
Bone Marrow Mesenchymal Stem Cells and Their Derived Extracellular Vesicles Attenuate Non-Alcoholic Steatohepatitis-Induced Cardiotoxicity via Modulating Cardiac Mechanisms


Abstract
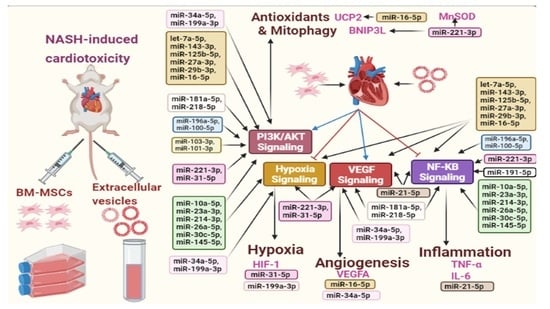
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolation and Characterization of BM-MSCs and BM-MSCs-EV
2.3. Labeling of BM-MSCs with PKH26
2.4. miRNA Sequencing and Association with Key Targets and Pathways
2.5. Experimental Design
2.6. Histopathological Examination and Scoring
2.7. Assessment of Serum Cardiotoxicity Markers
2.8. Immunohistochemical Analysis
2.9. Protein Assessment by ELISA
2.10. Reverse Transcription-Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
2.11. Statistical Analysis
3. Results
3.1. Characterization and Tracing of BM-MSCs-EV
3.2. BM-MSCs-EV and BM-MSCs Treatment Attenuate Histopathological Alterations of NASH and Its Associated Cardiotoxic Events
3.3. BM-MSCs-EV and BM-MSCs Treatment Alleviate Serum Cardiotoxic Markers Associated with NASH
3.4. BM-MSCs-EV and BM-MSCs Treatment Attenuated Cardiac Hypoxic Responses Induced by NASH
3.5. BM-MSCs-EV and BM-MSCs Treatment Improved Cardiac Vascular Endothelial Growth Factor in NASH
3.6. BM-MSCs-EV and BM-MSCs Treatment Restored Some Cardiac Autophagy Proteins
3.7. BM-MSCs-EV and BM-MSCs Treatment Improved Some Mitochondrial Antioxidant Markers and Some Mitochondrial Mitophagy Markers
3.8. BM-MSCs-EV and BM-MSCs Treatment Ameliorated the Cardiac Inflammatory Status Induced by NASH
3.9. BM-MSCs Secreted miRNAs and the Signaling Pathways in Cardiotoxic State Induced by NASH
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El-Derany, M.O.; El-Demerdash, E. Pyrvinium pamoate attenuates non-alcoholic steatohepatitis: Insight on hedgehog/Gli and Wnt/β-catenin signaling crosstalk. Biochem. Pharmacol. 2020, 177, 113942. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2020, 110, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Mahfood Haddad, T.; Hamdeh, S.; Kanmanthareddy, A.; Alla, V.M. Nonalcoholic fatty liver disease and the risk of clinical cardiovascular events: A systematic review and meta-analysis. Diabetes Metab. Syndr. 2017, 11 (Suppl. 1), S209–S216. [Google Scholar] [CrossRef]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Relationship between Heart Disease and Liver Disease: A Two-Way Street. Cells 2020, 9, 567. [Google Scholar] [CrossRef] [Green Version]
- Tana, C.; Ballestri, S.; Ricci, F.; Di Vincenzo, A.; Ticinesi, A.; Gallina, S.; Giamberardino, M.A.; Cipollone, F.; Sutton, R.; Vettor, R.; et al. Cardiovascular Risk in Non-Alcoholic Fatty Liver Disease: Mechanisms and Therapeutic Implications. Int. J. Environ. Res. Public Health 2019, 16, 3104. [Google Scholar] [CrossRef] [Green Version]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [Green Version]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. 2019, 99, 749–763. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, W.; Zhu, W.; He, W.; Zhao, H.; Xiang, Y.; Liu, C.; Wu, W. Chronic intermittent hypoxia promotes the development of experimental non-alcoholic steatohepatitis by modulating Treg/Th17 differentiation. Acta Biochim. Et Biophys. Sin. 2018, 50, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Xin, T.; Lv, W.; Liu, D.; Jing, Y.; Hu, F. Opa1 Reduces Hypoxia-Induced Cardiomyocyte Death by Improving Mitochondrial Quality Control. Front. Cell Dev. Biol. 2020, 8, 853. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharmacol. 2014, 9, 142–160. [Google Scholar] [CrossRef] [Green Version]
- White, I.A.; Sanina, C.; Balkan, W.; Hare, J.M. Mesenchymal Stem Cells in Cardiology. Methods Mol. Biol. 2016, 1416, 55–87. [Google Scholar] [CrossRef] [Green Version]
- Hare, J.M.; Fishman, J.E.; Gerstenblith, G.; DiFede Velazquez, D.L.; Zambrano, J.P.; Suncion, V.Y.; Tracy, M.; Ghersin, E.; Johnston, P.V.; Brinker, J.A.; et al. Comparison of allogeneic vs autologous bone marrow–derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: The POSEIDON randomized trial. Jama 2012, 308, 2369–2379. [Google Scholar] [CrossRef]
- Suzuki, E.; Fujita, D.; Takahashi, M.; Oba, S.; Nishimatsu, H. Therapeutic Effects of Mesenchymal Stem Cell-Derived Exosomes in Cardiovascular Disease. Adv. Exp. Med. Biol. 2017, 998, 179–185. [Google Scholar] [CrossRef]
- Hade, M.D.; Suire, C.N.; Suo, Z. Mesenchymal Stem Cell-Derived Exosomes: Applications in Regenerative Medicine. Cells 2021, 10, 1959. [Google Scholar] [CrossRef]
- Sun, S.J.; Wei, R.; Li, F.; Liao, S.Y.; Tse, H.F. Mesenchymal stromal cell-derived exosomes in cardiac regeneration and repair. Stem Cell Rep. 2021, 16, 1662–1673. [Google Scholar] [CrossRef]
- Taglauer, E.S.; Fernandez-Gonzalez, A.; Willis, G.R.; Reis, M.; Yeung, V.; Liu, X.; Prince, L.S.; Mitsialis, S.A.; Kourembanas, S. Antenatal Mesenchymal Stromal Cell Extracellular Vesicle Therapy Prevents Preeclamptic Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2021, 66, 86–95. [Google Scholar] [CrossRef]
- McKay, T.B.; Yeung, V.; Hutcheon, A.E.K.; Guo, X.; Zieske, J.D.; Ciolino, J.B. Extracellular Vesicles in the Cornea: Insights from Other Tissues. Anal. Cell. Pathol. 2021, 2021, 9983900. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, X.; Qian, H.; Zhu, W.; Sun, X.; Hu, J.; Zhou, H.; Chen, Y. Mesenchymal stem cells from adult human bone marrow differentiate into a cardiomyocyte phenotype in vitro. Exp. Biol. Med. 2004, 229, 623–631. [Google Scholar] [CrossRef]
- Yan, X.; Lv, A.; Xing, Y.; Liu, B.; Hou, J.; Huang, W.; Li, Y. Inhibition of p53-p21 pathway promotes the differentiation of rat bone marrow mesenchymal stem cells into cardiomyocytes. Mol. Cell. Biochem. 2011, 354, 21–28. [Google Scholar] [CrossRef]
- Martin-Rendon, E.; Brunskill, S.J.; Hyde, C.J.; Stanworth, S.J.; Mathur, A.; Watt, S.M. Autologous bone marrow stem cells to treat acute myocardial infarction: A systematic review. Eur. Heart J. 2008, 29, 1807–1818. [Google Scholar] [CrossRef] [Green Version]
