
Pathogenesis of Two Faces of DVT: New Identity of Venous Thromboembolism as Combined Micro-Macrothrombosis via Unifying Mechanism Based on “Two-Path Unifying Theory” of Hemostasis and “Two-Activation Theory of the Endothelium”


Abstract
:Simple Summary
Abstract
1. Introduction
2. Distal DVT vs. Proximal/Central DVT
3. Understanding of Thrombosis Phenotypes
- Macrothrombosis developing in DVT, PTE, VTE, CVST, SVT, IVCT, SVCT and Budd–Chiari syndrome, portal vein thrombosis, acute ischemic stroke, acute myocardial infarction, aortic thrombus and renal vein thrombosis, etc.
- Microthrombosis developing in TTP, TTP-like syndrome, ARDS, diffuse encephalopathic stroke, hemolytic-uremic syndrome, transient ischemic attack (TIA), microaneurysm and thrombosis of the retinal artery, multiorgan dysfunction syndrome (MODS), hepatic veno-occlusive disease (VOD) and non-occlusive mesenteric ischemia (NOMI)
- Heparin-induced thrombocytopenia with “white clot syndrome” (HIT with WCS)
- Fibrin clot disease occurring in acute promyelocytic leukemia (APL) and old term acute “disseminated intravascular coagulation (DIC)” occurring in EA-VMTD with hepatic coagulopathy
- Hematoma and hemarthrosis within the tissue or cavitary area
- Concurrent microthrombosis and macrothrombosis in both arterial and venous systems in paroxysmal nocturnal hemoglobinuria (PNH)
4. Physiological Mechanisms Involved in the Genesis of Thrombosis
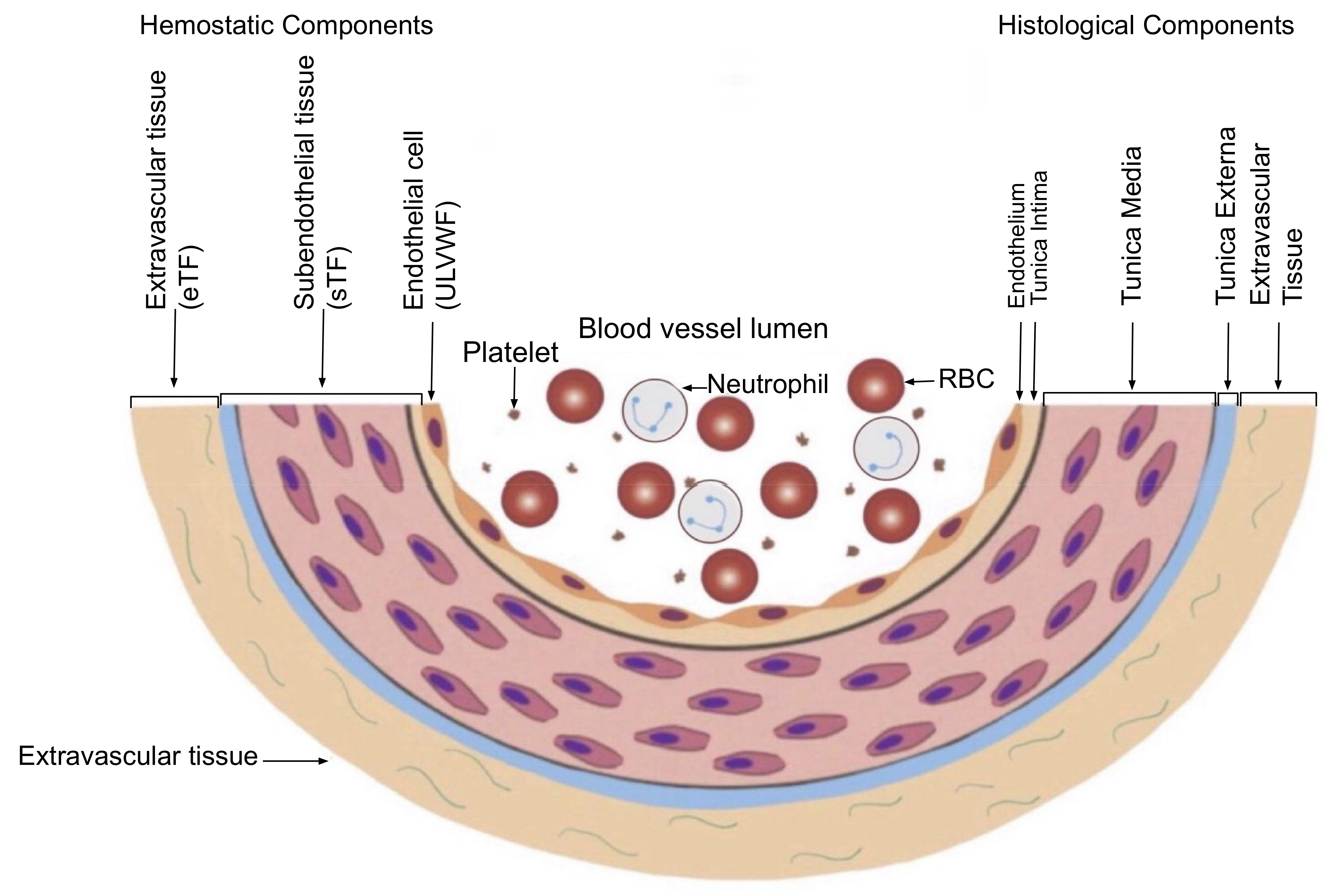
4.1. Vascular Wall Physiology in Venous Thrombosis
4.2. Hemostasis Leading to Microthrombosis and Macrothrombosis
4.3. Dissimilarity of VTE from Distal DVT
4.4. Pathogenesis of Combined Micro-Macrothrombosis Syndromes
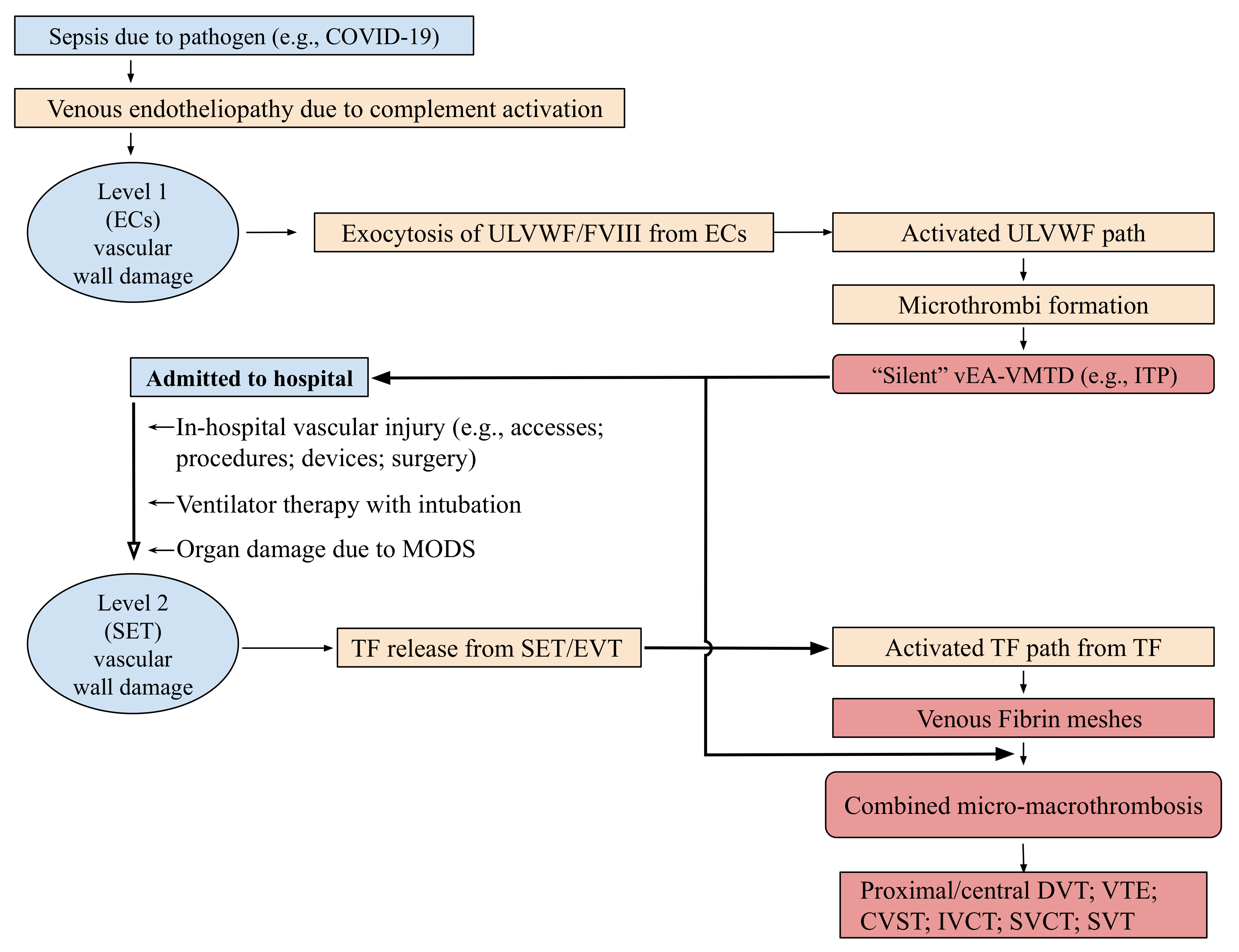
5. Additional Lessons Learned from COVID-19 Associated Thrombotic Syndromes
5.1. Role of In-Hospital and ICU Vascular Injury on the Pathogenesis of VTE
5.2. New Identity of Acute ITP as ITP-like Syndrome Triggered by Endotheliopathy
6. Consideration for the Diagnosis, Prevention and Treatment of Venous Combined Micro-Macrothrombosis
- Thrombocytopenia
- Increased activity of FVIII
- Overexpression of ULVWF/VWF/VWF antigen
- Markedly increased D-dimer level
- Decreased ADAMTS13 activity
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palareti, G.; Schellong, S. Isolated distal deep vein thrombosis: What we know and what we are doing. J. Thromb. Haemost. 2011, 10, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Galanaud, J.P.; Quenet, S.; Rivron-Guillot, K.; Quere, I.; Muã‘Oz-Torrero, J.F.S.; Tolosa, C.; Monreal, M.; The Riete Investigators. Comparison of the clinical history of symptomatic isolated distal deep-vein thrombosis vs. proximal deep vein thrombosis in 11 086 patients. J. Thromb. Haemost. 2009, 7, 2028–2034. [Google Scholar] [CrossRef]
- Chang, J.C. Sepsis and septic shock: Endothelial molecular pathogenesis associated with vascular microthrombotic disease. Thromb. J. 2019, 17, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.C. COVID-19 Sepsis: Pathogenesis and Endothelial Molecular Mechanisms Based on “Two-Path Unifying Theory” of Hemostasis and Endotheliopathy-Associated Vascular Microthrombotic Disease, and Proposed Therapeutic Approach with Antimicrothrombotic Therapy. Vasc. Heal. Risk Manag. 2021, 17, 273–298. [Google Scholar] [CrossRef]
- Chang, J.C. TTP-like syndrome: Novel concept and molecular pathogenesis of endotheliopathy-associated vascular microthrombotic disease. Thromb. J. 2018, 16, 20. [Google Scholar] [CrossRef] [Green Version]
- Shankar, A.; Varadan, B.; Ethiraj, D.; Sudarsanam, H.; Hakeem, A.R.; Kalyanasundaram, S. Systemic arterio-venous thrombosis in COVID-19: A pictorial review. World J. Radiol. 2020, 13, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Boonyawat, K.; Chantrathammachart, P.; Numthavej, P.; Nanthatanti, N.; Phusanti, S.; Phuphuakrat, A.; Niparuck, P.; Angchaisuksiri, P. Incidence of thromboembolism in patients with COVID-19: A systematic review and meta-analysis. Thrombosis. J. 2020, 18, 34. [Google Scholar] [CrossRef]
- Merrill, J.T.; Erkan, D.; Winakur, J.; James, J.A. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat. Rev. Rheumatol. 2020, 16, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, D.; Sperhake, J.P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann. Intern Med. 2020, 173, 268–277. [Google Scholar] [CrossRef]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.-D.; Sacco, C.; Bertuzzi, A.; et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef]
- Chang, J.C. Acute Respiratory Distress Syndrome as an Organ Phenotype of Vascular Microthrombotic Disease: Based on Hemostatic Theory and Endothelial Molecular Pathogenesis. Clin. Appl. Thromb. 2019, 25, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakay, K.; Cooper, J.; Bloomfield, J.; Overby, P.; Mayer, S.A.; Nuoman, R.; Sahni, R.; Gulko, E.; Kaur, G.; Santarelli, J.; et al. Cerebral Venous Sinus Thrombosis in COVID-19 Infection: A Case Series and Review of The Literature. J. Stroke Cerebrovasc. Dis. 2021, 30, 105434. [Google Scholar] [CrossRef]
