
Why Is the UAG (Amber) Stop Codon Almost Absent in Highly Expressed Bacterial Genes?



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data
2.2. Frequency of Stop Codons
2.3. Statistics
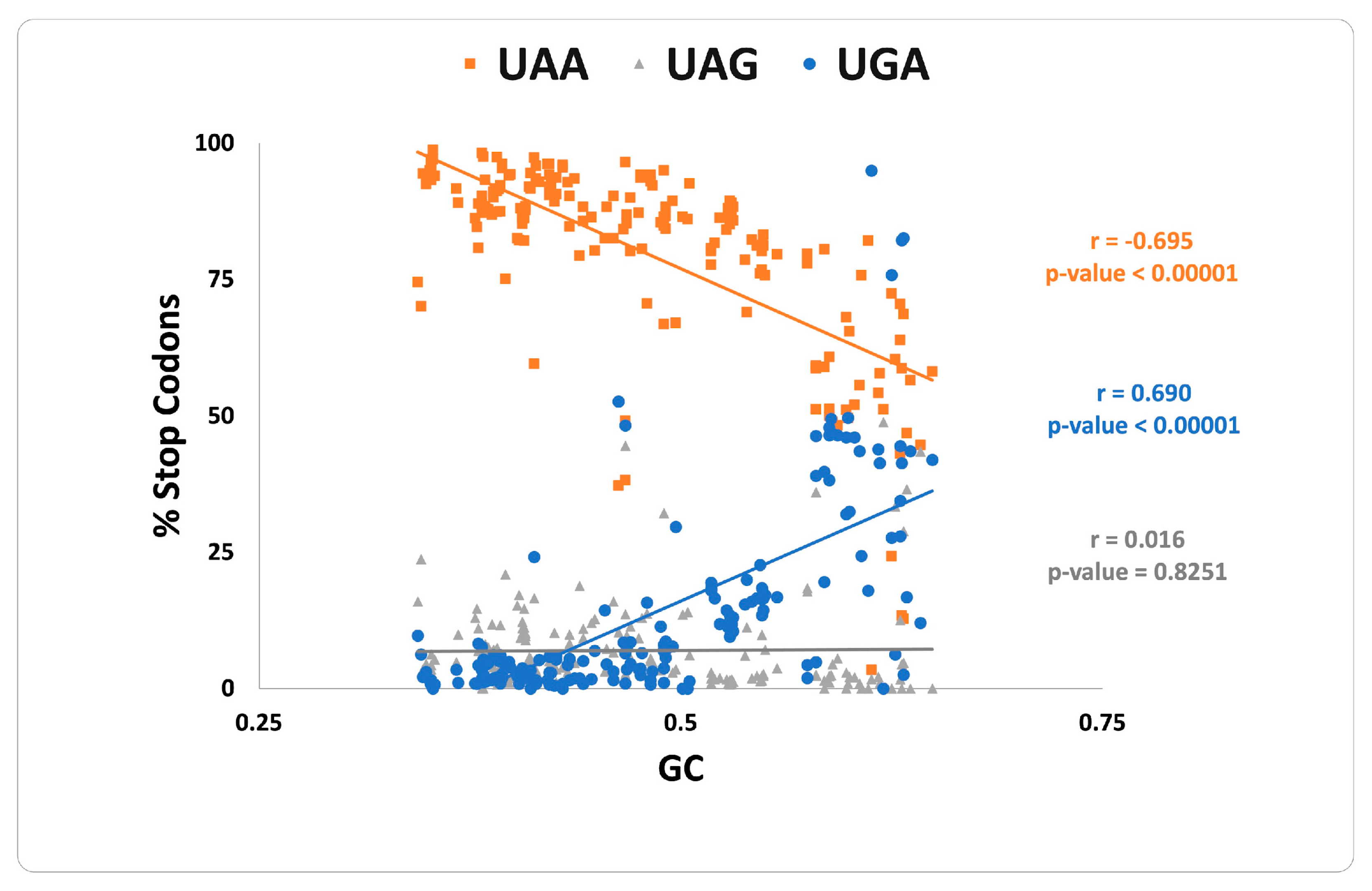
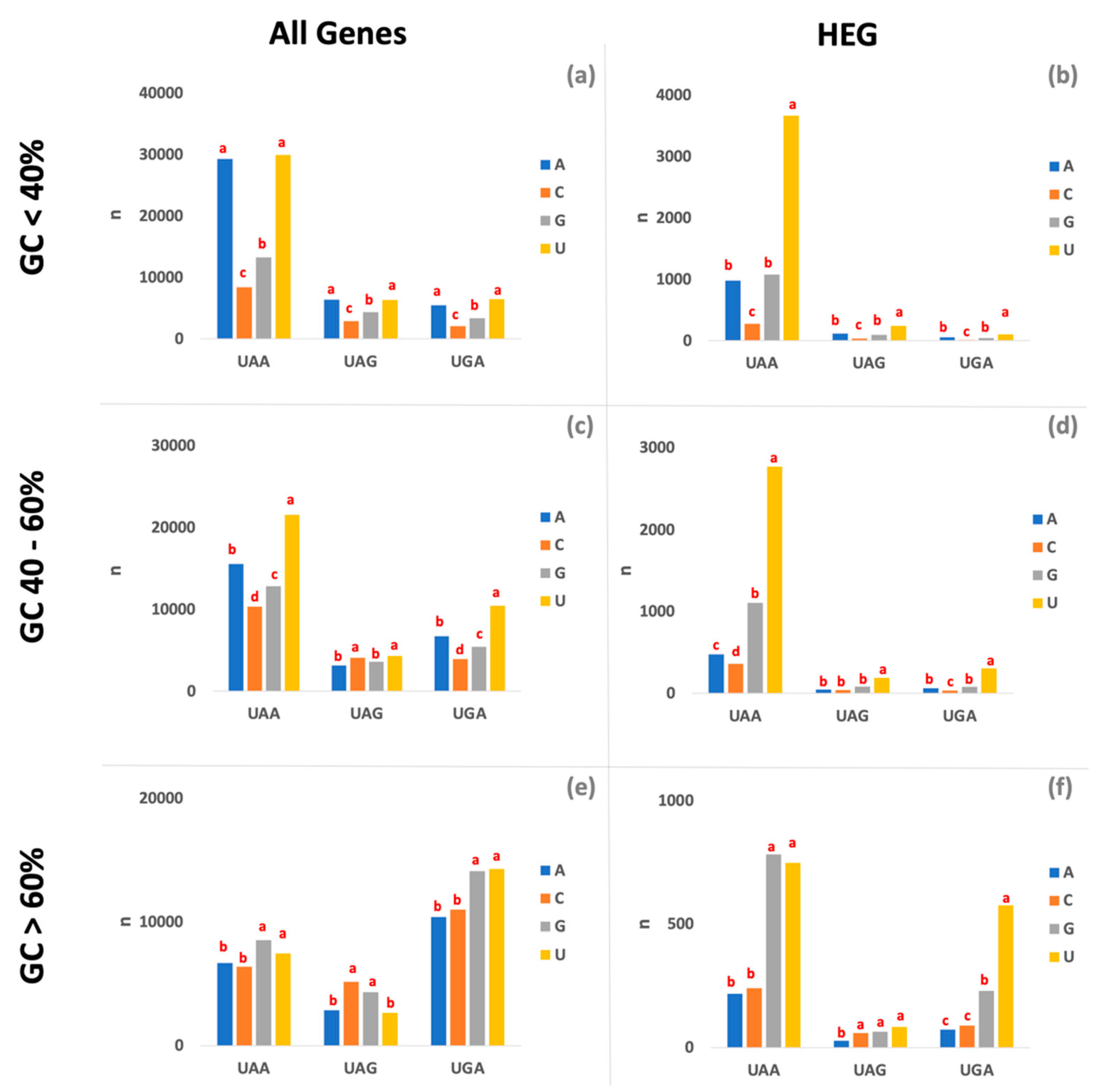
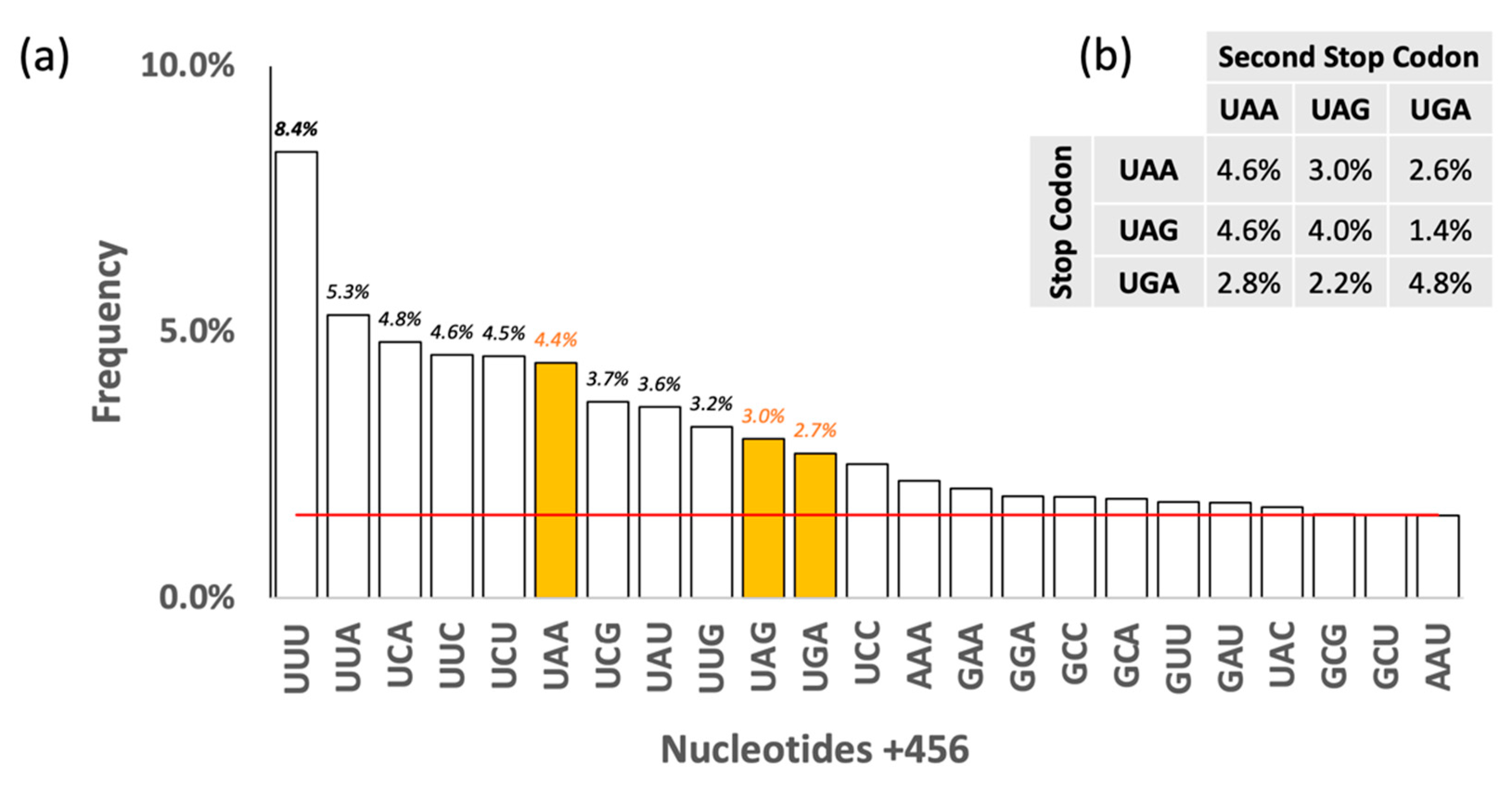
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crick, F.H.; Barnett, L.; Brenner, S.; Watts-Tobin, R.J. General nature of the genetic code for proteins. Nature 1961, 192, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R.; Gautier, C.; Gouy, M.; Mercier, R.; Pavé, A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980, 8, r49–r62. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from international DNA sequence databases: Status for the year 2000. Nucleic Acids Res. 2000, 28, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigbò, P.; Guzmán, E.; Romeu, A.; Garcia-Vallvé, S. OPTIMIZER: A web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007, 35, W126–W131. [Google Scholar] [CrossRef] [Green Version]
- Puigbò, P.; Romeu, A.; Garcia-Vallvé, S. HEG-DB: A database of predicted highly expressed genes in prokaryotic complete genomes under translational selection. Nucleic Acids Res. 2008, 36, D524–D527. [Google Scholar] [CrossRef] [Green Version]
- Benzer, S.; Champe, S.P. A change from nonsense to sense in the genetic code. Proc. Natl. Acad. Sci. USA 1962, 48, 1114–1121. [Google Scholar] [CrossRef] [Green Version]
- Garen, A.; Siddiqi, O. Suppression of mutations in the alkaline phosphatase structural cistron of E. coli. Proc. Natl. Acad. Sci. USA 1962, 48, 1121–1127. [Google Scholar] [CrossRef] [Green Version]
