
CRISPR-Cas Systems in Gut Microbiome of Children with Autism Spectrum Disorders

Abstract
:1. Introduction
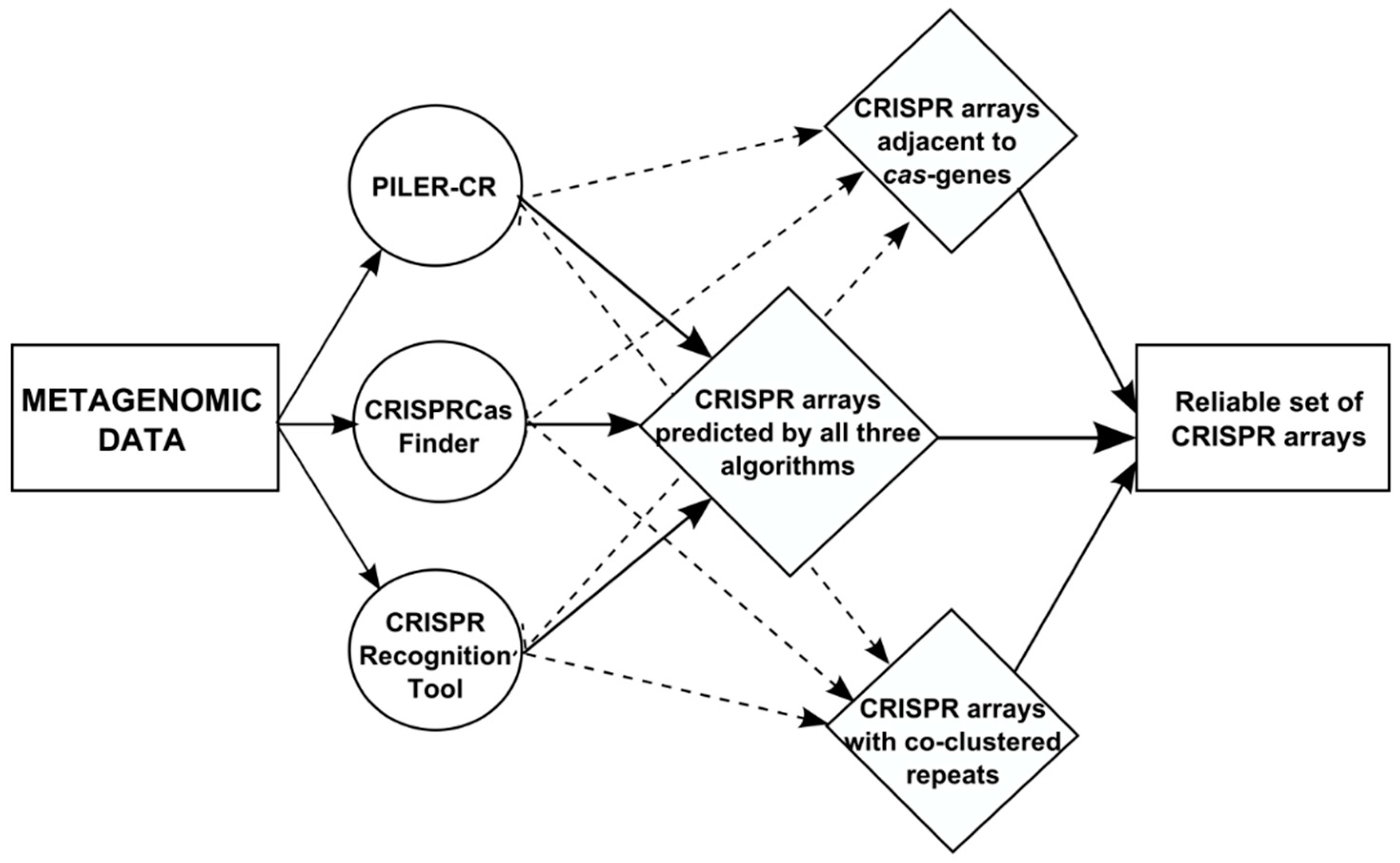
2. Data and Algorithms
3. Results
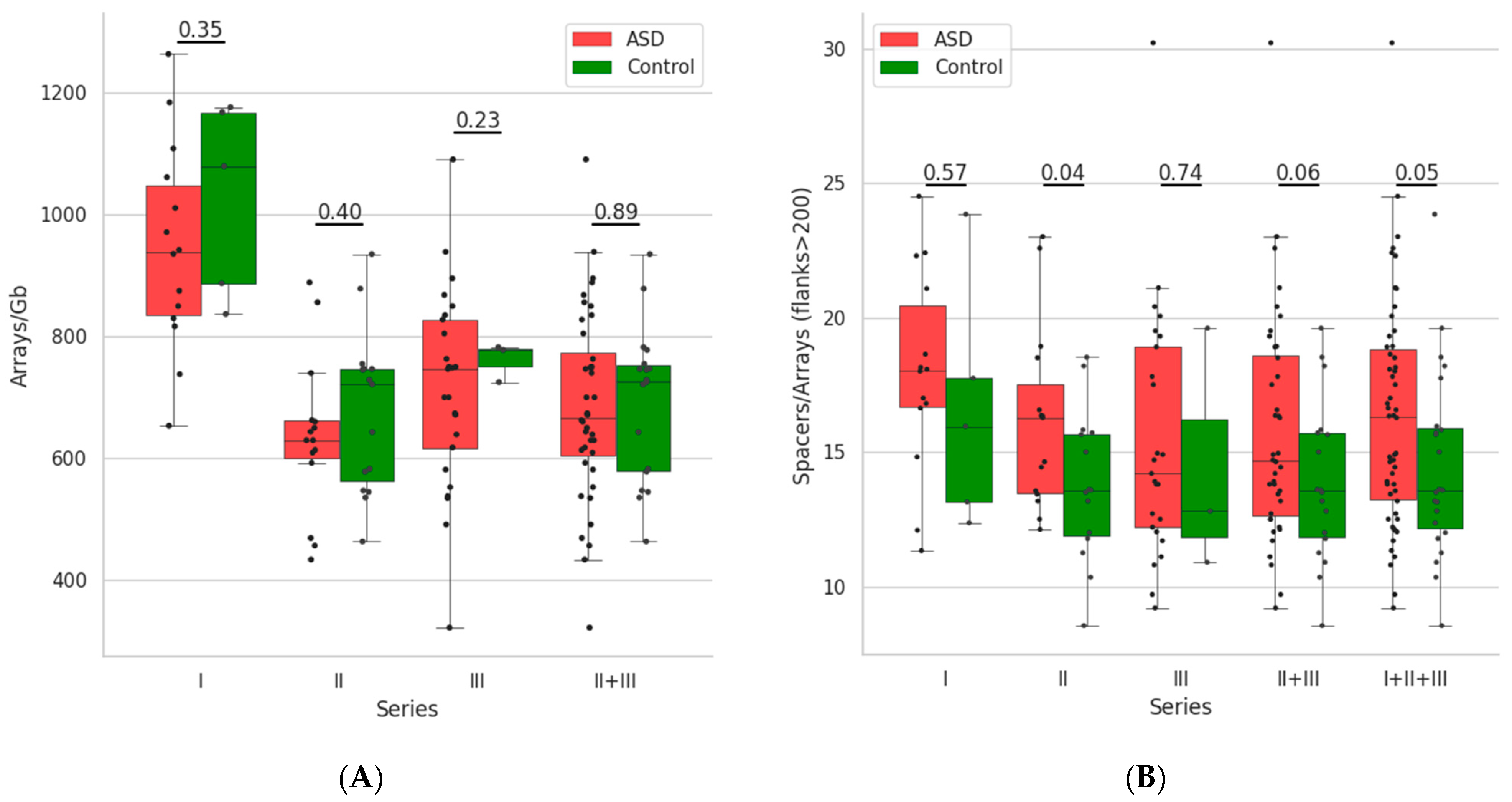
3.1. Distribution of the CRISPR-Cas Loci and Its Parameters
3.2. Search for Markers of the Disease among CRISPR-Cas Systems or Their Elements
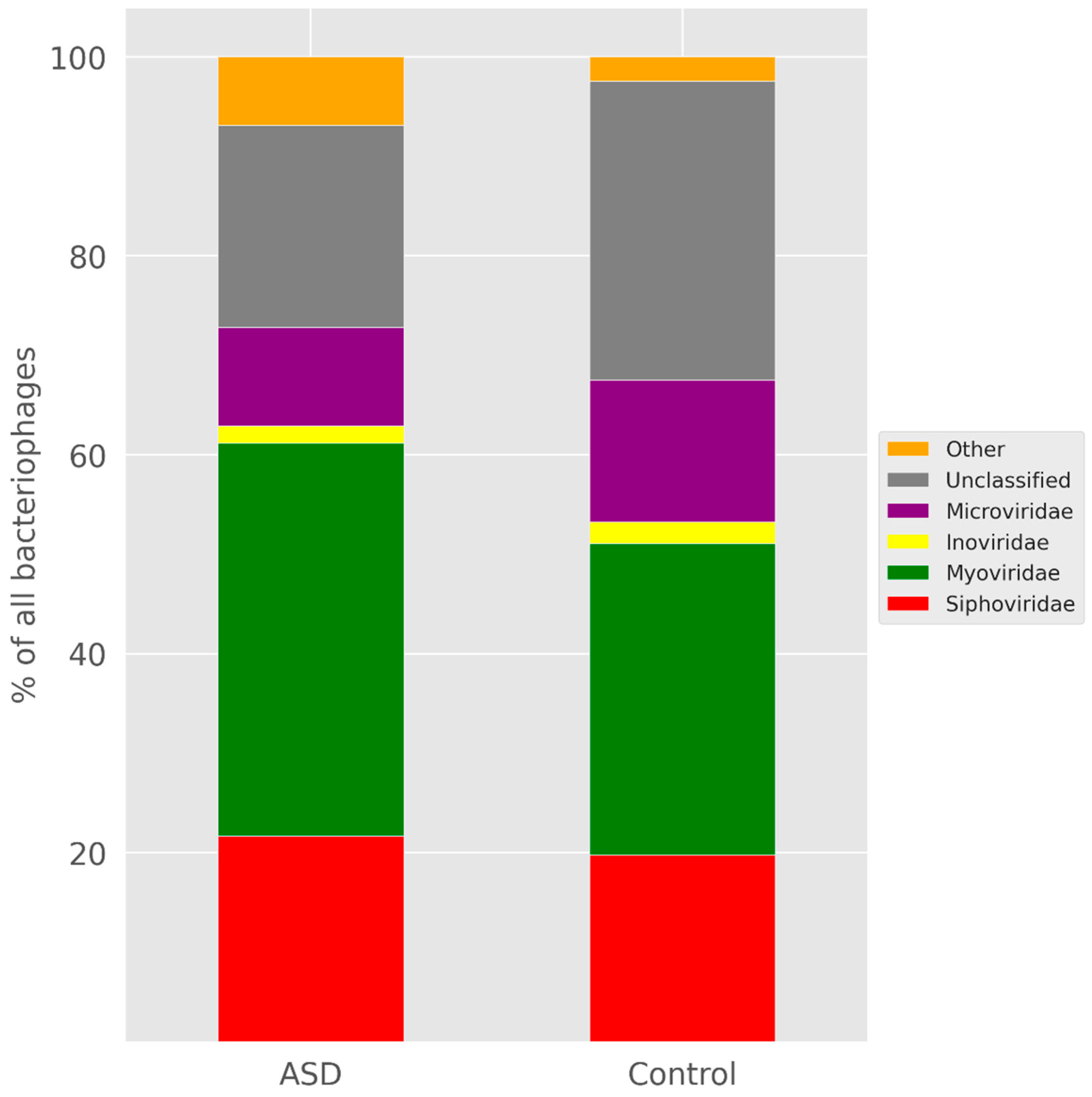
3.3. Search for Protospacers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; de Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 2019, 10, 4505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, T.R.; Mazmanian, S.K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015, 17, 565–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofer, U. The majority is uncultured. Nat. Rev. Microbiol. 2018, 16, 716–717. [Google Scholar] [CrossRef]
- Specht, T.; Szymanski, M.; Barciszewska, M.Z.; Barciszewski, J.; Erdmann, V.A. Compilation of 5S rRNA and 5S rRNA gene sequences. Nucleic Acids Res. 1997, 25, 96–97. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.B. 16S rRNA gene sequencing for bacterial pathogen identification in the clinical laboratory. Mol. Diagn. 2001, 6, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Teng, J.L.; Tse, H.; Yuen, K.Y. Then and now: Use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin. Microbiol. Infect. 2008, 14, 908–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Ehrlich, S.D.; Consortium, M. Metahit: The european union project on metagenomics of the human intestinal tract. Metagenom. Hum. Body 2011, 307–316. [Google Scholar] [CrossRef]
- Tyakht, A.V.; Alexeev, D.G.; Popenko, A.S.; Kostryukova, E.S.; Govorun, V.M. Rural and urban microbiota: To be or not to be? Gut Microbes 2014, 5, 351–356. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American gut: An open platform for citizen science microbiome research. mSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Guo, X.Y.; Liu, X.J.; Hao, J.Y. Gut microbiota in ulcerative colitis: Insights on pathogenesis and treatment. J. Dig. Dis. 2020, 21, 147–159. [Google Scholar] [CrossRef] [PubMed]