- Singh, B.; Kaur, P.; Maroules, M. Splanchnic vein thrombosis in COVID-19: A review of literature. Dig. Liver Dis. 2020, 52, 1407–1409. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.A.; Alsaleh, M.E.; Alsaleh, M.E.; Al Zaher, F.A.; Almajed, F.A.; Alkhudhair, A.M.; Alali, M.M.; Alzayer, H.A.; Alolayan, A.J. Budd-Chiari Syndrome: A Case Report of a Rare Presentation of COVID-19. Cureus 2021, 13, e12554. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, J.; Kaur, J.; Randhawa, H.S.; Kaur, S.; Singh, H. Thrombosis of the Portal Vein and Superior Mesenteric Vein in a Patient with Subclinical COVID-19 Infection. Cureus 2021, 13, e14366. [Google Scholar] [CrossRef]
- Chang, J.C. Thrombogenesis and thrombotic disorders based on ’two-path unifying theory of hemostasis’: Philosophical, physiological, and phenotypical interpretation. Blood Coagul Fibrinolysis 2018, 29, 585–595. [Google Scholar] [CrossRef]
- Chang, J.C. Stroke Classification: Critical Role of Unusually Large von Willebrand Factor Multimers and Tissue Factor on Clinical Phenotypes Based on Novel “Two-Path Unifying Theory” of Hemostasis. Clin. Appl. Thromb. 2020, 26, 1–23. [Google Scholar] [CrossRef]
- Chang, J.C.; Hawley, H.B. Vaccine-Associated Thrombocytopenia and Thrombosis: Venous Endotheliopathy Leading to Venous Combined Micro-Macrothrombosis. Medicina 2021, 57, 1163. [Google Scholar] [CrossRef]
- Robert-Ebadi, H.; Righini, M. Management of distal deep vein thrombosis. Thromb. Res. 2017, 149, 48–55. [Google Scholar] [CrossRef]
- Singer, A.J.; Zheng, H.; Francis, S.; Fermann, G.J.; Chang, A.M.; Parry, B.A.; Giordano, N.; Kabrhel, C. D-dimer levels in VTE patients with distal and proximal clots. Am. J. Emerg. Med. 2019, 37, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Debourdeau, P.; Farge, D.; Beckers, M.; Baglin, C.; Bauersachs, R.M.; Brenner, B.; Brilhante, D.; Falanga, A.; Gerotzafias, G.T.; Haim, N.; et al. International clinical practice guidelines for the treatment and prophylaxis of thrombosis associated with central venous catheters in patients with cancer. J. Thromb. Haemost. 2012, 11, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadam, S.; Popuri, A.; Hattab, Y.; Cheema, T. Controversies in Venous Thromboembolism. Crit. Care Nurs. Q. 2017, 40, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Kabashneh, S.; Singh, V.; Alkassis, S. A Comprehensive Literature Review on the Management of Distal Deep Vein Thrombosis. Cureus 2020, 12, e8048. [Google Scholar] [CrossRef] [PubMed]
- CDC. Hospitalization: A Major Risk Factor for Dangerous Blood Clots. Available online: https://www.cdc.gov/ncbddd/dvt/documents/hospitalization-infographic.pdf (accessed on 19 December 2021).
- National Blood Clot Alliance. DVT in the Hospital Setting. Available online: https://www.stoptheclot.org/learn_more/awareness_h/quick_facts_hosptial/ (accessed on 19 December 2021).
- Middeldorp, S.; Coppens, M.; Van Haaps, T.F.; Foppen, M.; Vlaar, A.P.; Müller, M.C.A.; Bouman, C.C.S.; Beenen, L.F.M.; Kootte, R.S.; Heijmans, J.; et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1995–2002. [Google Scholar] [CrossRef] [PubMed]
- Boddi, M.; Peris, A. Deep Vein Thrombosis in Intensive Care. Adv. Exp. Med. Biol. 2017, 906, 167–181. [Google Scholar] [PubMed]
- Minet, C.; Potton, L.; Bonadona, A.; Hamidfar-Roy, R.; Somohano, C.A.; Lugosi, M.; Cartier, J.-C.; Ferretti, G.; Schwebel, C.; Timsit, J.-F. Venous thromboembolism in the ICU: Main characteristics, diagnosis and thromboprophylaxis. Crit. Care 2015, 19, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Amin, A.N.; Varker, H.; Princic, N.; Lin, J.; Thompson, S.; Johnston, S. Duration of venous thromboembolism risk across a continuum in medically ill hospitalized patients. J. Hosp. Med. 2011, 7, 231–238. [Google Scholar] [CrossRef]
- Chang, J.C. Hemostasis based on a novel ‘two-path unifying theory’ and classification of hemostatic disorders. Blood Coagul. Fibrinolysis 2018, 29, 573–584. [Google Scholar] [CrossRef]
- Chang, J.C. Thrombocytopenia in critically ill patients due to vascular microthrombotic disease: Pathogenesis based on “two activation theory of the endothelium. Vasc. Dis. Ther. 2017, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Banerjee, M. Immune Thrombocytopenia Secondary to COVID-19: A Systematic Review. SN Compr. Clin. Med. 2020, 2, 2048–2058. [Google Scholar] [CrossRef]
- Rodeghiero, F. Is ITP a thrombophilic disorder? Am. J. Hematol. 2016, 91, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkata, C.; Kashyap, R.; Farmer, J.C.; Afessa, B. Thrombocytopenia in adult patients with sepsis: Incidence, risk factors, and its association with clinical outcome. J. Intensiv. Care 2013, 1, 9. [Google Scholar] [CrossRef]