- El-Derany, M.O.; Noureldein, M.H. Bone marrow mesenchymal stem cells and their derived exosomes resolve doxorubicin-induced chemobrain: Critical role of their miRNA cargo. Stem Cell Res. Ther. 2021, 12, 322. [Google Scholar] [CrossRef]
- El-Derany, M.O.; AbdelHamid, S.G. Upregulation of miR-96-5p by bone marrow mesenchymal stem cells and their exosomes alleviate non-alcoholic steatohepatitis: Emphasis on caspase-2 signaling inhibition. Biochem. Pharmacol. 2021, 190, 114624. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, B.; Yang, Y.; Jia, Q.; Qi, Z.; Zhang, A.; Lv, S.; Zhang, J. Exosomes in ischemic heart disease: Novel carriers for bioinformation. Biomed. Pharmacother. 2019, 120, 109451. [Google Scholar] [CrossRef]
- Fernandez-Gonzalez, A.; Willis, G.R.; Yeung, V.; Reis, M.; Liu, X.; Mitsialis, S.A.; Kourembanas, S. Therapeutic Effects of Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles in Oxygen-Induced Multi-Organ Disease: A Developmental Perspective. Front. Cell Dev. Biol. 2021, 9, 647025. [Google Scholar] [CrossRef] [PubMed]
- Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Yeung, V.; Liu, X.; Ericsson, M.; Andrews, N.A.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell-derived small extracellular vesicles restore lung architecture and improve exercise capacity in a model of neonatal hyperoxia-induced lung injury. J. Extracell. Vesicles 2020, 9, 1790874. [Google Scholar] [CrossRef]
- Willis, G.R.; Reis, M.; Gheinani, A.H.; Fernandez-Gonzalez, A.; Taglauer, E.S.; Yeung, V.; Liu, X.; Ericsson, M.; Haas, E.; Mitsialis, S.A.; et al. Extracellular Vesicles Protect the Neonatal Lung from Hyperoxic Injury through the Epigenetic and Transcriptomic Reprogramming of Myeloid Cells. Am. J. Respir. Crit. Care Med. 2021, 204, 1418–1432. [Google Scholar] [CrossRef]
- Chen, G.-H.; Xu, J.; Yang, Y.-J. Exosomes: Promising sacks for treating ischemic heart disease? Am. J. Physiol.-Heart Circ. Physiol. 2017, 313, H508–H523. [Google Scholar] [CrossRef]
- Mancuso, T.; Barone, A.; Salatino, A.; Molinaro, C.; Marino, F.; Scalise, M.; Torella, M.; De Angelis, A.; Urbanek, K.; Torella, D.; et al. Unravelling the Biology of Adult Cardiac Stem Cell-Derived Exosomes to Foster Endogenous Cardiac Regeneration and Repair. Int. J. Mol. Sci. 2020, 21, 3725. [Google Scholar] [CrossRef]
- van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [Green Version]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borràs, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef] [Green Version]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Shephard, A.P.; Yeung, V.; Clayton, A.; Webber, J.P. Prostate cancer exosomes as modulators of the tumor microenvironment. J. Cancer Metastasis Treat. 2017, 3, 288–301. [Google Scholar] [CrossRef]
- Shah, R.; Patel, T.; Freedman, J.E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 958–966. [Google Scholar] [CrossRef]