- John, G.K.; Mullin, G.E. The gut microbiome and obesity. Curr. Oncol. Rep. 2016, 18, 45. [Google Scholar] [CrossRef]
- Gupta, A.; Osadchiy, V.; Mayer, E.A. Brain-gut-microbiome interactions in obesity and food addiction. Nat. Rev. Gastroenterol Hepatol. 2020, 17, 655–672. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [PubMed]
- Caputi, V.; Giron, M.C. Microbiome-gut-brain axis and toll-like receptors in Parkinson’s disease. Int. J. Mol. Sci. 2018, 19, 1689. [Google Scholar] [CrossRef] [Green Version]
- van De Sande, M.M.; van Buul, V.J.; Brouns, F.J. Autism and nutrition: The role of the gut-brain axis. Nutr. Res. Rev. 2014, 27, 199–214. [Google Scholar] [CrossRef] [Green Version]
- Wimberley, T.; Agerbo, E.; Pedersen, C.B.; Dalsgaard, S.; Horsdal, H.T.; Mortensen, P.B.; Thompson, W.K.; Kohler-Forsberg, O.; Yolken, R.H. Otitis media, antibiotics, and risk of autism spectrum disorder. Autism Res. 2018, 11, 1432–1440. [Google Scholar] [CrossRef]
- Montecucco, C.; Schiavo, G. Mechanism of action of tetanus and botulinum neurotoxins. Mol. Microbiol. 1994, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Buckley, A.; Thurm, A.; Lee, L.C.; Azhagiri, A.; Neville, D.M.; Swedo, S.E. A pilot open-label trial of minocycline in patients with autism and regressive features. J. Neurodev. Disord. 2013, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 2002, 35, S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Sandler, R.H.; Finegold, S.M.; Bolte, E.R.; Buchanan, C.P.; Maxwell, A.P.; Vaisanen, M.L.; Nelson, M.N.; Wexler, H.M. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J. Child Neurol. 