
Pathogenic Molecular Mechanisms in Periodontitis and Peri-Implantitis: Role of Advanced Glycation End Products


Abstract
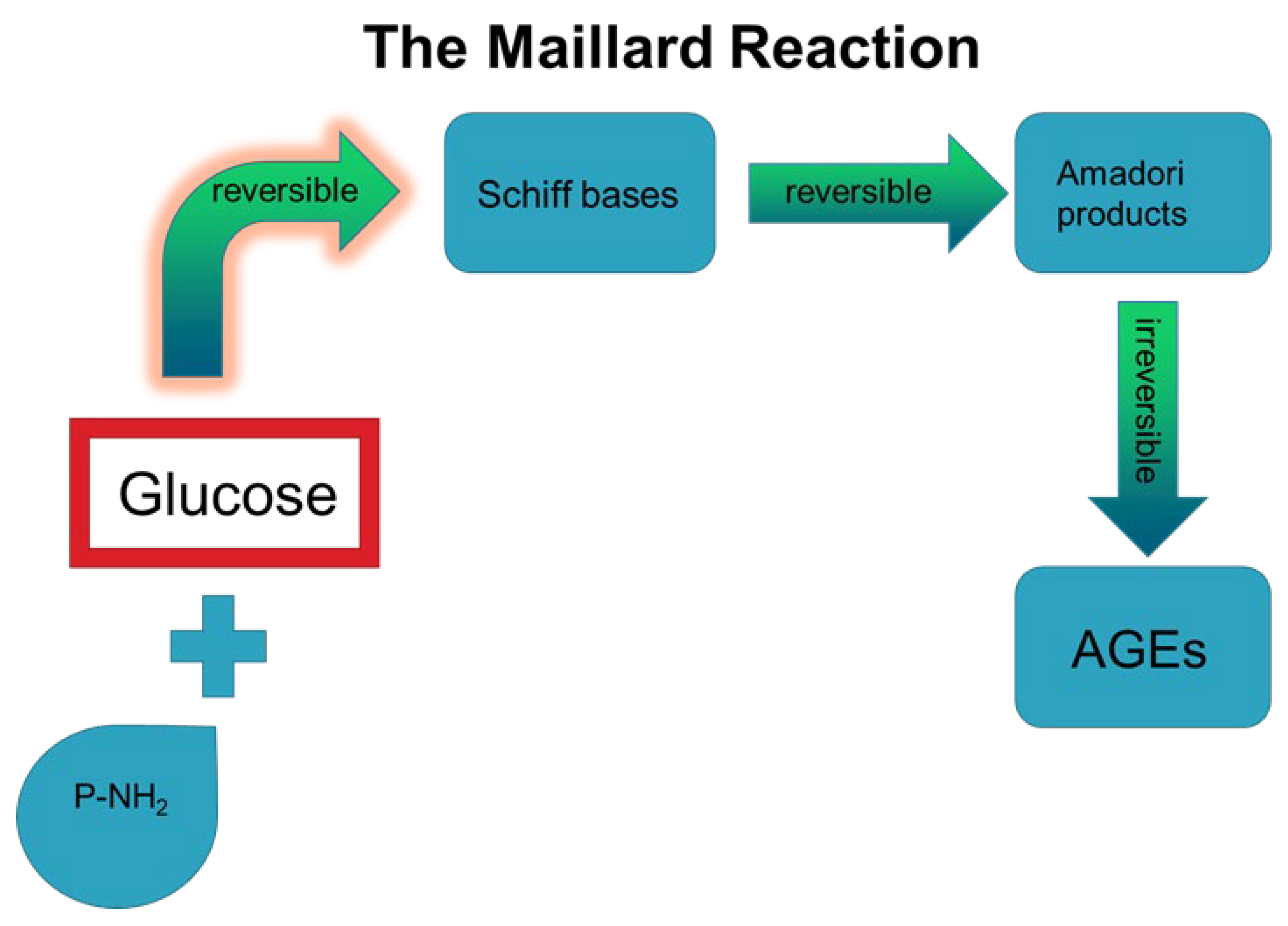
:1. Introduction
2. Direct Effects of AGEs in Periodontal Ligament Cells
2.1. AGE Effects on Osteoblasts-Osteocytes
2.1.1. In Vitro and In Vivo Studies
2.1.2. Clinical Studies
2.2. AGE Effects in Human Gingival Fibroblasts (HGFs)
2.2.1. In Vitro Studies
2.2.2. In Vivo Studies
2.3. AGE Effects in hPDL Cells
In Vitro Studies
2.4. AGE Effects in Gingival Epithelial Cells
In Vitro Studies
2.5. AGEs Effects in hPDL Stem Cells (hPDLSCs)
In Vitro Studies
2.6. AGE Effects in THP-1 Cells
3. Indirect Effects of AGEs in Periodontal Ligament Cells Mediated through Receptors
3.1. AGE–RAGE Axis in HGFs
In Vitro Studies
3.2. AGE–RAGE Axis in hPDL Cells
In Vitro Studies
3.3. AGE–RAGE Axis in Epithelial Cells
3.4. AGE–RAGE Axis in Gingival Tissues
3.5. AGE–RAGE Axis and Periradicular Tissues
3.6. AGE–RAGE Axis in Endothelial Cells
4. Effects of AGEs in Peri-Implantitis
5. AGE-Targeted Therapeutic Approaches in Periodontitis
5.1. In Vitro Studies
5.2. In Vivo Studies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AG | aminoguanidine |
| AGER1 | AGE receptor 1 |
| AGEs | Advanced Glycation End Products |
| ALP | alkaline phosphatase |
| CSF-1 | colony stimulating factor-1 |
| DM | Diabetes Mellitus |
| DM1/DM2 | Type 1 Diabetes Mellitus and Type 2 Diabetes Mellitus |
| ERK | extracellular signal-regulated kinase |
| GCF | gingival crevicular fluid |
| HbA1c | hemoglobin A1c |
| HGF | human gingival fibroblast |
| hPDL | human periodontal ligament |
| hPDLSCs | human periodontal ligament stem cells |
| IL-1β, 6, 10 | interleukins 1β, 6, 10 |
| JNK | c-Jun NH2-terminal kinase |
| LCN2 | neutrophil gelatinase-associated lipocalin |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| MG-H1 | hydroimidazolone |
| MMP | matrix metalloproteinase |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP1,3 | nucleotide-binding domain leucine-rich repeat proteins 1,3 |
| NOD2 | nucleotide-binding oligomerization domain-containing protein 2 |
| Pg LPS | Porphyromonas gingivalis lipopolysaccharide |
| PISF | peri-implant sulcus fluid |
| RAGE | receptor for AGEs |
| RANKL | receptor activator of nuclear factor κΒ ligand |
| ROS | reactive oxygen species |
| S100A8,9 | S100 calcium-binding protein A8,9 |
| sRAGE | soluble RAGE |
| TLR | Toll-like receptor |
| TNF-α | tumor necrosis factor-α |
| VEGF | vascular endothelial growth factor |
References
- Chapple, I.L.; Bouchard, P.; Cagetti, M.G.; Campus, G.; Carra, M.C.; Cocco, F.; Nibali, L.; Hujoel, P.; Laine, M.L.; Lingström, P.; et al. Interaction of lifestyle, behaviour or systemic diseases with dental caries and periodontal diseases: Consensus report of group 2 of the joint EFP/ORCA workshop on the boundaries between caries and periodontal diseases. J. Clin. Periodontol. 2017, 44 (Suppl. 18), S39–S51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hani, T.F. Studies on the Associations between Dental Caries, Periodontal Disease and Different Systemic Contidions. Ph.D. Thesis, University of Gothenburg, Gothenburg, Sweden, 2012. [Google Scholar]
- Pihlstrom, B.L.; Michalowicz, B.S.; Johnson, N.W. Periodontal diseases. Lancet 2005, 19, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Amir, J.; Waite, M.; Tobler, J.; Catalfamo, D.L.; Koutouzis, T.; Katz, J.; Wallet, S.M. The role of hyperglycemia in mechanisms of exacerbated inflammatory responses within the oral cavity. Cell Immunol. 2011, 272, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, D.T.; Ding, Z.; Yang, Y. The impact of diabetes on periodontal diseases. Periodontology 2000 2020, 82, 214–224. [Google Scholar] [CrossRef]