- Yeung, V.; Willis, G.R.; Taglauer, E.; Mitsialis, S.A.; Kourembanas, S. Paving the Road for Mesenchymal Stem Cell-Derived Exosome Therapy in Bronchopulmonary Dysplasia and Pulmonary Hypertension. In Stem Cell-Based Therapy for Lung Disease; Springer: Berlin, Germany, 2019; pp. 131–152. [Google Scholar] [CrossRef]
- Zou, L.; Ma, X.; Lin, S.; Wu, B.; Chen, Y.; Peng, C. Bone marrow mesenchymal stem cell-derived exosomes protect against myocardial infarction by promoting autophagy. Exp. Ther. Med. 2019, 18, 2574–2582. [Google Scholar] [CrossRef] [Green Version]
- Chong, S.Y.; Lee, C.K.; Huang, C.; Ou, Y.H.; Charles, C.J.; Richards, A.M.; Neupane, Y.R.; Pavon, M.V.; Zharkova, O.; Pastorin, G.; et al. Extracellular Vesicles in Cardiovascular Diseases: Alternative Biomarker Sources, Therapeutic Agents, and Drug Delivery Carriers. Int. J. Mol. Sci. 2019, 20, 3272. [Google Scholar] [CrossRef] [Green Version]
- Nazari-Shafti, T.Z.; Neuber, S.; Garcia Duran, A.; Xu, Z.; Beltsios, E.; Seifert, M.; Falk, V.; Stamm, C. Human mesenchymal stromal cells and derived extracellular vesicles: Translational strategies to increase their proangiogenic potential for the treatment of cardiovascular disease. Stem Cells Transl. Med. 2020, 9, 1558–1569. [Google Scholar] [CrossRef]
- Soleimani, M.; Nadri, S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat. Protoc. 2009, 4, 102–106. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Rikkert, L.G.; Nieuwland, R.; Terstappen, L.; Coumans, F.A.W. Quality of extracellular vesicle images by transmission electron microscopy is operator and protocol dependent. J. Extracell. Vesicles 2019, 8, 1555419. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Shi, H.; Yin, S.; Ji, C.; Zhang, X.; Zhang, B.; Wu, P.; Shi, Y.; Mao, F.; Yan, Y.; et al. Human Mesenchymal Stem Cell Derived Exosomes Alleviate Type 2 Diabetes Mellitus by Reversing Peripheral Insulin Resistance and Relieving β-Cell Destruction. ACS Nano 2018, 12, 7613–7628. [Google Scholar] [CrossRef]
- Ohara, M.; Ohnishi, S.; Hosono, H.; Yamamoto, K.; Yuyama, K.; Nakamura, H.; Fu, Q.; Maehara, O.; Suda, G.; Sakamoto, N. Extracellular Vesicles from Amnion-Derived Mesenchymal Stem Cells Ameliorate Hepatic Inflammation and Fibrosis in Rats. Stem Cells Int. 2018, 2018, 3212643. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Marrone, A.; Rinaldi, L. Non-alcoholic fatty liver disease: Beyond the liver is an emerging multifaceted systemic disease. Hepatobiliary Surg. Nutr. 2018, 7, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Ballestri, S.; Lonardo, A.; Bonapace, S.; Byrne, C.D.; Loria, P.; Targher, G. Risk of cardiovascular, cardiac and arrhythmic complications in patients with non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1724–1745. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Peng, K.-Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2007, 77, 334–343. [Google Scholar] [CrossRef]