2000, 15, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Dowd, S.E.; Gontcharova, V.; Liu, C.; Henley, K.E.; Wolcott, R.D.; Youn, E.; Summanen, P.H.; Granpeesheh, D.; Dixon, D.; et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 2010, 16, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Liu, C.X.; Finegold, S.A. Real-time PCR quantitation of Clostridia in feces of autistic children. Appl. Environ. Microb. 2004, 70, 6459–6465. [Google Scholar] [CrossRef] [Green Version]
- Tomova, A.; Soltys, K.; Repiska, G.; Palkova, L.; Filcikova, D.; Minarik, G.; Turna, J.; Prochotska, K.; Babinska, K.; Ostatnikova, D. Specificity of gut microbiota in children with autism spectrum disorder in slovakia and its correlation with astrocytes activity marker and specific behavioural patterns. Physiol. Behav. 2020, 214, 112745. [Google Scholar] [CrossRef]
- Zou, R.; Xu, F.; Wang, Y.; Duan, M.; Guo, M.; Zhang, Q.; Zhao, H.; Zheng, H. Changes in the gut microbiota of children with autism spectrum disorder. Autism Res. 2020, 13, 1614–1625. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism--comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; Labaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced incidence of prevotella and other fermenters in intestinal microflora of autistic children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Liang, J.; Dai, M.; Wang, J.; Luo, J.; Zhang, Z.; Jing, J. Altered gut microbiota in chinese children with autism spectrum disorders. Front. Cell. Infect. Microbiol. 2019, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Ilhan, Z.E.; Isern, N.G.; Hoyt, D.W.; Howsmon, D.P.; Shaffer, M.; Lozupone, C.A.; Hahn, J.; Adams, J.B.; Krajmalnik-Brown, R. Differences in fecal microbial metabolites and microbiota of children with autism spectrum disorders. Anaerobe 2018, 49, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Gondalia, S.V.; Palombo, E.A.; Knowles, S.R.; Cox, S.B.; Meyer, D.; Austin, D.W. Molecular characterisation of gastrointestinal microbiota of children with autism (with and without gastrointestinal dysfunction) and their neurotypical siblings. Autism