- Gurav, A.N. Management of diabolical diabetes mellitus and periodontitis nexus: Are we doing enough? World J. Diabetes 2016, 7, 50–66. [Google Scholar] [CrossRef]
- Shay, K. Infectious Complications of Dental and Periodontal Diseases in the Elderly Population. Clin. Infect. Dis. 2002, 34, 1215–1223. [Google Scholar] [CrossRef]
- Kornman, K.S.; Papapanou, P.N. Clinical application of the new classification of periodontal diseases: Ground rules, clarifications and “gray zones”. J. Periodontol. 2020, 91, 352–360. [Google Scholar] [CrossRef]
- Li, Y.; Du, Z.; Xie, X.; Zhang, Y.; Liu, H.; Zhou, Z.; Zhao, J.; Lee, R.S.; Xiao, Y.; Ivanoviski, S.; et al. Epigenetic changes caused by diabetes and their potential role in the development of periodontitis. J. Diabetes Investig. 2021, 12, 1326–1335. [Google Scholar] [CrossRef]
- Takeda, M.; Ojima, M.; Yoshioka, H.; Inaba, H.; Kogo, M.; Shizukuishi, S.; Nomura, M.; Amano, A. Relationship of serum advanced glycation end products with deterioration of periodontitis in type 2 diabetes patients. J. Periodontol. 2006, 77, 15–20. [Google Scholar] [CrossRef]
- Zizzi, A.; Tirabassi, G.; Aspriello, S.D.; Piemontese, M.; Rubini, C.; Lucarini, G. Gingival advanced glycation end-products in diabetes mellitus-associated chronic periodontitis: An immunohistochemical study. J. Periodontal Res. 2013, 48, 293–301. [Google Scholar] [CrossRef]
- Polak, D.; Shapira, L. An update on the evidence for pathogenic mechanisms that may link periodontitis and diabetes. J. Clin. Periodontol. 2018, 45, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Baque, V.; Garidou, L.; Pomié, C.; Escoula, Q.; Loubieres, P.; Le Gall-David, S.; Lemaitre, M.; Nicolas, S.; Klopp, P.; Waget, A.; et al. Periodontitis induced by Porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut 2017, 66, 872–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragno, M.; Mastrocola, R. Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients 2017, 14, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, I.; Morales, M.A.; Rojas, A. Polyphenols and AGEs/RAGE axis. Trends and challenges. Food Res. Int. 2020, 129, 108843. [Google Scholar] [CrossRef]
- Luevano-Contreras, C.; Chapman-Novakofski, K. Dietary advanced glycation end products and aging. Nutrients 2010, 2, 1247–1265. [Google Scholar] [CrossRef] [Green Version]
- Toraman, A.; Arabaci, T.; Aytekin, Z.; Albayrak, M.; Bayir, Y. Effects of vitamin C local application on ligature-induced periodontitis in diabetic rats. J. Appl. Oral Sci. Rev. FOB 2020, 28, e20200444. [Google Scholar] [CrossRef]
- Liu, C.M.; Chen, S.H.; Liao, Y.W.; Yu, C.H.; Yu, C.C.; Hsieh, P.L. Magnolol ameliorates the accumulation of reactive oxidative stress and inflammation in diabetic periodontitis. J. Formos. Med. Assoc. 2021, 120, 1452–1458. [Google Scholar] [CrossRef]
- Sakamoto, E.; Mihara, C.; Ikuta, T.; Inagaki, Y.; Kido, J.; Nagata, T. Inhibitory effects of advanced glycation end-products and Porphyromonas gingivalis lipopolysaccharide on the expression of osteoblastic markers of rat bone marrow cells in culture. J. Periodontal Res. 2016, 51, 313–320. [Google Scholar] [CrossRef]
- Sakamoto, E.; Kido, J.I.; Takagi, R.; Inagaki, Y.; Naruishi, K.; Nagata, T.; Yumoto, H. Advanced glycation end-product 2 and Porphyromonas gingivalis lipopolysaccharide increase sclerostin expression in mouse osteocyte-like cells. Bone 2019, 122, 22–30. [Google Scholar] [CrossRef]