- Tian, X.Y.; Ma, S.; Tse, G.; Wong, W.T.; Huang, Y. Uncoupling Protein 2 in Cardiovascular Health and Disease. Front. Physiol. 2018, 9, 1060. [Google Scholar] [CrossRef] [Green Version]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St Clair, D.K. Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, P.E.; Arias-Durán, C.; Ávalos-Guajardo, Y.; Aedo, G.; Verdejo, H.E.; Parra, V.; Lavandero, S. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol. Asp. Med. 2020, 71, 100822. [Google Scholar] [CrossRef] [PubMed]
- Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Marek-Iannucci, S.; Tucker, K.C.; Andres, A.M.; Gottlieb, R.A. Decrease of Cardiac Parkin Protein in Obese Mice. Front. Cardiovasc. Med. 2020, 6, 191. [Google Scholar] [CrossRef] [Green Version]
- Abudureyimu, M.; Yu, W.; Cao, R.Y.; Zhang, Y.; Liu, H.; Zheng, H. Berberine Promotes Cardiac Function by Upregulating PINK1/Parkin-Mediated Mitophagy in Heart Failure. Front. Physiol. 2020, 11, 1244. [Google Scholar] [CrossRef]
- An, M.; Ryu, D.-R.; Won Park, J.; Ha Choi, J.; Park, E.-M.; Eun Lee, K.; Woo, M.; Kim, M. ULK1 prevents cardiac dysfunction in obesity through autophagy-meditated regulation of lipid metabolism. Cardiovasc. Res. 2017, 113, 1137–1147. [Google Scholar] [CrossRef]
- Dorn, G.W. Mitochondrial pruning by Nix and BNip3: An essential function for cardiac-expressed death factors. J. Cardiovasc. Transl. Res. 2010, 3, 374–383. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wang, Y.; Li, W.; Chen, H.; Du, L.; Liu, D.; Wang, X.; Xu, T.; Liu, L.; Chen, Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019, 15, 1882–1898. [Google Scholar] [CrossRef]
- Ren, J.; Sun, M.; Zhou, H.; Ajoolabady, A.; Zhou, Y.; Tao, J.; Sowers, J.R.; Zhang, Y. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in Vivo. Circulation 2017, 136, 2248–2266. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Wu, G.; Wei, G.; Huang, J.; Pang, S.; Liu, L.; Yan, B. Decreased gene expression of LC3 in peripheral leucocytes of patients with coronary artery disease. Eur. J. Clin. Investig. 2011, 41, 958–963. [Google Scholar] [CrossRef]
- Feng, Z.; Jiang, H.X.; Chen, H.; Liu, Y.N.; Wang, Y.; Yang, R.B.; Han, X.; Xia, C.H.; Zhu, Z.B.; Shang, H.; et al. Adaptive Autophagy Offers Cardiorenal Protection in Rats with Acute Myocardial Infarction. Cardiol. Res. Pract. 2020, 2020, 7158975. [Google Scholar] [CrossRef]
- Liang, X.-f.; Guan, X.-r. p62/SQSTM1: A potential molecular target for treatment of atherosclerosis. Front. Lab. Med. 2017, 1, 104–106. [Google Scholar] [CrossRef]
- Song, H.P.; Chu, Z.G.; Zhang, D.X.; Dang, Y.M.; Zhang, Q. PI3K–AKT Pathway Protects Cardiomyocytes Against Hypoxia-Induced Apoptosis by MitoKATP-Mediated Mitochondrial Translocation of pAKT. Cell. Physiol. Biochem. 2018, 49, 717–727. [Google Scholar] [CrossRef]