Res. 2012, 5, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Xu, Y.; Zhang, X.; Zhang, L.; Duan, G.; Song, C.; Li, Z.; Yang, Y.; Wang, Y.; Wang, X.; et al. Gut microbiota changes in patients with autism spectrum disorders. J. Psychiatr. Res. 2020, 129, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Averina, O.V.; Kovtun, A.S.; Polyakova, S.I.; Savilova, A.M.; Rebrikov, D.V.; Danilenko, V.N. The bacterial neurometabolic signature of the gut microbiota of young children with autism spectrum disorders. J. Med. Microbiol. 2020, 69, 558–571. [Google Scholar] [CrossRef]
- Nussenzweig, P.M.; Marraffini, L.A. Molecular mechanisms of CRISPR-Cas immunity in bacteria. Annu. Rev. Genet. 2020, 54, 93–120. [Google Scholar] [CrossRef]
- McGinn, J.; Marraffini, L.A. Molecular mechanisms of CRISPR-Cas spacer acquisition. Nat. Rev. Microbiol. 2019, 17, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, V.A.; Gelfand, M.S.; Artamonova, I.I. Evolutionary dynamics of clustered regularly interspaced short palindromic repeat systems in the ocean metagenome. Appl. Environ. Microbiol. 2010, 76, 2136–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, D.; Harrington, L.B.; Strutt, S.C.; Probst, A.J.; Anantharaman, K.; Thomas, B.C.; Doudna, J.A.; Banfield, J.F. New CRISPR-Cas systems from uncultivated microbes. Nature 2017, 542, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munch, P.C.; Franzosa, E.A.; Stecher, B.; McHardy, A.C.; Huttenhower, C. Identification of natural CRISPR systems and targets in the human microbiome. Cell Host Microbe 2021, 29, 94–106.e4. [Google Scholar] [CrossRef]
- Gogleva, A.A.; Gelfand, M.S.; Artamonova, I.I. Comparative analysis of CRISPR cassettes from the human gut metagenomic contigs. BMC Genom. 2014, 15, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Neron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISPRFinder, includes a portable version, enhanced performance and integrates search for cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Ghodsi, M.; Liu, B.; Pop, M. DNACLUST: Accurate and efficient clustering of phylogenetic marker genes. BMC Bioinform. 