- Chiu, H.C.; Fu, M.M.; Yang, T.S.; Fu, E.; Chiang, C.Y.; Tu, H.P.; Chin, Y.T.; Lin, F.G.; Shih, K.C. Effect of high glucose, Porphyromonas gingivalis lipopolysaccharide and advanced glycation end-products on production of interleukin-6/-8 by gingival fibroblasts. J. Periodontal Res. 2017, 52, 268–276. [Google Scholar] [CrossRef]
- Huang, Y.; Guo, W.; Zeng, J.; Chen, G.; Sun, W.; Zhang, X.; Tian, W. Prediabetes Enhances Periodontal Inflammation Consistent with Activation of Toll-Like Receptor-Mediated Nuclear Factor-κB Pathway in Rats. J. Periodontol. 2016, 87, e64–e74. [Google Scholar] [CrossRef] [PubMed]
- Marinucci, L.; Balloni, S.; Fettucciari, K.; Bodo, M.; Talesa, V.N.; Antognelli, C. Nicotine induces apoptosis in human osteoblasts via a novel mechanism driven by H2O2 and entailing Glyoxalase 1-dependent MG-H1 accumulation leading to TG2-mediated NF-kB desensitization: Implication for smokers-related osteoporosis. Free Radic. Biol. Med. 2018, 117, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Akram, Z.; Alqahtani, F.; Alqahtani, M.; Al-Kheraif, A.A.; Javed, F. Levels of advanced glycation end products in gingival crevicular fluid of chronic periodontitis patients with and without type-2 diabetes mellitus. J. Periodontol. 2020, 91, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Sereti, M.; Roy, M.; Zekeridou, A.; Gastaldi, G.; Giannopoulou, C. Gingival crevicular fluid biomarkers in type 1 diabetes mellitus: A case-control study. Clin. Exp. Dent. Res. 2021, 7, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Alqerban, A. Levels of proinflammatory chemokines and advanced glycation end products in patients with type-2 diabetes mellitus undergoing fixed orthodontic treatment. Angle Orthod. 2021, 91, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Fu, Y.; Deng, Y.; Qi, L.; Jin, L. Advanced glycation end products inhibit the expression of collagens type I and III by human gingival fibroblasts. J. Periodontol. 2009, 80, 1166–1173. [Google Scholar] [CrossRef]
- Qi, L.Y.; Fu, Y.; Zhou, Y. Effect of advanced glycation end products on the human gingival fibroblast proliferation and type I collagen synthesis. Zhonghua Kou Qiang Yi Xue Za Zhi 2008, 43, 12–15. (In Chinese) [Google Scholar]
- Murillo, J.; Wang, Y.; Xu, X.; Klebe, R.J.; Chen, Z.; Zardeneta, G.; Pal, S.; Mikhailova, M.; Steffensen, B. Advanced glycation of type I collagen and fibronectin modifies periodontal cell behavior. J. Periodontol. 2008, 79, 2190–2199. [Google Scholar] [CrossRef]
- Xu, H.X.; Fu, Y.; Li, H.J. Mechanism of advanced glycation end-products in the inducement of apoptosis of human gingival fibroblast and related effect of puerarin in the process. Zhonghua Kou Qiang Yi Xue Za Zhi 2011, 46, 31–34. (In Chinese) [Google Scholar]
- Bender, O.; Weinberg, E.; Moses, O.; Nemcovsky, C.E.; Weinreb, M. Porphyromonas gingivalis lipopolysaccharide and glycated serum albumin increase the production of several pro-inflammatory molecules in human gingival fibroblasts via NFκB. Arch. Oral Biol. 2020, 116, 104766. [Google Scholar] [CrossRef]
- Nonaka, K.; Kajiura, Y.; Bando, M.; Sakamoto, E.; Inagaki, Y.; Lew, J.H.; Naruishi, K.; Ikuta, T.; Yoshida, K.; Kobayashi, T.; et al. Advanced glycation end-products increase IL-6 and ICAM-1 expression via RAGE, MAPK and NF-kappaB pathways in human gingival fibroblasts. J. Periodontal. Res. 2018, 53, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, Y.; Ye, C.; Wu, W.; Liao, G.; Lu, Y.; Huang, P. Changes in advanced glycation end products, beta-defensin-3, and interleukin-17 during diabetic periodontitis development in rhesus monkeys. Exp. Biol. Med. 2018, 243, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Weidman, E.; Lalla, E.; Yan, S.D.; Hori, O.; Cao, R.; Brett, J.G.; Lamster, I.B. Advanced glycation endproducts (AGEs) induce oxidant stress in the gingiva: A potential mechanism underlying accelerated periodontal disease associated with diabetes. J. Periodontal. Res. 1996, 31, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Lee, S.Y.; Kim, J.H.; Choi, Y.H.; Yang, D.W.; Kang, J.H.; Ko, H.M.; Cho, J.H.; Koh, J.T.; Kim, W.J.; et al. Synergistic alveolar bone resorption by diabetic advanced glycation end products and mechanical forces. J. Periodontol. 