- Agrawal, P.; Nawadkar, R.; Ojha, H.; Kumar, J.; Sahu, A. Complement Evasion Strategies of Viruses: An Overview. Front. Microbiol. 2017, 8, 1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, R.; Hamilton, K.K.; McEver, R.P.; Sims, P.J. Complement Proteins C5b-9 Induce Secretion of High Molecular Weight Multimers of Endothelial von Willebrand Factor and Translocation of Granule Membrane Protein GMP-140 to the Cell Surface. J. Biol. Chem. 1989, 264, 9053–9060. [Google Scholar] [CrossRef]
- Kudaravalli, P.; Pendela, V.S.; Gambhir, H.S.S. Acute on Chronic Immune Thrombocytopenia as a Cause of Thrombocytopenia in Sepsis. Cureus 2020, 12, e8168. [Google Scholar] [CrossRef]
- Zarychanski, R.; Houston, D.S. Assessing thrombocytopenia in the intensive care unit: The past, present, and future. Hematol. Am Soc Hematol Educ Program. 2017, 2017, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Luo, L.; Hu, Y. Thrombocytopenia and thrombosis in hospitalized patients with COVID-19. J. Hematol. Oncol. 2020, 13, 1–3. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, T.A.; Papatheou, D.; Melita, H. COVID-19 Infection: Viral Macro- and Micro-Vascular Coagulopathy and Thromboembolism/Prophylactic and Therapeutic Management. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 12–24. [Google Scholar] [CrossRef]
- Castro, R.A.; Frishman, W.H. Thrombotic Complications of COVID-19 Infection: A Review. Cardiol. Rev. 2021, 29, 43–47. [Google Scholar] [CrossRef]
- Rahi, M.S.; Jindal, V.; Reyes, S.-P.; Gunasekaran, K.; Gupta, R.; Jaiyesimi, I. Hematologic disorders associated with COVID-19: A review. Ann. Hematol. 2021, 100, 309–320. [Google Scholar] [CrossRef]
- Monreal, M.; Lafoz, E.; Casals, A.; Ruíz, J.; Arias, A. Platelet count and venous thromboembolism. A useful test for suspected pulmonary embolism. Chest 1991, 100, 1493–1496. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Lou, W.; He, X.; Liu, C.; Gu, J. The management of filter-related caval thrombosis complicated by heparin-induced thrombocytopenia and thrombosis. Int. J. Clin. Exp. Med. 2015, 8, 13078–13088. [Google Scholar] [PubMed]
- Baelum, J.K.; Moe, E.E.; Nybo, M.; Vinholt, P.J. Venous Thromboembolism in Patients with Thrombocytopenia: Risk Factors, Treatment, and Outcome. Clin. Appl. Thromb. 2015, 23, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Pagliari, M.T.; Boscarino, M.; Cairo, A.; Mancini, I.; Martinelli, I.; Bucciarelli, P.; Rossi, F.; Rosendaal, F.R.; Peyvandi, F. ADAMTS13 activity, high VWF and FVIII levels in the pathogenesis of deep vein thrombosis. Thromb. Res. 2021, 197, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Bittar, L.F.; de Paula, E.V.; Mello, T.B.T.; Siqueira, L.H.; Orsi, F.L.A.; Annichino-Bizzacchi, J.M. Polymorphisms and Mutations in vWF and ADAMTS13 Genes and Their Correlation with Plasma Levels of FVIII and vWF in Patients With Deep Venous Thrombosis. Clin. Appl. Thromb. 2011, 17, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Shahsavarzadeh, T.; Javanmard, S.; Saadatnia, M. Impact of Factor VIII and von Willebrand Factor Plasma Levels on Cerebral Venous and Sinus Thrombosis: Are They Independent Risk Factors? Eur. Neurol. 2011, 66, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Bugnicourt, J.-M.; Roussel, B.; Tramier, B.; Lamy, C.; Godefroy, O. Cerebral venous thrombosis and plasma concentrations of factor VIII and von Willebrand factor: A case control study. J. Neurol. Neurosurg. Psychiatry 2006, 78, 699–701. [Google Scholar] [CrossRef] [Green Version]
- Delrue, M.; Siguret, V.; Neuwirth, M.; Joly, B.; Beranger, N.; Sène, D.; Chousterman, B.G.; Voicu, S.; Bonnin, P.; Mégarbane, B.; et al. von Willebrand factor/ADAMTS13 axis and venous thromboembolism in moderate-to-severe COVID-19 patients. Br. J. Haematol. 2021, 192, 1097–1100. [Google Scholar] [CrossRef]
- Jennersjö, C.M.; Fagerberg, I.H.; Karlander, S.G.; Lindahl, T.L. Normal D-dimer concentration is a common finding in symptomatic outpatients with distal deep vein thrombosis. Blood Coagul. Fibrinolysis 2005, 16, 517–523. [Google Scholar] [CrossRef]
- Kearon, C.; Kahn, S.R. Long-term treatment of venous thromboembolism. Blood 2020, 135, 317–325. [Google Scholar] [CrossRef]
- Sartori, M.; Cosmi, B.; Legnani, C.; Favaretto, E.; Valdrè, L.; Guazzaloca, G.; Rodorigo, G.; Cini, M.; Palareti, G. The Wells rule and D-dimer for the diagnosis of isolated distal deep vein thrombosis. J. Thromb. Haemost. 2012, 10, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Natorska, J.; Celińska-Lowenhoff, M.; Undas, A.I. High prevalence of antinuclear antibodies in patients following venous thromboembolism. Adv. Clin. Exp. Med. 2018, 27, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Andrianova, I.A.; Ponomareva, A.A.; Mordakhanova, E.R.; Le Minh, G.; Daminova, A.G.; Nevzorova, T.A.; Rauova, L.; Litvinov, R.I.; Weisel, J.W. In systemic lupus erythematosus anti-dsDNA antibodies can promote thrombosis through direct platelet activation. J. Autoimmun. 2020, 107, 102355. [Google Scholar] [CrossRef]