2011, 12, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourcel, C.; Touchon, M.; Villeriot, N.; Vernadet, J.P.; Couvin, D.; Toffano-Nioche, C.; Vergnaud, G. CRISPRCasDB a successor of CRISPRdb containing CRISPR arrays and cas genes from complete genome sequences, and tools to download and query lists of repeats and spacers. Nucleic Acids Res. 2020, 48, D535–D544. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinegger, M.; Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Sun, H.X.; Zhang, C.; Cheng, L.; Peng, Y.; Deng, Z.; Wang, D.; Wang, Y.; Hu, M.; Liu, W.; et al. Prophage Hunter: An integrative hunting tool for active prophages. Nucleic Acids Res. 2019, 47, W74–W80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Horvath, P. CRISPR: New horizons in phage resistance and strain identification. Annu. Rev. Food Sci. Technol. 2012, 3, 143–162. [Google Scholar] [CrossRef] [PubMed]
- Haas, K.N.; Blanchard, J.L. Reclassification of the Clostridium clostridioforme and Clostridium sphenoides clades as Enterocloster gen. Nov. and Lacrimispora gen. Nov., including reclassification of 15 taxa. Int. J. Syst. Evol. Microbiol. 2020, 70, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Alkhnbashi, O.S.; Meier, T.; Mitrofanov, A.; Backofen, R.; Voss, B. CRISPR-Cas bioinformatics. Methods 2020, 172, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.G.; Liang, C. Metacrast: Reference-guided extraction of CRISPR spacers from unassembled metagenomes. PeerJ 2017, 5, e3788. [Google Scholar] [CrossRef]
- Skennerton, C.T.; Imelfort, M.; Tyson, G.W. CRASS: Identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res. 2013, 41, e105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CRISPRCasMeta. Available online: https://crisprcas.i2bc.paris-saclay.fr/CrisprCasMeta/Index (accessed on 30 December 2021).