2019, 90, 1457–1469. [Google Scholar] [CrossRef]
- Xu, J.; Xiong, M.; Huang, B.; Chen, H. Advanced glycation end products upregulate the endoplasmic reticulum stress in human periodontal ligament cells. J. Periodontol. 2015, 86, 440–447. [Google Scholar] [CrossRef]
- Gao, H.; Zhang, X.; Zheng, Y.; Peng, L.; Hou, J.; Meng, H. S100A9-induced release of interleukin (IL)-6 and IL-8 through toll-like receptor 4 (TLR4) in human periodontal ligament cells. Mol. Immunol. 2015, 67, 223–232. [Google Scholar] [CrossRef]
- Mei, Y.M.; Li, L.; Wang, X.Q.; Zhang, M.; Zhu, L.F.; Fu, Y.W.; Xu, Y. AGEs induces apoptosis and autophagy via reactive oxygen species in human periodontal ligament cells. J. Cell. Biochem. 2020, 121, 3764–3779. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Sakamoto, E.; Yoshida, K.; Abe, K.; Naruishi, K.; Yamamoto, T.; Shinohara, Y.; Kido, J.I.; Geczy, C.L. Advanced glycation end-products and Porphyromonas gingivalis lipopolysaccharide increase calprotectin expression in human gingival epithelial cells. J. Cell. Biochem. 2018, 119, 1591–1603. [Google Scholar] [CrossRef]
- Fang, H.; Yang, K.; Tang, P.; Zhao, N.; Ma, R.; Luo, X.; Liu, Q. Glycosylation end products mediate damage and apoptosis of periodontal ligament stem cells induced by the JNK-mitochondrial pathway. Aging 2020, 30, 12850–12868. [Google Scholar] [CrossRef]
- Yan, W.; Chao, D.; Kun, Y.; Xiaoxia, C.; Qi, L.; Yan, J. Canonical Wnt signaling pathway of the osteogenic differentiation of human periodontal ligament stem cells induced by advanced glycation end products. Hua Xi Kou Qiang Yi Xue Za Zhi 2015, 33, 627–632. (In Chinese) [Google Scholar]
- Chao, D.; Yan, W.; Kun, Y.; Xiaoxia, C.; Qi, L.; Yan, J. Effect of microRNA-17 on osteogenic differentiation of advanced glycation end products-stimulated human periodontal ligament stem cells. Hua Xi Kou Qiang Yi Xue Za Zhi 2015, 33, 21–24. (In Chinese) [Google Scholar] [PubMed]
- Zhang, L.N.; Wang, X.X.; Wang, Z.; Li, K.Y.; Xu, B.H.; Zhang, J. Berberine improves advanced glycation end products-induced osteogenic differentiation responses in human periodontal ligament stem cells through the canonical Wnt/β-catenin pathway. Mol. Med. Rep. 2019, 19, 5440–5452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settem, R.P.; Honma, K.; Shankar, M.; Li, M.; LaMonte, M.; Xu, D.; Genco, R.J.; Browne, R.W.; Sharma, A. Tannerella forsythia-produced methylglyoxal causes accumulation of advanced glycation endproducts to trigger cytokine secretion in human monocytes. Mol. Oral Microbiol. 2018, 33, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer—A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef] [PubMed]
- Rajeev, K.; Karthika, R.; Mythili, R.; Krishnan, V.; Nirmal, M. Role of receptors of advanced glycation end-products (RAGE) in type 2 diabetic and non-diabetic individuals with chronic periodontal disease: An immunohistochemical study. J. Investig. Clin. Dent. 2011, 2, 287–292. [Google Scholar] [CrossRef]
