homeostasis and Osteoarthritis. Curr. Rheumatol. Rep. 2016, 18, 52. [Google Scholar] [CrossRef]
- Huber, L.C.; Brock, M.; Hemmatazad, H.; Giger, O.T.; Moritz, F.; Trenkmann, M.; Distler, J.H.; Gay, R.E.; Kolling, C.; Moch, H.; et al. Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. 2007, 56, 1087–1093. [Google Scholar] [CrossRef]
- Zhang, H.; Ji, L.; Yang, Y.; Zhang, X.; Gang, Y.; Bai, L. The Role of HDACs and HDACi in Cartilage and Osteoarthritis. Front. Cell Dev. Biol. 2020, 8, 560117. [Google Scholar] [CrossRef]
- Li, Y.; Wen, H.; Xi, Y.; Tanaka, K.; Wang, H.; Peng, D.; Ren, Y.; Jin, Q.; Dent, S.Y.; Li, W.; et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 2014, 159, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Jesko, H.; Strosznajder, R.P. Sirtuins and their interactions with transcription factors and poly(ADP-ribose) polymerases. Folia Neuropathol. 2016, 54, 212–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, M.; Herrera, D.; Calvo-Calle, J.M.; Stern, L.J.; Parra-Lopez, C.A.; Butcher, E.; Franco, M.; Angel, J. Circulating human rotavirus specific CD4 T cells identified with a class II tetramer express the intestinal homing receptors alpha4beta7 and CCR9. Virology 2014, 452–453, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichert, N.; Choukrallah, M.A.; Matthias, P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell Mol. Life Sci. 2012, 69, 2173–2187. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E.; Dequiedt, F.; Kasler, H.G. Class II histone deacetylases: Versatile regulators. Trends Genet. 2003, 19, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, J.C.; Jiang, Q.; Lee, W.Y. Role of sirtuins in bone biology: Potential implications for novel therapeutic strategies for osteoporosis. Aging Cell 2021, 20, e13301. [Google Scholar] [CrossRef]
- Hong, S.; Derfoul, A.; Pereira-Mouries, L.; Hall, D.J. A novel domain in histone deacetylase 1 and 2 mediates repression of cartilage-specific genes in human chondrocytes. FASEB J. 2009, 23, 3539–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.J.; Prazak, L.; Fajardo, M.; Yu, S.; Tyagi, N.; Di Cesare, P.E. Leukemia/lymphoma-related factor, a POZ domain-containing transcriptional repressor, interacts with histone deacetylase-1 and inhibits cartilage oligomeric matrix protein gene expression and chondrogenesis. J. Biol. Chem. 2004, 279, 47081–47091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, K.; Wei, L.; Zhang, Z.; Guo, L.; Zhang, C.; Li, Y.; Sun, C.; Sun, X.; Wang, S.; Li, P.; et al. Decreased histone deacetylase 4 is associated with human osteoarthritis cartilage degeneration by releasing histone deacetylase 4 inhibition of runt-related transcription factor-2 and increasing osteoarthritis-related genes: A novel mechanism of human osteoarthritis cartilage degeneration. Arthritis Res. Ther. 2014, 16, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, L.; Zhang, S.; Gu, J.; Takarada, T.; Yoneda, Y.; Huang, J.; Zhao, L.; Oh, C.D.; Li, J.; Wang, B.; et al. Deletion of Runx2 in Articular Chondrocytes Decelerates the Progression of DMM-Induced Osteoarthritis in Adult Mice. Sci. Rep. 2017, 7, 2371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, S.; Wang, B.; Huang, J.; Lu, W.W.; Chen, D. Runx2 and microRNA regulation in bone and cartilage diseases. Ann. N. Y. Acad. Sci. 2016, 1383, 80–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashiyama, R.; Miyaki, S.; Yamashita, S.; Yoshitaka, T.; Lindman, G.; Ito, Y.; Sasho, T.; Takahashi, K.; Lotz, M.; Asahara, H. Correlation between MMP-13 and HDAC7 expression in human knee osteoarthritis. Mod. Rheumatol. 2010, 20, 11–17. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, L.; Wang, Y.; Li, W.; Lin, Y.; Yu, D.; Zhang, L.; Li, F.; Pan, Z. Overexpression of Sirtuin 6 suppresses cellular senescence and NF-kappaB mediated inflammatory responses in osteoarthritis development. Sci. Rep. 2015, 5, 17602. [Google Scholar] [CrossRef]
- Abed, É.; Couchourel, D.; Delalandre, A.; Duval, N.; Pelletier, J.-P.; Martel-Pelletier, J.; Lajeunesse, D. Low sirtuin 1 levels in human osteoarthritis subchondral osteoblasts lead to abnormal sclerostin expression which decreases Wnt/β-catenin activity. Bone 2014, 59, 28–36. [Google Scholar] [CrossRef]
- Dvir-Ginzberg, M.; Gagarina, V.; Lee, E.-J.; Hall, D.J. Regulation of cartilage-specific gene expression in human chondrocytes by SirT1 and nicotinamide phosphoribosyltransferase. J. Biol. Chem. 2008, 283, 36300–36310. [Google Scholar] [CrossRef] [Green Version]
- Bradley, E.W.; Carpio, L.R.; Olson, E.N.; Westendorf, J.J. Histone deacetylase 7 (Hdac7) suppresses chondrocyte proliferation and beta-catenin activity during endochondral ossification. J. Biol. Chem. 2015, 290, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Carpio, L.R.; Bradley, E.W.; McGee-Lawrence, M.E.; Weivoda, M.M.; Poston, D.D.; Dudakovic, A.; Xu, M.; Tchkonia, T.; Kirkland, J.L.; van Wijnen, A.J.; et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci. Signal. 2016, 9, ra79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldring, S.R.; Goldring, M.B. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin. Orthop. Relat. Res. 2004, 427, S27–S36. [Google Scholar] [CrossRef] [PubMed]