- Sarkar, K.; Cai, Z.; Gupta, R.; Parajuli, N.; Fox-Talbot, K.; Darshan, M.S.; Gonzalez, F.J.; Semenza, G.L. Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2012, 109, 10504–10509. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cox, S.R.; Morita, T.; Kourembanas, S. Hypoxia Regulates Vascular Endothelial Growth Factor Gene Expression in Endothelial Cells. Circ. Res. 1995, 77, 638–643. [Google Scholar] [CrossRef]
- Huang, A.; Qi, X.; Cui, Y.; Wu, Y.; Zhou, S.; Zhang, M. Serum VEGF: Diagnostic Value of Acute Coronary Syndrome from Stable Angina Pectoris and Prognostic Value of Coronary Artery Disease. Cardiol. Res. Pract. 2020, 2020, 6786302. [Google Scholar] [CrossRef]
- Braile, M.; Marcella, S.; Cristinziano, L.; Galdiero, M.R.; Modestino, L.; Ferrara, A.L.; Varricchi, G.; Marone, G.; Loffredo, S. VEGF-A in Cardiomyocytes and Heart Diseases. Int. J. Mol. Sci. 2020, 21, 5294. [Google Scholar] [CrossRef]
- Coulon, S.; Francque, S.; Colle, I.; Verrijken, A.; Blomme, B.; Heindryckx, F.; De Munter, S.; Prawitt, J.; Caron, S.; Staels, B.; et al. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine 2012, 59, 442–449. [Google Scholar] [CrossRef]
- Kessler, E.L.; Boulaksil, M.; van Rijen, H.V.M.; Vos, M.A.; van Veen, T.A.B. Passive ventricular remodeling in cardiac disease: Focus on heterogeneity. Front. Physiol. 2014, 5, 482. [Google Scholar] [CrossRef] [Green Version]
- Fioranelli, M.; Bottaccioli, A.G.; Bottaccioli, F.; Bianchi, M.; Rovesti, M.; Roccia, M.G. Stress and Inflammation in Coronary Artery Disease: A Review Psychoneuroendocrineimmunology-Based. Front. Immunol. 2018, 9, 31. [Google Scholar] [CrossRef]
- Simon, T.G.; Trejo, M.E.P.; McClelland, R.; Bradley, R.; Blaha, M.J.; Zeb, I.; Corey, K.E.; Budoff, M.J.; Chung, R.T. Circulating Interleukin-6 is a biomarker for coronary atherosclerosis in nonalcoholic fatty liver disease: Results from the Multi-Ethnic Study of Atherosclerosis. Int. J. Cardiol. 2018, 259, 198–204. [Google Scholar] [CrossRef]
- Caussy, C.; Aubin, A.; Loomba, R. The Relationship Between Type 2 Diabetes, NAFLD, and Cardiovascular Risk. Curr. Diabetes Rep. 2021, 21, 15. [Google Scholar] [CrossRef]
- Miettinen, J.A.; Ylitalo, K.; Hedberg, P.; Jokelainen, J.; Kervinen, K.; Niemelä, M.; Säily, M.; Koistinen, P.; Savolainen, E.R.; Ukkonen, H.; et al. Determinants of functional recovery after myocardial infarction of patients treated with bone marrow-derived stem cells after thrombolytic therapy. Heart Br. Card. Soc. 2010, 96, 362–367. [Google Scholar] [CrossRef]
- Chen, S.; Liu, Z.; Tian, N.; Zhang, J.; Yei, F.; Duan, B.; Zhu, Z.; Lin, S.; Kwan, T.W. Intracoronary transplantation of autologous bone marrow mesenchymal stem cells for ischemic cardiomyopathy due to isolated chronic occluded left anterior descending artery. J. Invasive Cardiol. 2006, 18, 552–556. [Google Scholar]
- Gnecchi, M.; He, H.; Melo, L.G.; Noiseaux, N.; Morello, F.; de Boer, R.A.; Zhang, L.; Pratt, R.E.; Dzau, V.J.; Ingwall, J.S. Early beneficial effects of bone marrow-derived mesenchymal stem cells overexpressing Akt on cardiac metabolism after myocardial infarction. Stem Cells 2009, 27, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikfarjam, S.; Rezaie, J.; Zolbanin, N.M.; Jafari, R. Mesenchymal stem cell derived-exosomes: A modern approach in translational medicine. J. Transl. Med. 2020, 18, 449. [Google Scholar] [CrossRef] [PubMed]