- Lalla, E.; Lamster, I.B.; Feit, M.; Huang, L.; Spessot, A.; Qu, W.; Kislinger, T.; Lu, Y.; Stern, D.M.; Schmidt, A.M. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J. Clin. Investig. 2000, 105, 1117–1124. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.C.; Jheng, Y.H.; Wang, C.Y.; Chen, Y.W.; Lin, Y.F.; Chen, C.C.; Chang, P.C. Osseous wound repair under inhibition of the axis of advanced glycation end-products and the advanced glycation end-products receptor. J. Formos. Med. Assoc. 2015, 114, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.C.; Chien, L.Y.; Yeo, J.F.; Wang, Y.P.; Chung, M.C.; Chong, L.Y.; Kuo, M.Y.; Chen, C.H.; Chiang, H.C.; Ng, B.N.; et al. Progression of periodontal destruction and the roles of advanced glycation end products in experimental diabetes. J. Periodontol. 2013, 84, 379–388. [Google Scholar] [CrossRef]
- Claudino, M.; Gennaro, G.; Cestari, T.M.; Spadella, C.T.; Garlet, G.P.; Assis, G.F. Spontaneous periodontitis development in diabetic rats involves an unrestricted expression of inflammatory cytokines and tissue destructive factors in the absence of major changes in commensal oral microbiota. Exp. Diabetes Res. 2012, 2012, 356841. [Google Scholar] [CrossRef]
- Bala, S.V.; Appukuttan, D.; Subramaniam, S.; Prakash, P.S.G.; Cholan, P.K.; Victor, D.J. Association of Receptor for advanced glycation end products G82S polymorphism with chronic periodontitis in type II diabetic and non-diabetic South Indians. Gene 2019, 5, 30–37. [Google Scholar] [CrossRef]
- Wu, Y.; Dong, G.; Xiao, W.; Xiao, E.; Miao, F.; Syverson, A.; Missaghian, N.; Vafa, R.; Cabrera-Ortega, A.A.; Rossa, C., Jr.; et al. Effect of Aging on Periodontal Inflammation, Microbial Colonization, and Disease Susceptibility. J. Dent. Res. 2016, 95, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altıngöz, S.M.; Kurgan, Ş.; Önder, C.; Serdar, M.A.; Ünlütürk, U.; Uyanık, M.; Başkal, N.; Tatakis, D.N.; Günhan, M. Salivary and serum oxidative stress biomarkers and advanced glycation end products in periodontitis patients with or without diabetes: A cross-sectional study. J. Periodontol. 2021, 92, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Pradeep, A.R.; Kanoriya, D.; Garg, V. Human soluble receptorfor advanced glycation end products and tumor necrosis factor-alphaas gingival crevicular fluid and serum markers of inflammation inchronic periodontitis and type 2 diabetes. J. Oral Sci. 2016, 58, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Li, H.; Ma, Y.; Fu, Y. Matrix metalloproteinase-1 of gingival fibroblasts influenced by advanced glycation end products (AGEs) and their association with receptor for AGEs and nuclear factor-κB in gingival connective tissue. J. Periodontol. 2012, 83, 119–126. [Google Scholar] [CrossRef]
- Li, D.X.; Deng, T.Z.; Lv, J.; Ke, J. Advanced glycation end products (AGEs) and their receptor (RAGE) induce apoptosis of periodontal ligament fibroblasts. Braz. J. Med. Biol. Res. 2014, 47, 1036–1043. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.C.; Chien, L.Y.; Chong, L.Y.; Kuo, Y.P.; Hsiao, J.K. Glycated matrix up-regulates inflammatory signaling similarly to Porphyromonas gingivalis lipopolysaccharide. J. Periodontal Res. 2013, 48, 184–193. [Google Scholar] [CrossRef]
- Yi, X.; Zhang, L.; Lu, W.; Tan, X.; Yue, J.; Wang, P.; Xu, W.; Ye, L.; Huang, D. The effect of NLRP inflammasome on the regulation of AGEs-induced inflammatory response in human periodontal ligament cells. J. Periodontal Res. 2019, 54, 681–689. [Google Scholar] [CrossRef]
- Kido, R.; Hiroshima, Y.; Kido, J.I.; Ikuta, T.; Sakamoto, E.; Inagaki, Y.; Naruishi, K.; Yumoto, H. Advanced glycation end-products increase lipocalin 2 expression in human oral epithelial cells. J Periodontal Res. 2020, 55, 539–550. [Google Scholar] [CrossRef]