- Sakao, K.; Takahashi, K.A.; Arai, Y.; Saito, M.; Honjo, K.; Hiraoka, N.; Asada, H.; Shin-Ya, M.; Imanishi, J.; Mazda, O. Osteoblasts derived from osteophytes produce interleukin-6, interleukin-8, and matrix metalloproteinase-13 in osteoarthritis. J. Bone Miner. Metab. 2009, 27, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhou, S.; Wang, C.; Huang, Y.; Li, H.; Wang, Y.; Zhu, Z.; Tang, J.; Yan, M. Epigenetic modifications of interleukin-6 in synovial fibroblasts from osteoarthritis patients. Sci. Rep. 2017, 7, 73592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochran, A.G. Chapter 5—Structure and Biochemistry of the Polycomb Repressive Complex 1 Ubiquitin Ligase Module. In Polycomb Group Proteins; Pirrotta, V., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 81–110. [Google Scholar]
- Marchesi, I.; Giordano, A.; Bagella, L. Roles of enhancer of zeste homolog 2: From skeletal muscle differentiation to rhabdomyosarcoma carcinogenesis. Cell Cycle 2014, 13, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Golbabapour, S.; Abdulla, M.A.; Hajrezaei, M. A concise review on epigenetic regulation: Insight into molecular mechanisms. Int. J. Mol. Sci. 2011, 12, 8661–8694. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.J.; Voruganti, S.; Nag, S.; Zhou, J.; Zhang, R. Polycomb Group (PcG) Proteins and Human Cancers: Multifaceted Functions and Therapeutic Implications. Med. Res. Rev. 2015, 35, 1220–1267. [Google Scholar] [CrossRef] [Green Version]
- Yuen, R.K.C.; Neumann, S.M.A.; Fok, A.K.; Peñaherrera, M.S.; McFadden, D.E.; Robinson, W.P.; Kobor, M.S. Extensive epigenetic reprogramming in human somatic tissues between fetus and adult. Epigenet. Chromatin 2011, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Tamburri, S.; Lavarone, E.; Fernández-Pérez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol. Cell 2020, 77, 840–856.e845. [Google Scholar] [CrossRef] [Green Version]
- Surface, L.E.; Thornton, S.R.; Boyer, L.A. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell 2010, 7, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, G. Post-translational modifications of PRC2: Signals directing its activity. Epigenet. Chromatin 2020, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Glancy, E.; Ciferri, C.; Bracken, A.P. Structural basis for PRC2 engagement with chromatin. Curr. Opin. Struct. Biol. 2021, 67, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsova, V.L.; Tchekanov, A.; Li, X.; Yan, X.; Tsigelny, I.F. Polycomb repressive 2 complex-Molecular mechanisms of function. Protein Sci. 2019, 28, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Bieluszewski, T.; Xiao, J.; Yang, Y.; Wagner, D. PRC2 activity, recruitment, and silencing: A comparative perspective. Trends Plant Sci. 2021, 26, 1186–1198. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.J.; Moorehead, R.A. Polycomb repressor complex 2 function in breast cancer (Review). Int. J. Oncol. 2020, 57, 1085–1094. [Google Scholar] [CrossRef]
- Kasinath, V.; Beck, C.; Sauer, P.; Poepsel, S.; Kosmatka, J.; Faini, M.; Toso, D.; Aebersold, R.; Nogales, E. JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science 2021, 371. [Google Scholar] [CrossRef]
- Allas, L.; Brochard, S.; Rochoux, Q.; Ribet, J.; Dujarrier, C.; Veyssiere, A.; Aury-Landas, J.; Grard, O.; Leclercq, S.; Vivien, D.; et al. EZH2 inhibition reduces cartilage loss and functional impairment related to osteoarthritis. Sci. Rep. 2020, 10, 19577. [Google Scholar] [CrossRef]
- Grandi, F.C.; Bhutani, N. Epigenetic Therapies for Osteoarthritis. Trends Pharmacol. Sci. 2020, 41, 557–569. [Google Scholar] [CrossRef]
- Culley, K.L.; Dragomir, C.L.; Chang, J.; Wondimu, E.B.; Coico, J.; Plumb, D.A.; Otero, M.; Goldring, M.B. Mouse models of osteoarthritis: Surgical model of posttraumatic osteoarthritis induced by destabilization of the medial meniscus. Methods Mol. Biol. 2015, 1226, 143–173. [Google Scholar] [CrossRef]
- Glasson, S.S.; Blanchet, T.J.; Morris, E.A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 2007, 15, 1061–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wu, Y.; Wu, Y.; Wang, Y.; Sun, L.; Li, F. The inhibition of EZH2 ameliorates osteoarthritis development through the Wnt/β-catenin pathway. Sci. Rep. 2016, 6, 29176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Chen, Y.; Zhang, Q.; Lin, J.; Yu, Y.; Pan, Z.; Sun, H.; Yuan, C.; Yu, D.; Wu, H. Ezh2 Ameliorates Osteoarthritis by Activating TNFSF13B. J. Bone Miner. Res. 2020, 35, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Dudakovic, A.; Camilleri, E.T.; Xu, F.; Riester, S.M.; McGee-Lawrence, M.E.; Bradley, E.W.; Paradise, C.R.; Lewallen, E.A.; Thaler, R.; Deyle, D.R.; et al. Epigenetic Control of Skeletal Development by the Histone Methyltransferase Ezh2. J. Biol. Chem. 2015, 290, 27604–27617. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Zhu, B. Chapter 10—Regulation of PRC2 Activity. In Polycomb Group Proteins; Pirrotta, V., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 225–258. [Google Scholar]
- Schmitges, F.W.; Prusty, A.B.; Faty, M.; Stützer, A.; Lingaraju, G.M.; Aiwazian, J.; Sack, R.; Hess, D.; Li, L.; Zhou, S.; et al. Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks. Mol. Cell 2011, 42, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Schipani, E.; Ryan, H.E.; Didrickson, S.; Kobayashi, T.; Knight, M.; Johnson, R.S. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001, 15, 2865–2876. [Google Scholar] [CrossRef]