- Farmer-Boatwright, M.K.; Roubey, R.A. Venous Thrombosis in the Antiphospholipid Syndrome. Arter. Thromb. Vasc. Biol. 2009, 29, 321–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karahan, S.; Erol, K.; Yuksel, R.C.; Artan, C.; Celik, I. Antiphospholipid antibodies in COVID-19-associated pneumonia patients in intensive care unit. Mod. Rheumatol. 2021, 32, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Roumen-Klappe, E.M.; den Heijer, M.; van Uum, S.H.; van der Ven-Jongekrijg, J.; van der Graaf, F.; Wollersheim, H. Inflammatory response in the acute phase of deep vein thrombosis. J. Vasc. Surg. 2002, 35, 701–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Garza, E.; Jerjes-Sanchez, C.; Navarrete, A.; Joya-Harrison, J.; Rodriguez, D. Venous thromboembolism: Thrombosis, inflammation, and immunothrombosis for clinicians. J. Thromb. Thrombolysis 2017, 44, 377–385. [Google Scholar] [CrossRef]
- Jezovnik, M.K.; Poredos, P. Idiopathic venous thrombosis is related to systemic inflammatory response and to increased levels of circulating markers of endothelial dysfunction. Int. Angiol. 2010, 29, 226–231. [Google Scholar]
- Chang, J.C. Viral hemorrhagic fevers due to endotheliopathy-associated disseminated intravascular microthrombosis and hepatic coagulopathy: Pathogenesis based on “two activation theory of the endothelium. Clin. Microbiol. Infect. Dis. 2017, 2, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Garabet, L.; Henriksson, C.E.; Lozano, M.L.; Ghanima, W.; Bussel, J.; Brodin, E.; Fernández-Pérez, M.P.; Martínez, C.; González-Conejero, R.; Mowinckel, M.-C.; et al. Markers of endothelial cell activation and neutrophil extracellular traps are elevated in immune thrombocytopenia but are not enhanced by thrombopoietin receptor agonists. Thromb. Res. 2020, 185, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Al-Mufti, F.; Amuluru, K.; Sahni, R.; Bekelis, K.; Karimi, R.; Ogulnick, J.; Cooper, J.; Overby, P.; Nuoman, R.; Tiwari, A.; et al. Cerebral Venous Thrombosis in COVID-19: A New York Metropolitan Cohort Study. Am. J. Neuroradiol. 2021, 42, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- McAree, B.J.; O’Donnell, M.E.; Fitzmaurice, G.J.; Reid, J.A.; Spence, R.A.J.; Lee, B. Inferior vena cava thrombosis: A review of current practice. Vasc. Med. 2013, 18, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.A.L.; Júnior, J.E.; Miranda, C.H. Atypical COVID–19 presentation with Budd-Chiari syndrome leading to an outbreak in the emergency department. Am. J. Emerg. Med. 2021, 46, 800.e5–800.e7. [Google Scholar] [CrossRef]
- Novara, E.; Molinaro, E.; Benedetti, I.; Bonometti, R.; Lauritano, E.C.; Boverio, R. Severe acute dried gangrene in COVID-19 infection: A case report. Eur. Rev. Med Pharmacol. Sci. 2020, 24, 5769–5771. [Google Scholar] [PubMed]
- Wang, J.S.; Pasieka, H.B.; Petronic-Rosic, V.; Sharif-Askary, B.; Evans, K.K. Digital Gangrene as a Sign of Catastrophic Coronavirus Disease 2019-related Microangiopathy. Plast. Reconstr. Surg. Glob. Open 2020, 8, e3025. [Google Scholar] [CrossRef] [PubMed]
- Martino, G.P.; Bitti, G. COVID Fingers: Another Severe Vascular Manifestation. Eur. J. Vasc. Endovasc. Surg. 2020, 61, 97. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.-Z.; Pan, J.-Y. COVID-19 With Limb Ischemic Necrosis. J. Cardiothorac. Vasc. Anesthesia 2020, 34, 2846–2847. [Google Scholar] [CrossRef] [PubMed]
- Gheisari, M.; Baghani, M.; Ganji, R.; Forouzanfar, M. Huge carbuncle leading to necrotizing fasciitis in the COVID-19 pandemic era. Clin. Case Rep. 2021, 9, 1583–1586. [Google Scholar] [CrossRef]
- Alonso, M.N.; Mata-Forte, T.; García-León, N.; Vullo, P.A.; Ramírez-Olivencia, G.; Estébanez, M.; Álvarez-Marcos, F. Incidence, Characteristics, Laboratory Findings and Outcomes in Acro-Ischemia in COVID-19 Patients. Vasc. Health Risk Manag. 2020, 16, 467–478. [Google Scholar] [CrossRef]
- European Medicines Agency. Signal Assessment Report on Embolic and Thrombotic Events (SMQ) with COVID-19 Vaccine (ChAdOx1-S [recombinant])—COVID-19 Vaccine AstraZeneca (Other Viral Vaccines). 2021. Available online: https://www.ema.europa.eu/en/documents/pracrecommendation/signal-assessment-report- (accessed on 19 December 2021).
- See, I.; Su, J.R.; Lale, A.; Woo, E.J.; Guh, A.Y.; Shimabukuro, T.T.; Streiff, M.B.; Rao, A.K.; Wheeler, A.P.; Beavers, S.F.; et al. US Case Reports of Cerebral Venous Sinus Thrombosis with Thrombocytopenia After Ad26.COV2.S Vaccination. JAMA 2021, 325, 2448–2456. [Google Scholar] [CrossRef]