- Abbass, M.M.; Korany, N.S.; Salama, A.H.; Dmytryk, J.J.; Safiejko-Mroczka, B. The relationship between receptor for advanced glycation end products expression and the severity of periodontal disease in the gingiva of diabetic and non diabetic periodontitis patients. Arch. Oral Biol. 2012, 57, 1342–1354. [Google Scholar] [CrossRef]
- Katz, J.; Bhattacharyya, I.; Farkhondeh-Kish, F.; Perez, F.M.; Caudle, R.M.; Heft, M.W. Expression of the receptor of advanced glycation end products in gingival tissues of type 2 diabetes patients with chronic periodontal disease: A study utilizing immunohistochemistry and RT-PCR. J. Clin. Periodontol. 2005, 32, 40–44. [Google Scholar] [CrossRef]
- Katz, J.; Caudle, R.M.; Bhattacharyya, I.; Stewart, C.M.; Cohen, D.M. Receptor for advanced glycation end product (RAGE) upregulation in human gingival fibroblasts incubated with nornicotine. J. Periodontol. 2005, 76, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Detzen, L.; Cheng, B.; Chen, C.Y.; Papapanou, P.N.; Lalla, E. Soluble Forms of the Receptor for Advanced Glycation Endproducts (RAGE) in Periodontitis. Sci. Rep. 2019, 9, 8170. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.; Yoon, T.Y.; Mao, S.; Lamont, R.J.; Caudle, R.M. Expression of the receptor of advanced glycation end products in the gingival tissue of smokers with generalized periodontal disease and after nornicotine induction in primary gingival epithelial cells. J. Periodontol. 2007, 78, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Koutouzis, T.; Catania, D.; Neiva, K.; Wallet, S.M. Innate immune receptor expression in peri-implant tissues of patients with different susceptibility to periodontal diseases. J. Periodontol. 2013, 84, 221–229. [Google Scholar] [CrossRef]
- Crabtree, M.; Pileggi, R.; Bhattacharyya, I.; Caudle, R.; Perez, F.; Riley, J.; Vertucci, F.; Katz, J. RAGE mRNA expression and its correlation with nuclear factor kappa beta mRNA expression in inflamed human periradicular tissues. J. Endod. 2008, 34, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Pollreisz, A.; Hudson, B.I.; Chang, J.S.; Qu, W.; Cheng, B.; Papapanou, P.N.; Schmidt, A.M.; Lalla, E. Receptor for advanced glycation endproducts mediates pro-atherogenic responses to periodontal infection in vascular endothelial cells. Atherosclerosis 2010, 212, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; Hori, O.; Chen, J.X.; Li, J.F.; Crandall, J.; Zhang, J.; Cao, R.; Yan, S.D.; Brett, J.; Stern, D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Investig. 1995, 96, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- Alqahtani, F.; Alqhtani, N.; Alkhtani, F.; Devang Divakar, D.; Al-Kheraif, A.A.; Javed, F. Clinicoradiographic markers of peri-implantitis in cigarette-smokers and never-smokers with type 2 diabetes mellitus at 7-years follow-up. J. Periodontol. 2020, 91, 1132–1138. [Google Scholar] [CrossRef]
- Monje, A.; Catena, A.; Borgnakke, W.S. Association between diabetes mellitus/hyperglycaemia and peri-implant diseases: Systematic review and meta-analysis. J. Clin. Periodontol. 2017, 44, 636–648. [Google Scholar] [CrossRef]
- Guo, M.; Liu, L.; Zhang, J.; Liu, M. Role of Reactive Oxygen Species and Advanced Glycation End Products in the Malfunctioning of Dental Implants. West Indian Med. J. 2015, 64, 419–423. [Google Scholar]