- Mirzamohammadi, F.; Papaioannou, G.; Inloes, J.; Rankin, E.; Xie, H.; Schipani, E.; Orkin, S.; Kobayashi, T. Polycomb repressive complex 2 regulates skeletal growth by suppressing Wnt and TGF-β signalling. Nat. Commun. 2016, 7, 12047. [Google Scholar] [CrossRef]
- Lunter, G.; Ponting, C.P.; Hein, J. Genome-Wide Identification of Human Functional DNA Using a Neutral Indel Model. PLoS Comput. Biol. 2006, 2, e5. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [Green Version]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Chaumeil, J.; Le Baccon, P.; Wutz, A.; Heard, E. A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Gene Dev. 2006, 20, 2223–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.; Guttman, M. RNA and dynamic nuclear organization. Science 2014, 345, 1240–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayne, E.H.; Allshire, R.C. RNA-directed transcriptional gene silencing in mammals. Trends Genet. 2005, 21, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Corey, D.R. Regulating mammalian transcription with RNA. Trends Biochem. Sci. 2005, 30, 655–658. [Google Scholar] [CrossRef]
- Sondag, G.R.; Haqqi, T.M. The Role of MicroRNAs and Their Targets in Osteoarthritis. Curr. Rheumatol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef]
- Jiang, S.-D.; Lu, J.; Deng, Z.-H.; Li, Y.-S.; Lei, G.-H. Long noncoding RNAs in osteoarthritis. Jt. Bone Spine 2017, 84, 553–556. [Google Scholar] [CrossRef]
- Mao, X.; Cao, Y.; Guo, Z.; Wang, L.; Xiang, C. Biological roles and therapeutic potential of circular RNAs in osteoarthritis. Mol. Ther. Nucleic Acids 2021, 24, 856–867. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [Green Version]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Baulcombe, D. RNA silencing. Trends Biochem Sci 2005, 30, 290–293. [Google Scholar] [CrossRef]
- Akagi, R.; Sasho, T.; Saito, M.; Endo, J.; Yamaguchi, S.; Muramatsu, Y.; Mukoyama, S.; Akatsu, Y.; Katsuragi, J.; Fukawa, T. Effective knock down of matrix metalloproteinase-13 by an intra-articular injection of small interfering RNA (siRNA) in a murine surgically-induced osteoarthritis model. J. Orthop. Res. 2014, 32, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, R.; Akagi, R.; Yamaguchi, S.; Enomoto, T.; Sato, Y.; Kimura, S.; Ogawa, Y.; Sadamasu, A.; Ohtori, S.; Sasho, T. Single vs. repeated matrix metalloproteinase-13 knockdown with intra-articular short interfering RNA administration in a murine osteoarthritis model. Connect. Tissue Res. 2019, 60, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Bedingfield, S.K.; Colazo, J.M.; Yu, F.; Liu, D.D.; Jackson, M.A.; Himmel, L.E.; Cho, H.; Crofford, L.J.; Hasty, K.A.; Duvall, C.L. Amelioration of post-traumatic osteoarthritis via nanoparticle depots delivering small interfering RNA to damaged cartilage. Nat. Biomed. Eng. 2021, 5, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Schiffelers, R.M.; Xu, J.; Storm, G.; Woodle, M.C.; Scaria, P.V. Effects of treatment with small interfering RNA on joint inflammation in mice with collagen-induced arthritis. Arthritis Rheum. 2005, 52, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Rai, M.F.; Pan, H.; Yan, H.; Sandell, L.J.; Pham, C.T.N.; Wickline, S.A. Applications of RNA interference in the treatment of arthritis. Transl. Res. 2019, 214, 1–16. [Google Scholar] [CrossRef]
- Tang, X.; Muhammad, H.; McLean, C.; Miotla-Zarebska, J.; Fleming, J.; Didangelos, A.; Onnerfjord, P.; Leask, A.; Saklatvala, J.; Vincent, T.L. Connective tissue growth factor contributes to joint homeostasis and osteoarthritis severity by controlling the matrix sequestration and activation of latent TGFbeta. Ann. Rheum. Dis. 2018, 77, 1372–1380. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Duan, X.; Pan, H.; Holguin, N.; Rai, M.F.; Akk, A.; Springer, L.E.; Wickline, S.A.; Sandell, L.J.; Pham, C.T.N. Suppression of NF-κB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc. Natl. Acad. Sci. USA 2016, 113, E6199–E6208. [Google Scholar] [CrossRef] [Green Version]
- Felekkis, K.; Touvana, E.; Stefanou, C.; Deltas, C. microRNAs: A newly described class of encoded molecules that play a role in health and disease. Hippokratia 2010, 14, 236–240. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Li, L.; Jia, J.; Liu, X.; Yang, S.; Ye, S.; Yang, W.; Zhang, Y. MicroRNA-16-5p controls development of osteoarthritis by targeting SMAD3 in chondrocytes. Curr. Pharm. Des. 2015, 21, 5160–5167. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Kim, D.; Chun, C.-H.; Jin, E.-J. MicroRNA-9 regulates survival of chondroblasts and cartilage integrity by targeting protogenin. Cell Commun. Signal. 2013, 11, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Jones, S.; Watkins, G.; Le Good, N.; Roberts, S.; Murphy, C.; Brockbank, S.; Needham, M.; Read, S.; Newham, P. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-α and MMP13. Osteoarthr. Cartil. 2009, 17, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Makki, M.S.; Haseeb, A.; Haqqi, T.M. MicroRNA-9 promotes IL-6 expression by inhibiting MCPIP1 expression in IL-1β-stimulated human chondrocytes. Arthritis Rheumatol. 2015, 67, 2117. [Google Scholar] [CrossRef]
- Araldi, E.; Schipani, E. MicroRNA-140 and the silencing of osteoarthritis. Genes Dev. 2010, 24, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Min, Z. MicroRNAs associated with osteoarthritis differently expressed in bone matrix gelatin (BMG) rat model. Int. J. Clin. Exp. Med. 2015, 8, 1009. [Google Scholar]