- Brenner, S.; Stretton, A.O.; Kaplan, S. Genetic code: The “nonsense” triplets for chain termination and their suppression. Nature 1965, 206, 994–998. [Google Scholar] [CrossRef]
- Belin, D. Why are suppressors of amber mutations so frequent among Escherichia coli K12 strains? A plausible explanation for a long-lasting puzzle. Genetics 2003, 165, 455–456. [Google Scholar] [CrossRef]
- Brenner, S.; Beckwith, J.R. Ochre mutants, a new class of suppressible nonsense mutants. J. Mol. Biol. 1965, 13, 629–637. [Google Scholar] [CrossRef]
- Brenner, S.; Barnett, L.; Katz, E.R.; Crick, F.H. UGA: A third nonsense triplet in the genetic code. Nature 1967, 213, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.F.; Fan, D.P.; Brenner, S. A strong suppressor specific for UGA. Nature 1967, 214, 452–453. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Köhrer, C.; Mandal, D.; RajBhandary, U.L. Nonsense suppression in archaea. Proc. Natl. Acad. Sci. USA 2015, 112, 6015–6020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhang, J. Stop-codon read-through arises largely from molecular errors and is generally nonadaptive. PLoS Genet. 2019, 15, e1008141. [Google Scholar] [CrossRef] [PubMed]
- Namy, O.; Hatin, I.; Rousset, J.P. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001, 2, 787–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povolotskaya, I.S.; Kondrashov, F.A.; Ledda, A.; Vlasov, P.K. Stop codons in bacteria are not selectively equivalent. Biol. Direct 2012, 7, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belinky, F.; Babenko, V.N.; Rogozin, I.B.; Koonin, E.V. Purifying and positive selection in the evolution of stop codons. Sci. Rep. 2018, 8, 9260. [Google Scholar] [CrossRef] [PubMed]