- Pietropaoli, D.; Ortu, E.; Severino, M.; Ciarrocchi, I.; Gatto, R.; Monaco, A. Glycation and oxidative stress in the failure of dental implants: A case series. BMC Res. Notes 2013, 6, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Sowygh, Z.H.; Ghani, S.M.A.; Sergis, K.; Vohra, F.; Akram, Z. Peri-implant conditions and levels of advanced glycation end products among patients with different glycemic control. Clin. Implant. Dent. Relat. Res. 2018, 20, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Alrabiah, M.; Al-Aali, K.A.; Al-Sowygh, Z.H.; Binmahfooz, A.M.; Mokeem, S.A.; Abduljabbar, T. Association of advanced glycation end products with peri-implant inflammation in prediabetes and type 2 diabetes mellitus patients. Clin. Implant. Dent. Relat. Res. 2018, 20, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Al-Aali, K.A.; AlHelal, A.; Alhamoudi, N.; Alhenaki, A.M.; Javed, F.; Abduljabbar, T. Assessment of advanced glycation end products in the peri-implant sulcular fluid among moderate cigarette-smokers and nonsmokers with peri-implantitis. Clin. Implant. Dent. Relat. Res. 2020, 22, 380–386. [Google Scholar] [CrossRef]
- Butera, A.; Gallo, S.; Maiorani, C.; Preda, C.; Chiesa, A.; Esposito, F.; Pascadopoli, M.; Scribante, A. Management of Gingival Bleeding in Periodontal Patients with Domiciliary Use of Toothpastes Containing Hyaluronic Acid, Lactoferrin, or Paraprobiotics: A Randomized Controlled Clinical Trial. Appl. Sci. 2021, 11, 8586. [Google Scholar] [CrossRef]
- Tiwari, S.; Avinash, A.; Katiyar, S.; Iyer, A.A.; Jain, S. Dental applications of ozone therapy: A review of literature. Saudi J. Dent. Res. 2017, 8, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, X.; Zhang, L.; Wang, B.; Xu, B.; Zhang, J. GLP-1 inhibits PKCβ2 phosphorylation to improve the osteogenic differentiation potential of hPDLSCs in the AGE microenvironment. J. Diabetes Its Complicat. 2020, 34, 107495. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, C.H.; Zhou, C.H.; Cui, X.X.; Yang, K.; Deng, C.; Xia, J.J.; Wu, Y.; Liu, L.C.; Jin, Y. DKK1 rescues osteogenic differentiation of mesenchymal stem cells isolated from periodontal ligaments of patients with diabetes mellitus induced periodontitis. Sci. Rep. 2015, 5, 13142. [Google Scholar] [CrossRef]
- Chang, P.C.; Tsai, S.C.; Jheng, Y.H.; Lin, Y.F.; Chen, C.C. Soft-tissue wound healing by anti-advanced glycation end-products agents. J. Dent. Res. 2014, 93, 388–393. [Google Scholar] [CrossRef]
- Chang, P.C.; Chong, L.Y.; Tsai, S.C.; Lim, L.P. Aminoguanidine inhibits the AGE-RAGE axis to modulate the induction of periodontitis but has limited effects on the progression and recovery of experimental periodontitis: A preliminary study. J. Periodontol. 2014, 85, 729–739. [Google Scholar] [CrossRef]
- Chang, P.C.; Tsai, S.C.; Chong, L.Y.; Kao, M.J. N-Phenacylthiazolium bromide inhibits the advanced glycation end product (AGE)-AGE receptor axis to modulate experimental periodontitis in rats. J. Periodontol. 2014, 85, e268–e276. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.C.; Chang, C.Y.; Chao, Y.C.; Jheng, Y.H.; Yang, C.; Lee, N.; Yu, S.H.; Yu, X.H.; Liu, D.M.; Chang, P.C. pH-Responsive Hydrogel with an Anti-Glycation Agent for Modulating Experimental Periodontitis. J. Periodontol. 2016, 87, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, G.; So, H.S.; Kieu, T.; Kook, S.H.; Lee, J.C.; Jeon, Y.M. Astaxanthin Inhibits Diabetes-Triggered Peridontal Destruction, Ameliorates Oxidative Complications in STZ-Injected Mice, and Recovers Nrf2-Dependent Antioxidant System. Nutrients 2021, 13, 3575. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type of Study | Cell Type/Tissue/Biological Fluid | AGE Effects | Reference |
|---|---|---|---|
| In vitro | Rat bone marrow cells | Decreased collagen type I, core-binding protein-a and osteocalcin production Increased IL-1β and S1008A production | [19] |