- Miyaki, S.; Sato, T.; Inoue, A.; Otsuki, S.; Ito, Y.; Yokoyama, S.; Kato, Y.; Takemoto, F.; Nakasa, T.; Yamashita, S. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010, 24, 1173–1185. [Google Scholar] [CrossRef] [Green Version]
- Miyaki, S.; Nakasa, T.; Otsuki, S.; Grogan, S.P.; Higashiyama, R.; Inoue, A.; Kato, Y.; Sato, T.; Lotz, M.K.; Asahara, H. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 2009, 60, 2723–2730. [Google Scholar] [CrossRef] [Green Version]
- Nugent, M. MicroRNAs: Exploring new horizons in osteoarthritis. Osteoarthr. Cartil. 2016, 24, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonk, L.A.; Kragten, A.H.; Dhert, W.; Saris, D.B.; Creemers, L.B. Overexpression of hsa-miR-148a promotes cartilage production and inhibits cartilage degradation by osteoarthritic chondrocytes. Osteoarthr. Cartil. 2014, 22, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yu, Q.; Ye, Y.; Yan, Y.; Chen, X. Abnormal expression of miR-4784 in chondrocytes of osteoarthritis and associations with chondrocyte hyperplasia. Exp. Ther. Med. 2018, 16, 4690–4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Liu, Y.L.; Chen, X.Y.; Li, Q.; Zhong, J.; Dai, B.Y.; Shao, X.F.; Wu, G.B. Role of MicroRNA, LncRNA, and exosomes in the progression of osteoarthritis: A review of recent literature. Orthop. Surg. 2020, 12, 708–716. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Jin, S.; Lin, J.; Zheng, H.; Zhang, H.; Fan, H.; He, F.; Ma, S.; Li, Q. Altered expression of microRNA-98 in IL-1β-induced cartilage degradation and its role in chondrocyte apoptosis Corrigendum in/10.3892/mmr. 2018.8794. Mol. Med. Rep. 2017, 16, 3208–3216. [Google Scholar] [CrossRef] [Green Version]
- Zhai, X.; Meng, R.; Li, H.; Li, J.; Jing, L.; Qin, L.; Gao, Y. miR-181a modulates chondrocyte apoptosis by targeting glycerol-3-phosphate dehydrogenase 1-like protein (GPD1L) in osteoarthritis. Med. Sci. Monit. 2017, 23, 1224. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Zhang, X.; Hu, X.; Zhou, C.; Ao, Y. Silencing of microRNA-101 prevents IL-1β-induced extracellular matrix degradation in chondrocytes. Arthritis Res. Ther. 2012, 14, R268. [Google Scholar] [CrossRef] [Green Version]
- Kung, L.H.; Ravi, V.; Rowley, L.; Bell, K.M.; Little, C.B.; Bateman, J.F. Comprehensive expression analysis of microRNAs and mRNAs in synovial tissue from a mouse model of early post-traumatic osteoarthritis. Sci. Rep. 2017, 7, 17701. [Google Scholar]
- Cheleschi, S.; Gallo, I.; Barbarino, M.; Giannotti, S.; Mondanelli, N.; Giordano, A.; Tenti, S.; Fioravanti, A. MicroRNA mediate visfatin and resistin induction of oxidative stress in human osteoarthritic synovial fibroblasts via NF-κB pathway. Int. J. Mol. Sci. 2019, 20, 5200. [Google Scholar] [CrossRef] [Green Version]
- Tavallaee, G.; Rockel, J.S.; Lively, S.; Kapoor, M. MicroRNAs in synovial pathology associated with osteoarthritis. Front. Med. 2020, 7, 376. [Google Scholar] [CrossRef]
- Wade, S.M.; Ohnesorge, N.; McLoughlin, H.; Biniecka, M.; Carter, S.P.; Trenkman, M.; Cunningham, C.C.; McGarry, T.; Canavan, M.; Kennedy, B.N. Dysregulated miR-125a promotes angiogenesis through enhanced glycolysis. EBioMedicine 2019, 47, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Yan, K.; Wang, Y. Increased miR-381a-3p contributes to osteoarthritis by targeting IkBα. Ann. Clin. Lab. Sci. 2016, 46, 247–253. [Google Scholar] [PubMed]
- Yang, C.-R.; Shih, K.-S.; Liou, J.-P.; Wu, Y.-W.; Hsieh, I.-N.; Lee, H.-Y.; Lin, T.-C.; Wang, J.-H. Denbinobin upregulates miR-146a expression and attenuates IL-1β-induced upregulation of ICAM-1 and VCAM-1 expressions in osteoarthritis fibroblast-like synoviocytes. J. Mol. Med. 2014, 92, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Ren, J.; Qi, S. Human bone mesenchymal stem cells-derived exosomes overexpressing microRNA-26a-5p alleviate osteoarthritis via down-regulation of PTGS2. Int. Immunopharmacol. 2020, 78, 105946. [Google Scholar] [CrossRef]
- Wang, J.; Shih, K.; Wu, Y.; Wang, A.; Yang, C. Histone deacetylase inhibitors increase microRNA-146a expression and enhance negative regulation of interleukin-1β signaling in osteoarthritis fibroblast-like synoviocytes. Osteoarthr. Cartil. 2013, 21, 1987–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wang, W.; Zhang, F.; Deng, Y.; Long, Z. NEAT1/miR-181c regulates osteopontin (OPN)-mediated synoviocyte proliferation in osteoarthritis. J. Cell. Biochem. 2017, 118, 3775–3784. [Google Scholar] [CrossRef]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 924–933. [Google Scholar] [CrossRef]
- Li, C.H.; Chen, Y. Targeting long non-coding RNAs in cancers: Progress and prospects. Int. J. Biochem. Cell Biol. 2013, 45, 1895–1910. [Google Scholar] [CrossRef]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Marques-Rocha, J.L.; Samblas, M.; Milagro, F.I.; Bressan, J.; Martínez, J.A.; Marti, A. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 2015, 29, 3595–3611. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long non-coding RNAs in the regulation of gene expression: Physiology and disease. Non-Coding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aigner, T.; Söder, S.; Gebhard, P.M.; McAlinden, A.; Haag, J. Mechanisms of disease: Role of chondrocytes in the pathogenesis of osteoarthritis—structure, chaos and senescence. Nat. Clin. Pract. Rheumatol. 2007, 3, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Y.; Liu, J.; Yang, B.; Wang, T.; Zhang, Z.; Jiang, X.; Guo, Y.; Zhang, Y. Roles of long non-coding RNA in osteoarthritis. Int. J. Mol. Med. 2021, 48, 133. [Google Scholar] [CrossRef]