- Haider, K.H.; Aramini, B. Mircrining the injured heart with stem cell-derived exosomes: An emerging strategy of cell-free therapy. Stem Cell Res. Ther. 2020, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Stavely, R.; Nurgali, K. The emerging antioxidant paradigm of mesenchymal stem cell therapy. Stem Cells Transl. Med. 2020, 9, 985–1006. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, R.; Liu, D.; Deng, W.; Xu, G.; Liu, W.; Rong, J.; Long, X.; Ge, J.; Shi, B. Exosomes Derived from miR-214-Enriched Bone Marrow-Derived Mesenchymal Stem Cells Regulate Oxidative Damage in Cardiac Stem Cells by Targeting CaMKII. Oxidative Med. Cell. Longev. 2018, 2018, 4971261. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, Y.; Hu, S.; Chen, Y.; Shen, Z. The therapeutic potential of mesenchymal stem cells for cardiovascular diseases. Cell Death Dis. 2020, 11, 349. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, M.; Deng, S.; Lu, J.; Huang, H.; Zhang, Y.; Gong, P.; Shen, X.; Ruan, H.; Jin, M.; et al. miR-93-5p-Containing Exosomes Treatment Attenuates Acute Myocardial Infarction-Induced Myocardial Damage. Mol. Ther. Nucleic Acids 2018, 11, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Batsali, A.K.; Georgopoulou, A.; Mavroudi, I.; Matheakakis, A.; Pontikoglou, C.G.; Papadaki, H.A. The Role of Bone Marrow Mesenchymal Stem Cell Derived Extracellular Vesicles (MSC-EVs) in Normal and Abnormal Hematopoiesis and Their Therapeutic Potential. J. Clin. Med. 2020, 9, 856. [Google Scholar] [CrossRef] [Green Version]
- Bian, X.; Ma, K.; Zhang, C.; Fu, X. Therapeutic angiogenesis using stem cell-derived extracellular vesicles: An emerging approach for treatment of ischemic diseases. Stem Cell Res. Ther. 2019, 10, 158. [Google Scholar] [CrossRef]
- Leavitt, R.J.; Limoli, C.L.; Baulch, J.E. miRNA-based therapeutic potential of stem cell-derived extracellular vesicles: A safe cell-free treatment to ameliorate radiation-induced brain injury. Int. J. Radiat. Biol. 2019, 95, 427–435. [Google Scholar] [CrossRef]
- Asgarpour, K.; Shojaei, Z.; Amiri, F.; Ai, J.; Mahjoubin-Tehran, M.; Ghasemi, F.; ArefNezhad, R.; Hamblin, M.R.; Mirzaei, H. Exosomal microRNAs derived from mesenchymal stem cells: Cell-to-cell messages. Cell Commun. Signal. 2020, 18, 149. [Google Scholar] [CrossRef]
- Soni, N.; Gupta, S.; Rawat, S.; Krishnakumar, V.; Mohanty, S.; Banerjee, A. MicroRNA-Enriched Exosomes from Different Sources of Mesenchymal Stem Cells Can Differentially Modulate Functions of Immune Cells and Neurogenesis. Biomedicines 2021, 10, 69. [Google Scholar] [CrossRef]
- Ferguson, S.W.; Wang, J.; Lee, C.J.; Liu, M.; Neelamegham, S.; Canty, J.M.; Nguyen, J. The microRNA regulatory landscape of MSC-derived exosomes: A systems view. Sci. Rep. 2018, 8, 1419. [Google Scholar] [CrossRef]