- Saadatnia, M.; Fatehi, F.; Basiri, K.; Mousavi, S.A.; Mehr, G.K. Cerebral Venous Sinus Thrombosis Risk Factors. Int. J. Stroke 2009, 4, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Giladi, O.; Steinberg, D.M.; Peleg, K.; Tanne, D.; Givon, A.; Grossman, E.; Klein, Y.; Avigdori, S.; Greenberg, G.; Katz, R.; et al. Head trauma is the major risk factor for cerebral sinus-vein thrombosis. Thromb. Res. 2016, 137, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Matsushige, T.; Nakaoka, M.; Kiya, K.; Takeda, T.; Kurisu, K. Cerebral Sinovenous Thrombosis After Closed Head Injury. J. Trauma Inj. Infect. Crit. Care 2009, 66, 1599–1604. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-M. ADAMTS13 and microvascular thrombosis. Expert Rev. Cardiovasc. Ther. 2006, 4, 813–825. [Google Scholar] [CrossRef]
- Calabrò, P.; Gragnano, F.; Golia, E.; Grove, E.L. von Willebrand Factor and Venous Thromboembolism: Pathogenic Link and Therapeutic Implications. Semin. Thromb. Hemost. 2017, 44, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, B.; Tombak, A.; Serin, M.S.; Tiftik, N. Change in plasma a disintegrin and metalloprotease with thrombospondin type-1 repeats-13 and von Willebrand factor levels in venous thromboembolic patients. Hematology 2016, 21, 295–299. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, C.J.; Wooster, L.; Sigurslid, H.H.; Li, R.H.; Jiang, W.; Tian, W.; Cardenas, C.L.L.; Malhotra, R. Estimating risk of mechanical ventilation and in-hospital mortality among adult COVID-19 patients admitted to Mass General Brigham: The VICE and DICE scores. EClinicalMedicine 2021, 33, 100765. [Google Scholar] [CrossRef]
- Divatia, J.V.; Myatra, S.; Khan, P.U. Tracheal intubation in the ICU: Life saving or life threatening? Indian J. Anaesth. 2011, 55, 470–475. [Google Scholar] [CrossRef]
- McGuinness, G.; Zhan, C.; Rosenberg, N.; Azour, L.; Wickstrom, M.; Mason, D.M.; Thomas, K.M.; Moore, W.H. Increased Incidence of Barotrauma in Patients with COVID-19 on Invasive Mechanical Ventilation. Radiology 2020, 297, E252–E262. [Google Scholar] [CrossRef]
- Guglielmetti, G.; Quaglia, M.; Sainaghi, P.P.; Castello, L.M.; Vaschetto, R.; Pirisi, M.; Corte, F.D.; Avanzi, G.C.; Stratta, P.; Cantaluppi, V. “War to the knife” against thromboinflammation to protect endothelial function of COVID-19 patients. Crit. Care 2020, 24, 1–4. [Google Scholar] [CrossRef]
- Lefrancais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.; David, T.; Coughlin, T.D.S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Kroll, M.H.; Kharghan, V.A. Platelets in Pulmonary Vascular Physiology and Pathology. Pulm. Circ. 2012, 2, 291–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.; Charytan, D.; Gasmi, B.; et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef]
- Zufferey, A.; Kapur, R.; Semple, J.W. Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP). J. Clin. Med. 2017, 6, 16. [Google Scholar] [CrossRef]
- Audia, S.; Mahévas, M.; Samson, M.; Godeau, B.; Bonnotte, B. Pathogenesis of immune thrombocytopenia. Autoimmun. Rev. 2017, 16, 620–632. [Google Scholar] [CrossRef] [PubMed]
- LeVine, D.N.; Brooks, M.B. Immune thrombocytopenia (ITP): Pathophysiology update and diagnostic dilemmas. Vet. Clin. Pathol. 2019, 48, 17–28. [Google Scholar]
- Johnsen, J. Pathogenesis in immune thrombocytopenia: New insights. Hematol. Am. Soc. Hematol. Educ. Program. 2012, 2012, 306–312. [Google Scholar] [CrossRef]
- Rasheed, M.; Soliman, M.A.T.; Yassin, M.M.A. Thrombosis in Patients with Immune Thrombocytopenia, Review of Literature. Blood 2020, 136, 9–10. [Google Scholar] [CrossRef]
- Rodeghiero, F. ITP and thrombosis: An intriguing association. Blood Adv. 2017, 1, 2280. [Google Scholar] [CrossRef]
- Moulis, G.; Audemard-Verger, A.; Arnaud, L.; Luxembourger, C.; Montastruc, F.; Gaman, A.M.; Svenungsson, E.; Ruggeri, M.; Mahévas, M.; Gerfaud-Valentin, M.; et al. Risk of thrombosis in patients with primary immune thrombocytopenia and antiphospholipid antibodies: A systematic review and meta-analysis. Autoimmun. Rev. 2016, 15, 203–209. [Google Scholar] [CrossRef]
- Schultz, N.H.; Sørvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.-H.; Skattør, T.H.; Tjønnfjord, G.E.; et al. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.E.; Luz, B.; Niehaus, L.; Bhogal, P.; Bäzner, H.; Henkes, H. Thrombocytopenia and Intracranial Venous Sinus Thrombosis after “COVID-19 Vaccine AstraZeneca” Exposure. J. Clin. Med. 2021, 10, 1599. [Google Scholar] [CrossRef]
- Portuguese, A.J.; Sunga, C.; Kruse-Jarres, R.; Gernsheimer, T.; Abkowitz, J. Autoimmune- and complement-mediated hematologic condition recrudescence following SARS-CoV-2 vaccination. Blood Adv. 2021, 5, 2794–2798. [Google Scholar] [CrossRef] [PubMed]