- Xing, D.; Liang, J.-Q.; Li, Y.; Lu, J.; Jia, H.-B.; Xu, L.-Y.; Ma, X.-L. Identification of Long Noncoding RNA Associated with Osteoarthritis in Humans. Orthop. Surg. 2014, 6, 288–293. [Google Scholar] [CrossRef]
- Song, J.; Ahn, C.; Chun, C.H.; Jin, E.J. A long non-coding RNA, GAS5, plays a critical role in the regulation of miR-21 during osteoarthritis. J. Orthop. Res. 2014, 32, 1628–1635. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, P.; Jiang, P.; Lv, Y.; Dong, C.; Dai, X.; Tan, L.; Wang, Z. Upregulation of lncRNA HOTAIR contributes to IL-1β-induced MMP overexpression and chondrocytes apoptosis in temporomandibular joint osteoarthritis. Gene 2016, 586, 248–253. [Google Scholar] [CrossRef] [Green Version]
- Nanus, D.E.; Wijesinghe, S.N.; Pearson, M.J.; Hadjicharalambous, M.R.; Rosser, A.; Davis, E.T.; Lindsay, M.; Jones, S.W. Obese osteoarthritis patients exhibit an inflammatory synovial fibroblast phenotype, which is regulated by the long non coding RNA MALAT1. Arthritis Rheumatol. 2019, 72, 1–29. [Google Scholar]
- Zhang, Y.; Wang, F.; Chen, G.; He, R.; Yang, L. LncRNA MALAT1 promotes osteoarthritis by modulating miR-150-5p/AKT3 axis. Cell Biosci. 2019, 9, 54. [Google Scholar] [CrossRef]
- Gómez, R.; Villalvilla, A.; Largo, R.; Gualillo, O.; Herrero-Beaumont, G. TLR4 signalling in osteoarthritis—Finding targets for candidate DMOADs. Nat. Rev. Rheumatol. 2015, 11, 159–170. [Google Scholar] [CrossRef]
- Fu, M.; Huang, G.; Zhang, Z.; Liu, J.; Zhang, Z.; Huang, Z.; Yu, B.; Meng, F. Expression profile of long noncoding RNAs in cartilage from knee osteoarthritis patients. Osteoarthr. Cartil. 2015, 23, 423–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, N.; Matsumoto, K.; Nishihara, M.; Nakano, Y.; Shibata, A.; Maruyama, H.; Shuto, S.; Matsuda, A.; Yoshida, M.; Ito, Y.; et al. Rolling Circle Translation of Circular RNA in Living Human Cells. Sci. Rep. 2015, 5, 16435. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liang, Y.-H.; Yang, Y.-T.; He, M.; Cai, Z.-J.; Xiao, W.-F.; Li, Y.-S. Circular RNA in osteoarthritis: An updated insight into the pathophysiology and therapeutics. Am. J. Transl. Res. 2021, 13, 11–23. [Google Scholar]
- Ni, J.; Dang, X.; Shi, Z. CircPSM3 inhibits the proliferation and differentiation of OA chondrocytes by targeting miRNA-296-5p. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3467–3475. [Google Scholar]
- Sui, C.; Liu, D.; Que, Y.; Xu, S.; Hu, Y. Knockdown of hsa_circ_0037658 inhibits the progression of osteoarthritis via inducing autophagy. Hum. Cell 2021, 34, 76–85. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, Y.; Wang, C.; Zhang, X.; He, D. CircGCN1L1 promotes synoviocyte proliferation and chondrocyte apoptosis by targeting miR-330-3p and TNF-α in TMJ osteoarthritis. Cell Death Dis. 2020, 11, 284. [Google Scholar] [CrossRef]
- Shen, S.; Wu, Y.; Chen, J.; Xie, Z.; Huang, K.; Wang, G.; Yang, Y.; Ni, W.; Chen, Z.; Shi, P.; et al. CircSERPINE2 protects against osteoarthritis by targeting miR-1271 and ETS-related gene. Ann. Rheum. Dis. 2019, 78, 826–836. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Yang, Y.; Liu, G.; Chen, W.; Chen, J.; Wang, Q.; Gao, H.; Fan, S.; Shen, S.; Zhao, X. CircCDK14 protects against osteoarthritis by sponging miR-125a-5p and promoting the expression of Smad2. Theranostics 2020, 10, 9113. [Google Scholar] [CrossRef]
- Li, H.-Z.; Lin, Z.; Xu, X.-H.; Lin, N.; Lu, H.-D. The potential roles of circRNAs in osteoarthritis: A coming journey to find a treasure. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Lv, X. Revealing new landscape of cardiovascular disease through circular RNA-miRNA-mRNA axis. Genomics 2020, 112, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.J.; Felson, D.T. Factors associated with osteoarthritis of the knee in the first national Health and Nutrition Examination Survey (HANES I) evidence for an association with overweight, race, and physical demands of work. Am. J. Epidemiol. 1988, 128, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Ayral, X.; Pickering, E.; Woodworth, T.; Mackillop, N.; Dougados, M. Synovitis: A potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis–results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthr. Cartil. 2005, 13, 361–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Gibson, A.A.; Gale, J.; Schneuer, F.; Ding, D.; March, L.; Sainsbury, A.; Nassar, N. Does weight loss reduce the incidence of total knee and hip replacement for osteoarthritis?—A prospective cohort study among middle-aged and older adults with overweight or obesity. Int. J. Obes. 2021, 45, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Panunzi, S.; Maltese, S.; De Gaetano, A.; Capristo, E.; Bornstein, S.R.; Mingrone, G. Comparative efficacy of different weight loss treatments on knee osteoarthritis: A network meta-analysis. Obes. Rev. 2021, 22, e13230. [Google Scholar] [CrossRef]
- Webb, E.J.; Osmotherly, P.G.; Baines, S.K. Physical function after dietary weight loss in overweight and obese adults with osteoarthritis: A systematic review and meta-analysis. Public Health Nutr. 2021, 24, 338–353. [Google Scholar] [CrossRef]