- Gupta, D.; Zickler, A.M.; El Andaloussi, S. Dosing extracellular vesicles. Adv. Drug Deliv. Rev. 2021, 178, 113961. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Primer Sequence | GenBank Accession Number |
|---|---|---|
| MnSOD F: MnSOD R: | 5′-GTGCAGGTAAGTGGCAGGG-3′ 5′-TCGTGGTACTTCTCCTCGGT-3′ | NM_017051.2 |
| UCP2 F: UCP2 R: | 5′-CGTCTGCACTCCTGTGTTCT-3′ 5′-TGTTGAGTGGGGCATTGTGT-3′ | NM_019354.3 |
| PINK1 F: PINK1 R: | 5′-GACCTCAGCCTGCTCTTCTG-3′ 5′-GCTTTGCTGGGACACCTTTG-3′ | NM_001106694.1 |
| Parkin F: Parkin R: | 5′-CCAGGTACAATCTCTCCGCG-3′ 5′-AAGTTGCGATCTCCACCAGG-3′ | NM_020093.1 |
| FUNDC 1 F: FUNDC 1 R: | 5’-ACATTGTGATATCCAGCGGC-3’ 5’-AAGCTTCAGGTGGCAAAGTG-3’ | NM_001025027.1 |
| ULK1 F: ULK1 R: | 5’-GTTGCTGACTCCAAGCCAAA-3’ 5’-ATCTTGGAGGACGAAAGCCA-3’ | NM_001108341.1 |
| BNIP3L F: BNIP3L R: | 5’-TGCCCTATCACAAAGCAGGA-3’ 5’-GGTCCAACAAACGCTTCACT-3’ | NM_080888.2 |
| β-actin F: β-actin R: | 5′-TGTCACCAACTGGGACGATA-3′ 5′-GGGGTGTTGAAGGTCTCAAA-3′ | NM_031144.3 |
| Groups | CK-MB (U/L) | LDH (U/L) | cTnI (ng/mL) |
|---|---|---|---|
| Control | 29.2 ± 0.4 | 39.7 ± 4.4 | 1.42 ± 0.2 |
| Cardiotoxicity of NASH | 40 ± 0.6 a | 64 ± 5.7 a | 2.89 ± 0.09 a |
| BM-MSC-EV | 31.1 ± 0.38 b | 39.5 ± 3.08 b | 1.72 ± 0.3 b |
| BM-MSCs | 32.7 ± 1.9 b | 47.8 ± 7.1 | 1.79 ± 0.3 |
| miRNAs | Signaling Pathways and Targets |
|---|---|
| let-7a-5p, miR-143-3p, miR-125b-5p, miR-27a-3p, miR-29b-3p, miR-16-5p | PI3K/AKT, NF-κB, VEGF Hypoxia |
| miR-34a-5p, miR-199a-3p | PI3K/AKT, VEGF Hypoxia |
| miR-26a-5p, miR-30c-5p, miR-145-5p, miR-23a-3p, miR-10a-5p, miR-214-3p | PI3K/AKT, NF-κB, Hypoxia |
| miR-181a-5p, miR-218-5p | PI3K/AKT, NF-κB, VEGF |
| miR-196a-5p, miR-100-5p | PI3K/AKT, NF-κB |
| miR-221-3p, miR-31-5p | PI3K/AKT, VEGF Hypoxia |
| miR-21-5p | NF-κB, VEGF |
| miR-103-3p, miR-101-3p | PI3K/AKT |
| miR-221-3p | MnSOD |
| miR-16-5p | UCP2 |
| miR-34a-5p, miR-16-5p | VEGFA |
| miR-221-3p | BNIP3L |
| miR-31-5p, miR-199a-5p | HIF-1 |
| miR-21-5p | TNF-α, IL-6 |
| miR-191-5p, miR-221-3p | NF-κB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Derany, M.O.; AbdelHamid, S.G. Bone Marrow Mesenchymal Stem Cells and Their Derived Extracellular Vesicles Attenuate Non-Alcoholic Steatohepatitis-Induced Cardiotoxicity via Modulating Cardiac Mechanisms. Life 2022, 12, 355. https://doi.org/10.3390/life12030355
El-Derany MO, AbdelHamid SG. Bone Marrow Mesenchymal Stem Cells and Their Derived Extracellular Vesicles Attenuate Non-Alcoholic Steatohepatitis-Induced Cardiotoxicity via Modulating Cardiac Mechanisms. Life. 2022; 12(3):355. https://doi.org/10.3390/life12030355
Chicago/Turabian StyleEl-Derany, Marwa O., and Sherihan G. AbdelHamid. 2022. "Bone Marrow Mesenchymal Stem Cells and Their Derived Extracellular Vesicles Attenuate Non-Alcoholic Steatohepatitis-Induced Cardiotoxicity via Modulating Cardiac Mechanisms" Life 12, no. 3: 355. https://doi.org/10.3390/life12030355