- Keseler, I.M.; Gama-Castro, S.; Mackie, A.; Billington, R.; Bonavides-Martínez, C.; Caspi, R.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Muñiz-Rascado, L.; et al. The ecocyc database in 2021. Front. Microbiol. 2021, 12, 711077. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, G.; Holm, M.; Wiens, T.; Sanyal, S. Comprehensive analysis of stop codon usage in bacteria and its correlation with release factor abundance. J. Biol. Chem. 2014, 289, 30334–30342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, M.; Rydén-Aulin, M. Glutamine is incorporated at the nonsense codons UAG and UAA in a suppressor-free Escherichia coli strain. Biochim. Biophys. Acta 2003, 1627, 1–6. [Google Scholar] [CrossRef]
- Fluck, M.M.; Epstein, R.H. Isolation and characterization of context mutations affecting the suppressibility of nonsense mutations. Mol. Gen. Genet. 1980, 177, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Engelberg-Kulka, H. UGA suppression by normal tRNA Trp in Escherichia coli: Codon context effects. Nucleic Acids Res. 1981, 9, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.H.; Albertini, A.M. Effects of surrounding sequence on the suppression of nonsense codons. J. Mol. Biol. 1983, 164, 59–71. [Google Scholar] [CrossRef]
- Bossi, L. Context effects: Translation of UAG codon by suppressor tRNA is affected by the sequence following UAG in the message. J. Mol. Biol. 1983, 164, 73–87. [Google Scholar] [CrossRef]
- Poole, E.S.; Brown, C.M.; Tate, W.P. The identity of the base following the stop codon determines the efficiency of in vivo translational termination in Escherichia coli. EMBO J. 1995, 14, 151–158. [Google Scholar] [CrossRef]