- Conway, R.; McCarthy, G.M. Obesity and osteoarthritis: More than just mechanics. EMJ Rheumatol. 2015, 2, 75–83. [Google Scholar]
- Goldstein, B.J.; Scalia, R. Adiponectin: A novel adipokine linking adipocytes and vascular function. J. Clin. Endocrinol. Metab. 2004, 89, 2563–2568. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.; Pessin, J.E. Adipokines mediate inflammation and insulin resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, J.C.; Ljubicic, S.; Leibiger, B.; Kern, M.; Leibiger, I.B.; Moede, T.; Kelly, M.E.; Bhowmick, D.C.; Murano, I.; Cohen, P. Adipsin is an adipokine that improves β cell function in diabetes. Cell 2014, 158, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, S.; Bachmann, A.; Lossner, U.; Kratzsch, J.R.; BLuher, M.; Stumvoll, M.; Fasshauer, M. Serum levels of the adipokine FGF21 depend on renal function. Diabetes Care 2009, 32, 126–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, A.K.; Abraham, J.; Bonner, D.; Gilleland, D.; Hobbs, G.; Kurian, S.; Yanosik, M.A.; Vona-Davis, L. Exercise and dietary advice intervention for survivors of triple-negative breast cancer: Effects on body fat, physical function, quality of life, and adipokine profile. Support. Care Cancer 2015, 23, 2995–3003. [Google Scholar] [CrossRef] [Green Version]
- Conde, J.; Scotece, M.; López, V.; Abella, V.; Hermida, M.; Pino, J.; Lago, F.; Gómez-Reino, J.J.; Gualillo, O. Differential expression of adipokines in infrapatellar fat pad (IPFP) and synovium of osteoarthritis patients and healthy individuals. Ann. Rheum. Dis. 2014, 73, 631–633. [Google Scholar] [CrossRef]
- Fowler-Brown, A.; Kim, D.H.; Shi, L.; Marcantonio, E.; Wee, C.C.; Shmerling, R.H.; Leveille, S. The mediating effect of leptin on the relationship between body weight and knee osteoarthritis in older adults. Arthritis Rheumatol. 2015, 67, 169–175. [Google Scholar] [CrossRef]
- Honsawek, S.; Chayanupatkul, M. Correlation of plasma and synovial fluid adiponectin with knee osteoarthritis severity. Arch. Med. Res. 2010, 41, 593–598. [Google Scholar] [CrossRef]
- Lübbert, M.; Suciu, S.; Baila, L.; Rüter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R. Low-dose decitabine versus best supportive care in elderly patients with intermediate-or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: Final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J. Clin. Oncol. 2011, 29, 1987–1996. [Google Scholar]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Yang, S.; Ryu, J.-H.; Oh, H.; Jeon, J.; Kwak, J.-S.; Kim, J.-H.; Kim, H.A.; Chun, C.-H.; Chun, J.-S. NAMPT (visfatin), a direct target of hypoxia-inducible factor-2α, is an essential catabolic regulator of osteoarthritis. Ann. Rheum. Dis. 2015, 74, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xing, X.; Hensley, G.; Chang, L.W.; Liao, W.; Abu-Amer, Y.; Sandell, L.J. Resistin induces expression of proinflammatory cytokines and chemokines in human articular chondrocytes via transcription and messenger RNA stabilization. Arthritis Rheum. 2010, 62, 1993–2003. [Google Scholar] [PubMed] [Green Version]
- Milagro, F.I.; Miranda, J.; Portillo, M.P.; Fernandez-Quintela, A.; Campion, J.; Martínez, J.A. High-throughput sequencing of microRNAs in peripheral blood mononuclear cells: Identification of potential weight loss biomarkers. PLoS ONE 2013, 8, e54319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, F.J.; Mercader, J.M.; Catalan, V.; Moreno-Navarrete, J.M.; Pueyo, N.; Sabater, M.; Gomez-Ambrosi, J.; Anglada, R.; Fernández-Formoso, J.A.; Ricart, W. Targeting the circulating microRNA signature of obesity. Clin. Chem. 2013, 59, 781–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maegdefessel, L. The emerging role of micro RNA s in cardiovascular disease. J. Intern. Med. 2014, 276, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, T.; Hattori, T.; Takahashi, K.; Kuboki, T.; Yamashita, A.; Takigawa, M. Cyclic mechanical stress induces extracellular matrix degradation in cultured chondrocytes via gene expression of matrix metalloproteinases and interleukin-1. J. Biochem. 1999, 125, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, R.; Iannelli, D. Role of epigenetics in type 2 diabetes and obesity. Biomedicines 2021, 9, 977. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Liu, J.-L.; Lu, X.; Yang, Q. Epigenetic regulation of energy metabolism in obesity. J. Mol. Cell Biol. 2021, 13, 480–499. [Google Scholar] [CrossRef]
- Masi, S.; Ambrosini, S.; Mohammed, S.A.; Sciarretta, S.; Lüscher, T.F.; Paneni, F.; Costantino, S. Epigenetic remodeling in obesity-related vascular disease. Antioxid. Redox Signal. 2021, 34, 1165–1199. [Google Scholar] [CrossRef]
- Cheong, Y.; Nishitani, S.; Yu, J.; Habata, K.; Kamiya, T.; Shiotsu, D.; Omori, I.M.; Okazawa, H.; Tomoda, A.; Kosaka, H. The effects of epigenetic age and its acceleration on surface area, cortical thickness, and volume in young adults. Cereb. Cortex 2022, bhac043. [Google Scholar] [CrossRef]
- Galow, A.-M.; Peleg, S. How to Slow down the Ticking Clock: Age-Associated Epigenetic Alterations and Related Interventions to Extend Life Span. Cells 2022, 11, 468. [Google Scholar] [CrossRef]