- Casonato, A.; Fabris, F.; Boscaro, M.; Girolami, A. Increased factor VIII/vWf levels in patients with reduced platelet number. Ann. Hematol. 1987, 54, 281–288. [Google Scholar] [CrossRef]
- Moore, J.C.; Hayward, C.; Warkentin, T.E.; Kelton, J.G. Decreased von Willebrand factor protease activity associated with thrombocytopenic disorders. Blood 2001, 98, 1842–1846. [Google Scholar] [CrossRef] [Green Version]
- Pluta, J.; Trzebicki, J. Thrombocytopenia: The most frequent haemostatic disorder in the ICU. Anestezjol. Intensywna Ter. 2019, 51, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, A.B.; Rygård, S.L.; Russell, L.; Perner, A.; Møller, M.H. Bleeding and thrombosis in intensive care patients with thrombocytopenia-Protocol for a topical systematic review. Acta Anaesthesiol. Scand. 2019, 63, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, S.; Holmes, A.; Loscalzo, J. Platelets and cardiovascular disease. Eur. J. Cardiovasc. Nurs. 2002, 1, 273–288. [Google Scholar] [CrossRef]
- Sharma, S.P.; Chaudhary, R.; Gupta, P.; Kondur, S.; Gatla, N.; Blaceri, S.; Choksi, N.; Kassab, E.; Sareen, N.; Kondur, A. Baseline thrombocytopenia in women with coronary heart disease increases incident acute coronary syndrome: Insights from national inpatient database. J. Thromb. Thrombolysis 2020, 50, 462–467. [Google Scholar] [CrossRef]
- Chandan, J.S.; Thomas, T.; Lee, S.; Marshall, T.; Willis, B.; Nirantharakumar, K.; Gill, P. The association between idiopathic thrombocytopenic purpura and cardiovascular disease: A retrospective cohort study. J. Thromb. Haemost. 2018, 16, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-K.; Lee, S.-H. Ischemic stroke associated with immune thrombocytopenia: Lesion patterns and characteristics. Neurol. Sci. 2014, 35, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Kirkilesis, G.; Kakkos, S.K.; Bicknell, C.; Salim, S.; Kakavia, K. Treatment of distal deep vein thrombosis. Cochrane Database Syst. Rev. 2019, 4, CD013422. [Google Scholar] [CrossRef]
- Palareti, G. How I treat isolated distal deep vein thrombosis (IDDVT). Blood 2014, 123, 1802–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squires, J.E. Indications for platelet transfusion in patients with thrombocytopenia. High Speed Blood and Transfusion Equipment 2015, 13, 221–226. [Google Scholar] [CrossRef]
- Goel, R.; Ness, P.M.; Takemoto, C.M.; Krishnamurti, L.; King, K.E.; Tobian, A.A.R. Platelet transfusions in platelet consumptive disorders are associated with arterial thrombosis and in-hospital mortality. Blood 2015, 125, 1470–1476. [Google Scholar] [CrossRef] [Green Version]
- Wiernek, S.L.; Jiang, B.; Gustafson, G.M.; Dai, X. Cardiac implications of thrombotic thrombocytopenic purpura. World J. Cardiol. 2018, 10, 254–266. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Examples of Phenotypes | Distal DVT (De Novo Venous Macrothrombosis) | Proximal/Central DVT (Combined Venous Micro-Macrothrombosis) |
|---|---|---|
| Disease Examples | ||
| Distal DVT | ||
| Solitary DVT | ||
| Complex DVT | ||
| Proximal/central DVT | ||
| VTE/PTE | ||
| CVST | ||
| IVCT/SVCT | ||
| Budd–Chiari syndrome | ||
| Splanchnic vein thrombosis | ||
| Clinical Features Example in DVT | ||
| Size | Small | Large and extended |
| Shape | Simple | Irregular |
| Multiplicity/character of thrombosis | Single, and isolated | Multiple, and connected |
| Embolic complication | Absent or unusual | Common |
| Prognosis | Excellent | Very unfavorable |
| Mechanisms of Vascular Injury | ||
| Event/Underlying disease | Local trauma (rarely with surgery/vascular access) | Underlying vEA-VMTD (e.g., sepsis) + local trauma (commonly with surgery/vascular access) |
| Extent of vascular damage | Local ECs/SET injury | Disseminated ECs injury + local/regional ECs/SET injury |
| 1—Genesis of Primary Phenotypes of Thrombosis |
|---|
| (1) The phenotype is determined by the functional character of the injured vessel (i.e., vascular pressure and oxygen/CO2 carrying function: arterial or venous vasculature) (2) The phenotype is determined by the location of the involved vessel (i.e., involved organ or tissue: brain, lungs, heart, liver, nerve, muscle etc.) (3) The phenotype is determined by the size of the vasculature (i.e., microvasculature and larger vessel: capillary/arteriole, venule, artery, vein) (4) The phenotype is determined by the depth of vascular wall injury (i.e., ECs, SET, and/or EVT) |
| 2—Genesis of Secondary Phenotype of Thrombosis |
| (1) The secondary phenotype is influenced by the extent of vascular tree involvement (e.g., localized injury vs. disseminated injury: e.g., sepsis vs. local trauma) (2) The secondary phenotype is influenced by the underlying genetic disorder (e.g., thrombophilia: e.g., ADAMTS13 deficiency, protein C deficiency, FV-Leiden) (3) The secondary phenotype is influenced by the iatrogenic vascular injury (e.g., hospitalization related vascular injury such as surgery, and vascular device/access) (4) The secondary phenotype is influenced by inappropriate treatment (e.g., platelet transfusion [e.g., for ITP], or recombinant FVIIa treatment causing fibrin clot disease) |