- Tate, W.P.; Poole, E.S.; Dalphin, M.E.; Major, L.L.; Crawford, D.J.; Mannering, S.A. The translational stop signal: Codon with a context, or extended factor recognition element? Biochimie 1996, 78, 945–952. [Google Scholar] [CrossRef]
- Wei, Y.; Xia, X. The role of +4U as an extended translation termination signal in bacteria. Genetics 2017, 205, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Cavalcanti, A.R.O.; Landweber, L.F. Conservation of tandem stop codons in yeasts. Genome Biol. 2005, 6, R31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Major, L.L.; Edgar, T.D.; Yee Yip, P.; Isaksson, L.A.; Tate, W.P. Tandem termination signals: Myth or reality? FEBS Lett. 2002, 514, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.T.; Hurst, L.D. In eubacteria, unlike eukaryotes, there is no evidence for selection favouring fail-safe 3′ additional stop codons. PLoS Genet. 2019, 15, e1008386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, J.L. Nucleotide sequence from the polypeptide chain termination region of the coat protein cistron in bacteriophage R17 RNA. Nature 1970, 225, 147–151. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belin, D.; Puigbò, P. Why Is the UAG (Amber) Stop Codon Almost Absent in Highly Expressed Bacterial Genes? Life 2022, 12, 431. https://doi.org/10.3390/life12030431
Belin D, Puigbò P. Why Is the UAG (Amber) Stop Codon Almost Absent in Highly Expressed Bacterial Genes? Life. 2022; 12(3):431. https://doi.org/10.3390/life12030431
Chicago/Turabian StyleBelin, Dominique, and Pere Puigbò. 2022. "Why Is the UAG (Amber) Stop Codon Almost Absent in Highly Expressed Bacterial Genes?" Life 12, no. 3: 431. https://doi.org/10.3390/life12030431