- Zabransky, D.J.; Jaffee, E.M.; Weeraratna, A.T. Shared genetic and epigenetic changes link aging and cancer. Trends Cell Biol. 2022, 32, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Ramos, Y.F.; den Hollander, W.; Bovee, J.V.; Bomer, N.; van der Breggen, R.; Lakenberg, N.; Keurentjes, J.C.; Goeman, J.J.; Slagboom, P.E.; Nelissen, R.G.; et al. Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage; the RAAK study. PLoS ONE 2014, 9, e103056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdes, A.M.; Spector, T.D. Genetic epidemiology of hip and knee osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Ecsedi, S.; Rodríguez-Aguilera, J.R.; Hernandez-Vargas, H. 5-Hydroxymethylcytosine (5hmC), or how to identify your favorite cell. Epigenomes 2018, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.E.; Li, Y.H.; Smeriglio, P.; Rath, M.; Wong, W.H.; Bhutani, N. Stable 5-hydroxymethylcytosine (5hmC) acquisition marks gene activation during chondrogenic differentiation. J. Bone Mineral. Res. 2016, 31, 524–534. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Ratneswaran, A.; Kapoor, M. Osteoarthritis year in review: Genetics, genomics, epigenetics. Osteoarthr. Cartil. 2021, 29, 151–160. [Google Scholar] [CrossRef]
- Santiago, M.; Antunes, C.; Guedes, M.; Sousa, N.; Marques, C.J. TET enzymes and DNA hydroxymethylation in neural development and function—How critical are they? Genomics 2014, 104, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Smeriglio, P.; Grandi, F.C.; Davala, S.; Masarapu, V.; Indelli, P.F.; Goodman, S.B.; Bhutani, N. Inhibition of TET1 prevents the development of osteoarthritis and reveals the 5hmC landscape that orchestrates pathogenesis. Sci. Transl. Med. 2020, 12, eaax2332. [Google Scholar] [CrossRef]
- McHugh, J. TET1: An epigenetic controller of OA. Nat. Rev. Rheumatol. 2020, 16, 351. [Google Scholar] [CrossRef]
- Buocikova, V.; Longhin, E.M.; Pilalis, E.; Mastrokalou, C.; Miklikova, S.; Cihova, M.; Poturnayova, A.; Mackova, K.; Babelova, A.; Trnkova, L. Decitabine potentiates efficacy of doxorubicin in a preclinical trastuzumab-resistant HER2-positive breast cancer models. Biomed. Pharmacother. 2022, 147, 112662. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Zuo, Q.; Pilrose, J.; Wang, Y.; Shen, C.; Li, M.; Wulfridge, P.; Matei, D.; Nephew, K.P. Decitabine reactivated pathways in platinum resistant ovarian cancer. Oncotarget 2014, 5, 3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, S.; Heil, O.; Lyko, F.; Brueckner, B. Azacytidine and decitabine induce gene-specific and non-random DNA demethylation in human cancer cell lines. PLoS ONE 2011, 6, e17388. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wu, Y.; Wang, W.; Xu, J.; Lv, X.; Cao, X.; Wan, T. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell. Mol. Immunol. 2019, 16, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Q.; Zhang, C.; Huang, C. Genome-wide DNA methylation analysis of articular chondrocytes identifies TRAF1, CTGF, and CX3CL1 genes as hypomethylated in osteoarthritis. Clin. Rheumatol. 2017, 36, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 1–12. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Schoffski, P.; Jones, R.; Agulnik, M.; Villalobos, V.M.; Jahan, T.M.; Chen, T.W.-W.; Italiano, A.; Demetri, G.D.; Cote, G.M. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients (pts) with epithelioid sarcoma (ES)(NCT02601950). J. Clin. Oncol. 2019, 37, 11003. [Google Scholar] [CrossRef]
- Tremblay-LeMay, R.; Rastgoo, N.; Pourabdollah, M.; Chang, H. EZH2 as a therapeutic target for multiple myeloma and other haematological malignancies. Biomark. Res. 2018, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-P.; Bao, J.-P.; Hu, P.-F.; Feng, J.; Wu, L.-D. Alleviation of osteoarthritis by Trichostatin A, a histone deacetylase inhibitor, in experimental osteoarthritis. Mol. Biol. Rep. 2010, 37, 3967–3972. [Google Scholar] [CrossRef]
- Qu, H.; Li, J.; Wu, L.D.; Chen, W.P. Trichostatin A increases the TIMP-1/MMP ratio to protect against osteoarthritis in an animal model of the disease. Mol. Med. Rep. 2016, 14, 2423–2430. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Sun, Y.; Ge, Q.; Teng, H.; Jiang, Q. Histone deacetylase 4 alters cartilage homeostasis in human osteoarthritis. BMC Musculoskelet. Disord. 2014, 15, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.M.; Haqqi, T.M. Epigenetics in osteoarthritis: Potential of HDAC inhibitors as therapeutics. Pharmacol. Res. 2018, 128, 73–79. [Google Scholar] [CrossRef]
- Bradley, E.W.; Carpio, L.R.; Van Wijnen, A.J.; McGee-Lawrence, M.E.; Westendorf, J.J. Histone deacetylases in bone development and skeletal disorders. Physiol. Rev. 2015, 95, 1359–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batshon, G.; Elayyan, J.; Qiq, O.; Reich, E.; Ben-Aderet, L.; Kandel, L.; Haze, A.; Steinmeyer, J.; Lefebvre, V.; Zhang, H. Serum NT/CT SIRT1 ratio reflects early osteoarthritis and chondrosenescence. Ann. Rheum. Dis. 2020, 79, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Matsushita, T.; Takayama, K.; Matsumoto, T.; Nishida, K.; Kuroda, R.; Kurosaka, M. Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis. 2014, 73, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Feng, M.; Peng, H.; Qian, Z.; Zhao, L.; Wu, S. Epigenetic regulation of chondrocyte hypertrophy and apoptosis through Sirt1/P53/P21 pathway in surgery-induced osteoarthritis. Biochem. Biophys. Res. Commun. 2020, 528, 179–185. [Google Scholar] [CrossRef]