- Pavlova, Y.S.; Paez-Espino, D.; Morozov, A.Y.; Belalov, I.S. Searching for fat tails in CRISPR-Cas systems: Data analysis and mathematical modeling. PLoS Comput. Biol. 2021, 17, e1008841. [Google Scholar] [CrossRef] [PubMed]
- Huntemann, M.; Ivanova, N.N.; Mavromatis, K.; Tripp, H.J.; Paez-Espino, D.; Palaniappan, K.; Szeto, E.; Pillay, M.; Chen, I.M.; Pati, A.; et al. The standard operating procedure of the DOE-JGI microbial genome annotation pipeline (MGAP v.4). Stand. Genomic. Sci. 2015, 10, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.C.; Lombardo, M.V.; Chakrabarti, B.; Baron-Cohen, S. Subgrouping the autism “spectrum”: Reflections on DSM-5. PLoS Biol. 2013, 11, e1001544. [Google Scholar] [CrossRef]
- Gong, T.; Zeng, J.; Tang, B.; Zhou, X.; Li, Y. CRISPR-Cas systems in oral microbiome: From immune defense to physiological regulation. Mol. Oral Microbiol. 2020, 35, 41–48. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Human Gut Metagenome Samples (NCBI BioProject ID: PRJNA516054) | |||
|---|---|---|---|
| Series I | Series II | Series III | |
| Sex | both sexes | ||
| Age (y.o.) | 1–9 (ASD) 3–4 (control) | 2–4 (ASD) 2–4 (control) | 2–6 (ASD) 3 (control) |
| Number of samples | 14 (ASD) 5 (control) | 15 (ASD) 15 (control) | 25 (ASD) 3 (control) |
| Platform and type of sequencing | Illumina HiSeq 2500, paired-end | Illumina HiSeq 4000, paired-end | Illumina NovaSeq 6000, paired-end |
| Read length, nt | 135 | 150 | 150 |
| Range of assembly size, Gb | 0.06–0.26 (ASD) 0.13–0.19 (control) | 0.11–0.30 (ASD) 0.14–0.28 (control) | 0.11–0.37 (ASD) 0.17–0.27 (control) |
| Parameters | Series I | Series II | Series III | |
|---|---|---|---|---|
| ASD | Age | 4.50 ± 2.47 | 3.20 ± 0.77 | 3.60 ± 0.96 |
| Assembly size (Gb) | 0.17 ± 0.05 | 0.18 ± 0.06 | 0.21 ± 0.07 | |
| Arrays | 161.14 ± 58.66 | 115.93 ± 45.23 | 155.08 ± 63.86 | |
| Complete arrays (flanks > 200) | 22.21 ± 10.24 | 22.00 ± 9.58 | 33.04 ± 15.34 | |