| 3—Basic and Combined Phenotypes of Thrombosis |
| (1) Microthrombosis, macrothrombosis, fibrin clot disease, and hematoma a (e.g., EA-VMTD, TTP-like syndrome, MODS, AT, ITP, isolated DVT, isolated PT, APL, hemarthrosis a) (2) Multiple combined micro-macrothrombosis (i.e., micro-macrothrombosis with gangrene: e.g., SPG, PDIS, ANF, acrocyanosis, Fournier’s disease; micro-macrothrombosis with VCCS: e.g., VTE, PTE, CVST) (3) Complex phenotypes with underlying VMTD and additional localized vascular injury and genetic thrombophilia (e.g., various phenotypes of “DIC” with microthrombosis and hepatic coagulopathy with/without thrombophilia, Kawasaki disease with inflammation, purpura fulminans) |
| Phenotypes | Distal DVT (De Novo Venous Macrothrombosis) | Proximal/Central DVT (Combined Venous Micro-Macrothrombosis) |
|---|---|---|
| Disease Examples | ||
| Venous thrombosis | ||
| DVT | Distal DVT | |
| VTE | Proximal/central, multiple and/or large/long DVT | |
| Multiple VTE; PTE | ||
| IVCT; SVT; PVT; BCS; SVCT; CVST | ||
| Other complex venous thrombosis | ||
| Mechanisms of Vascular Injury | ||
| Events | Local trauma (rarely with surgery/vascular access) | Underlying disease (vEA-VMTD [e.g., sepsis]) + local trauma (commonly with surgery/vascular access) |
| Extent of vascular damage | Local ECs/SET injury | Disseminated ECs injury + local/regional ECs/SET injury |
| Pathogenesis | ||
| Activated thrombosis path | ULVWF and TF paths from local trauma | ULVWF path from vEA-VMTD and TF path from regional trauma |
| Thrombi character | Macrothrombus | Combined “microthrombi strings–fibrin meshes” |
| Severity | Typically, solitary and self-limited | Serious and often with multiple/large thrombi |
| Severe inflammation | Absent | May be present and can be severe |
| Venous disseminated EA-VMTD | Absent | Commonly present (e.g., ITP-like syndrome) |
| MOIS | Absent | Commonly present |
| Diagnostic Findings/Markers | ||
| ITP-like syndrome | Does not occur | Sometimes occurs |
| ULVWF/VWF/VWF antigen | Normal | Overexpressed |
| FVIII activity | Normal | Increased |
| ADAMTS13 activity | Normal | Mild to moderately decreased |
| D-dimer | Normal | Markedly increased |
| Immune: ANA; APLA; Anti- | Negative | May be positive |
| dsDNA; Anti-PF4A | ||
| Therapeutic Approach per Theory | No treatment or short-term anticoagulant | Anticoagulant and antimicrothrombotic agent (?) |
| Clinical Phenotype | Arterial Endotheliopathy | Venous Endothelipathy |
|---|---|---|
| aEA-VMTD | vEA-VMTD | |
| Underlying pathology | Efferent circulation from the heart (oxygen delivery) | Afferent circulation into the heart and lungs (CO2 disposal) |
| Physiological/hemodynamic difference | High pressure flow | Low pressure flow |
| High shear stress | Low shear stress | |
| Capillary and arteriolar microvascular event | Venous and pulmonary microvascular event | |
| Primary cause | ||
| Vascular injury (ECs) | Sepsis-induced microvascular endotheliopathy | Sepsis-induced venous endotheliopathy |
| Vaccine-induced venous endotheliopathy | ||
| Vascular pathology site | Disseminated aEA-VMTD at microvasculature | Transient or “silent” vEA-VMTD at venous system |
| Activated hemostatic path | ULVWF path | ULVWF path |
| Thrombosis component | Microthrombi strings | Microthrombi strings |
| Clinical phenotypes | TTP-like syndrome | ITP-like syndrome |
|
|
| Character Phenotypes | Microthrombosis | Macrothrombosis | Combined Micro-macrothrombosis |
|---|---|---|---|
| Vascular Tree System | |||
| Arterial | aEA-VMTD (e.g., TIA, TCIP, TTP-like syndrome, MODS such as DES, HUS, ANP, RML) | Arterial macrothrombosis (e.g., AIS, aortic thrombosis) | Gangrene (arterial combined MMT) (e.g., SPG, multiple digit gangrene, limb gangrene, PF, acrocyanosis, Burger’s disease, Fournier’s disease, necrotizing fasciitis, diabetic gangrene) |
| Venous | vEA-VMTD (e.g., ITP, TCIP, PNH, ARDS, hepatic VOD) | Venous macrothrombosis (e.g., distal DVT, solitary DVT) | Multiple DVT (venous combined MMT) (e.g., proximal/central DVT, VTE, PTE, CVST, IVCT, SVCT, SVT, PVT, Budd-Chiari syndrome) |
| Vascular Wall System | |||
| Involved vessel | Capillary/arteriole Venule Vasa vasorum | Small artery Vein - | Multiple small arterial vasculatures Multiple extended venous vasculatures |
| Thrombus character | Microthrombi strings | Macrothrombus | Microthrombi strings-fibrin meshes Combined micro-macrothrmbi |