- Hecht, J.T.; Veerisetty, A.C.; Wu, J.; Coustry, F.; Hossain, M.G.; Chiu, F.; Gannon, F.H.; Posey, K.L. Primary Osteoarthritis Early Joint Degeneration Induced by Endoplasmic Reticulum Stress Is Mitigated by Resveratrol. Am. J. Pathol. 2021, 191, 1624–1637. [Google Scholar] [CrossRef]
- Nishida, K.; Matsushita, T.; Takayama, K.; Tanaka, T.; Miyaji, N.; Ibaraki, K.; Araki, D.; Kanzaki, N.; Matsumoto, T.; Kuroda, R. Intraperitoneal injection of the SIRT1 activator SRT1720 attenuates the progression of experimental osteoarthritis in mice. Bone Jt. Res. 2018, 7, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Jia, J.; Jin, X.; Tong, W.; Tian, H. Resveratrol ameliorates inflammatory damage and protects against osteoarthritis in a rat model of osteoarthritis. Mol. Med. Rep. 2018, 17, 1493–1498. [Google Scholar] [CrossRef]
- Cheng, C.; Shan, W.; Huang, W.; Ding, Z.; Cui, G.; Liu, F.; Lu, W.; Xu, J.; He, W.; Yin, Z. ACY-1215 exhibits anti-inflammatory and chondroprotective effects in human osteoarthritis chondrocytes via inhibition of STAT3 and NF-κB signaling pathways. Biomed. Pharmacother. 2019, 109, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ji, L.; Yang, Y.; Wei, Y.; Zhang, X.; Gang, Y.; Lu, J.; Bai, L. The therapeutic effects of treadmill exercise on osteoarthritis in rats by inhibiting the HDAC3/NF-KappaB pathway in vivo and in vitro. Front. Physiol. 2019, 1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delplace, V.; Boutet, M.A.; Le Visage, C.; Maugars, Y.; Guicheux, J.; Vinatier, C. Osteoarthritis: From upcoming treatments to treatments yet to come. Jt. Bone Spine 2021, 88, 105206. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Kolahi, A.A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; Hoy, D.; Ashrafi-Asgarabad, A.; Sepidarkish, M.; Almasi-Hashiani, A.; et al. Global, regional and national burden of osteoarthritis 1990–2017: A systematic analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, J.; Xu, H.; Lin, Z.; Chang, H.; Liu, W.; Kong, L. Mesenchymal stem cells in knee osteoarthritis treatment: A systematic review and meta-analysis. J. Orthop. Translat. 2020, 24, 121–130. [Google Scholar] [CrossRef]
- Ankrum, J.A.; Ong, J.F.; Karp, J.M. Mesenchymal stem cells: Immune evasive, not immune privileged. Nat. Biotechnol. 2014, 32, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Xing, D.; Liu, W.; Wang, B.; Li, J.J.; Zhao, Y.; Li, H.; Liu, A.; Du, Y.; Lin, J. Intra-articular injection of cell-laden 3D microcryogels empower low-dose cell therapy for osteoarthritis in a rat model. Cell Transplant. 2020, 29, 0963689720932142. [Google Scholar] [CrossRef]
- Gong, Y.; Li, S.J.; Liu, R.; Zhan, J.F.; Tan, C.; Fang, Y.F.; Chen, Y.; Yu, B. Inhibition of YAP with siRNA prevents cartilage degradation and ameliorates osteoarthritis development. J. Mol. Med. 2019, 97, 103–114. [Google Scholar] [CrossRef]
- Hoshi, H.; Akagi, R.; Yamaguchi, S.; Muramatsu, Y.; Akatsu, Y.; Yamamoto, Y.; Sasaki, T.; Takahashi, K.; Sasho, T. Effect of inhibiting MMP13 and ADAMTS5 by intra-articular injection of small interfering RNA in a surgically induced osteoarthritis model of mice. Cell Tissue Res. 2017, 368, 379–387. [Google Scholar] [CrossRef]
- Si, H.B.; Zeng, Y.; Liu, S.Y.; Zhou, Z.K.; Chen, Y.N.; Cheng, J.Q.; Lu, Y.R.; Shen, B. Intra-articular injection of microRNA-140 (miRNA-140) alleviates osteoarthritis (OA) progression by modulating extracellular matrix (ECM) homeostasis in rats. Osteoarthr. Cartil. 2017, 25, 1698–1707. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.-Y.; Lee, M.S.; Lian, W.-S.; Weng, W.-T.; Sun, Y.-C.; Chen, Y.-S.; Wang, F.-S. MicroRNA-29a Counteracts Synovitis in Knee Osteoarthritis Pathogenesis by Targeting VEGF. Sci. Rep. 2017, 7, 3584. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Song, J.; Han, J.; Kim, Y.; Chun, C.-H.; Jin, E.-J. Two non-coding RNAs, MicroRNA-101 and HOTTIP contribute cartilage integrity by epigenetic and homeotic regulation of integrin-α1. Cell. Signal. 2013, 25, 2878–2887. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, X.; Dai, L.; Hu, X.; Zhu, J.; Li, L.; Zhou, C.; Ao, Y. Long Noncoding RNA Related to Cartilage Injury Promotes Chondrocyte Extracellular Matrix Degradation in Osteoarthritis. Arthritis Rheumatol. 2014, 66, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, C.; Zhang, F.; Zhang, Y.; Ren, Z.; Lammi, M.J.; Guo, X. Screening for Differentially Expressed Circular RNAs in the Cartilage of Osteoarthritis Patients for Their Diagnostic Value. Genet. Test Mol. Biomark. 2019, 23, 706–716. [Google Scholar] [CrossRef] [PubMed]
- United Nations; Department of Economic. World Population Ageing, 1950–2050; United Nations Publications: New York, NY, USA, 2002. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ball, H.C.; Alejo, A.L.; Samson, T.K.; Alejo, A.M.; Safadi, F.F. Epigenetic Regulation of Chondrocytes and Subchondral Bone in Osteoarthritis. Life 2022, 12, 582. https://doi.org/10.3390/life12040582
Ball HC, Alejo AL, Samson TK, Alejo AM, Safadi FF. Epigenetic Regulation of Chondrocytes and Subchondral Bone in Osteoarthritis. Life. 2022; 12(4):582. https://doi.org/10.3390/life12040582
Chicago/Turabian StyleBall, Hope C., Andrew L. Alejo, Trinity K. Samson, Amanda M. Alejo, and Fayez F. Safadi. 2022. "Epigenetic Regulation of Chondrocytes and Subchondral Bone in Osteoarthritis" Life 12, no. 4: 582. https://doi.org/10.3390/life12040582