| Arrays near cas | 23.86 ± 9.99 | 25.87 ± 10.38 | 37.12 ± 16.69 | |
| Spacers | 1247.57 ± 488.94 | 895.07 ± 321.10 | 1241.16 ± 511.32 | |
| Protospacers/Spacers (%) | 6.09 ± 2.06 | 6.73 ± 2.42 | 4.64 ± 1.77 | |
| Control | Age | 3.40 ± 0.55 | 2.87 ± 0.52 | 3.0 ± 0.0 |
| Assembly size (Gb) | 0.16 ± 0.02 | 0.18 ± 0.04 | 0.23 ± 0.05 | |
| Arrays | 160.60 ± 16.62 | 122.13 ± 31.64 | 172.33 ± 38.53 | |
| Complete arrays (flanks > 200) | 22.00 ± 8.03 | 23.47 ± 5.74 | 37.00 ± 19.08 | |
| Arrays near cas | 24.80 ± 8.84 | 26.07 ± 5.90 | 39.00 ± 13.45 | |
| Spacers | 1265.40 ± 226.49 | 923.73 ± 277.29 | 1381.67 ± 465.66 | |
| Protospacers/Spacers (%) | 5.62 ± 2.17 | 6.36 ± 2.66 | 8.62 ± 2.34 | |
| Localisation in Relation to cas | ATCC BAA-613 DR | CBBP-2 DR | Series I | Series II | Series III |
|---|---|---|---|---|---|
| ASD/ Control | ASD/ Control | ASD/ Control | |||
| Adjacent to cas | GTCTCCGTCCTCGCGGGCGGAGTGGGTTGAAAT | ATTTCAACCCACTCCGCCCACGAGGACGGAGAC | 3/0 | 4/2 | 4/1 |
| Distal from cas | ATTTCAATCCACAAGGCTCTCGCGAGCCTCGAC | GTCGAGGCTCGCGAGAGCCTTGTGGATTGAAAT | 3/0 | 4/2 | 8/0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakharevich, N.V.; Nikitin, M.S.; Kovtun, A.S.; Malov, V.O.; Averina, O.V.; Danilenko, V.N.; Artamonova, I.I. CRISPR-Cas Systems in Gut Microbiome of Children with Autism Spectrum Disorders. Life 2022, 12, 367. https://doi.org/10.3390/life12030367
Zakharevich NV, Nikitin MS, Kovtun AS, Malov VO, Averina OV, Danilenko VN, Artamonova II. CRISPR-Cas Systems in Gut Microbiome of Children with Autism Spectrum Disorders. Life. 2022; 12(3):367. https://doi.org/10.3390/life12030367
Chicago/Turabian StyleZakharevich, Natalia V., Mikhail S. Nikitin, Alexey S. Kovtun, Vsevolod O. Malov, Olga V. Averina, Valery N. Danilenko, and Irena I. Artamonova. 2022. "CRISPR-Cas Systems in Gut Microbiome of Children with Autism Spectrum Disorders" Life 12, no. 3: 367. https://doi.org/10.3390/life12030367