| Pathogenetic factors | Endotheliopathy (ECs) (e.g., sepsis, diabetes) | Trauma (ECs +SET +/− EVT) (e.g., vascular access, surgery) | Endotheliopathy + traumatic vascular injury (e.g., sepsis + vascular access) |
| Damaged Vessel | |||
| Depth of injury | ECs | ECs + SET +/− EVT | ECs + SET +/− EVT |
| Location | Generalized or localized | Localized or solitary | Multiple or regionalized |
| Hemostatic components | ULVWF + platelet | ULVWF+TF + fibrin | ULVWF + TF + fibrin + NETs |
| Involved hemostatic path | ULVWF | ULVWF and TF | ULVWF, TF with NETosis |
| Clinical Phenotype | Arterial Combined Micro-Macrothrombosis | Venous Combined Micro-Macrothrombosis |
|---|---|---|
| Pathologic nature of thrombosis | ||
| (Examples) | Gangrene: | Venous circulatory congestion syndrome: |
| SPG, limb gangrene, Fournier’s disease, PF, diabetic gangrene, necrotizing fasciitis | Proximal/central DVT, VTE, PTE, CVST, PVT, IVCT/SVCT, SVT, Budd-Chiari syndrome | |
| Primary event | ||
| Vascular injury (ECs) | Diseases causing microvascular endotheliopathy (e.g., sepsis, diabetes, pregnancy) | Diseases causing venous endotheliopathy (e.g., sepsis, diabetes, pregnancy, autoimmunity) |
| Vascular pathology site | Disseminated aEA-VMTD at terminal arterial tree | Regional vEA-VMTD at vascular injury site |
| Activated hemostatic path | ULVWF path | ULVWF path |
| Thrombosis component | Microthrombi strings | Microthrombi strings |
| Secondary event | ||
| Vascular injury (SET) | Arterial vascular damage (e.g., surgery, vascular accesses/devices) | Venous vascular damage (e.g., surgery, vascular accesses/devices) |
| Pathology | Fibrin clots in arterial system | Fibrin clots in venous system |
| Vascular pathology site | Terminal arterial vasculature tree | Large vein or pulmonary artery |
| Activated hemostatic path | TF path | TF path |
| Thrombosis component | Fibrin meshes | Fibrin meshes |
| Pathogenesis | ||
| Mechanism | Unifying of microthrombi string and fibrin meshes in arterial system as minute macrothrombi | Unifying of microthrombi strings and fibrin meshes in venous system often as connected macrothrombi |
| Thrombosis character | Microthrombi strings-fibrin meshes complex | Microthrombi strings-fibrin meshes complex |
| Thrombosis form and effect | Multiple terminal efferent digit anoxia and peripheral gangrene | Connected and regional, with circulatory afferent flow with venous circulatory congestion syndrome |
| Benign DVT | Serious DVT | ||||
|---|---|---|---|---|---|
| Phenotype | Distal DVT | “Silent” vEA-VMTD | VTE | PTE | “Serious” vEA-VMTD |
| Example | Calf DVT (local) | ITP-like syndrome | PVT; SVC;/IVCT; SVT | PTE from VTE | Hepatic VOD |
| CVST | PNH | ||||
| Clinical characteristics | |||||
| Cause | Trauma | Septic endotheliopathy | Endotheliopathy + vascular access | Endotheliopathy + vascular access | Endotheliopathy |
| Genesis | Local vascular injury | vEA-VMTD | vEA-VMTD + injury | vEA-VMTD +injury | Transplant (VOD) PIGA gene mutation (PNH) |
| Thrombosis character | Macrothrombus | Microthrombi strings | Combined micro-macrothrombi | Combined micro-macrothrombi | Microthrombi strings |
| Activated hemostatic path | ULVWF + TF | ULVWF | ULVWF + TF | ULVWF + TF | ULVWF |
| Laboratory characteristics | |||||
| Thrombocytopenia | Absent | Common | Common | May be present | Always present |
| VWF/FVIII activity | Normal | Mildly increased | Increased | May be increased | Markedly increased |
| ANA | Negative | May be positive | May be positive | May be positive | Unknown |
| Anti-PLA/Anti-PF4A | Negative | May be positive | May be positive | May be positive | Unknown |
| ADAMTS13 activity | Normal | May be decreased | Decreased | May be decreased | Decreased (?) |
| Proposed treatment | None/short-term anticoagulation | No treatment or IVIG | Anticoagulant + antimicrothrombotic agent | Anticoagulant + antimicrothrombotic agent | Antimicrothrombotic Agent (?) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.C. Pathogenesis of Two Faces of DVT: New Identity of Venous Thromboembolism as Combined Micro-Macrothrombosis via Unifying Mechanism Based on “Two-Path Unifying Theory” of Hemostasis and “Two-Activation Theory of the Endothelium”. Life 2022, 12, 220. https://doi.org/10.3390/life12020220
Chang JC. Pathogenesis of Two Faces of DVT: New Identity of Venous Thromboembolism as Combined Micro-Macrothrombosis via Unifying Mechanism Based on “Two-Path Unifying Theory” of Hemostasis and “Two-Activation Theory of the Endothelium”. Life. 2022; 12(2):220. https://doi.org/10.3390/life12020220
Chicago/Turabian StyleChang, Jae C. 2022. "Pathogenesis of Two Faces of DVT: New Identity of Venous Thromboembolism as Combined Micro-Macrothrombosis via Unifying Mechanism Based on “Two-Path Unifying Theory” of Hemostasis and “Two-Activation Theory of the Endothelium”" Life 12, no. 2: 220. https://doi.org/10.3390/life12020220