| Mouse osteocytic-like cells | Increased sclerostin, IL-6, and TNF-α production | [20] | |
| Human gingival fibroblasts | Increase IL-6 and IL-8 production | [21] | |
| Human osteoblasts | Nicotine enhances AGEs-related actions by increasing H2O2 production | [23] | |
| Human gingival fibroblasts | Reduced HGF cell viability and decreased collagen type I and III production | [27] | |
| Human gingival fibroblasts | Decreased collagen type I production | [28] | |
| Human gingival fibroblasts and periodontal ligament fibroblasts | Fibronectin and collage type I glycation | [29] | |
| Human gingival fibroblasts | Increased HGF cell apoptosis | [30] | |
| Human gingival fibroblasts | Increased MMP-1, MCP-1, IL-6, and IL-8 production | [31] | |
| Human gingival fibroblasts | Increased IL-6, RAGE, and ICAM-1 production | [32] | |
| Human periodontal ligament cells | Increased IL-6 and IL-8 production | [36] | |
| Human periodontal ligament cells | Increased IL-6 and IL-8 production | [37] | |
| Human periodontal ligament cells | Human periodontal ligament cells autophagy | [38] | |
| Human gingival epithelial cells | Increased RAGE expression | [39] | |
| Periodontal ligament stem cells | Increased endogenous ROS | [40] | |
| Human periodontal ligament stem cells | Less-calcified osteogenic nodules | [41] | |
| Human periodontal ligament stem cells | Inhibition of osteogenic differentiation | [42] | |
| Human periodontal ligament cells | Inhibition of differentiation | [43] | |
| Human monocytes (THP-1 cells) | Increased IL-1β and TNF-α production | [44] | |
| Gingival fibroblasts | Increased MMP-1 production | [55] | |
| Periodontal ligament fibroblasts | Increased RAGE and cell apoptosis | [56] | |
| Human periodontal ligament cells | Increased IL-6, IL-1β, NLRP1, and NLRP3 production | [58] | |
| Human oral epithelial cells | Increased LCN-2 production | [59] | |
| Human gingival fibroblasts | RAGE expression increased by nornicotine | [62] | |
| Vascular endothelial cells | Increased MCP-1 production | [67] | |
| In vivo | Serum | Secretion of MMP-1, increased IL-17 expression, and decreased beta-defensine-3 production | [33] |
| Gingival connective tissue | Increased oxidative stress and IL-6 production | [34] | |
| Human periodontal ligament cells | Increased CSF-1 and VEGF production | [35] | |
| Gingival tissues | Anti-inflammatory effect of sRAGE | [47] | |
| Wound healing assessment | Anti-inflammatory effect of aminoguanidine | [48] | |
| Periodontal tissues | Increased IL-1 and TNF-α production | [50] | |
| Dendritic cells and osteoclasts | Reduction in dendritic cell migration | [52] | |
| Human periodontal ligament cells and mesenchymal stem cells | Increased RAGE and TLRs production | [57] | |
| Cultured human endothelial cells and murine vasculature | Increased VCAM-1 production | [68] | |
| Clinical | GCF | Increased AGEs levels in T2DM patients | [24] |
| GCF | Did not significantly increase AGEs levels between T1DM patients and healthy | [25] | |
| Serum | Increased AGEs levels in T2DM patients | [26] | |
| Gingival tissues | Increased RAGE levels in T2DM patients | [46] | |
| Periodontal tissues | RAGEG82S gene polymorphism as a risk factor of periodontitis | [51] | |
| Saliva, serum | Increased AGEs levels in periodontal patients rather than non-periodontal | [53] | |
| Serum, GCF | Increased TNF-α and reduced sRAGE levels | [54] | |
| Gingival tissues | Increased RAGE levels in DM patients | [60] | |
| Gingival tissues | Main location of RAGE is the basal epithelial membrane | [61] | |
| Gingival tissues | Anti-inflammatory effect of AGER1 | [63] | |
| Gingival tissues | Increased RAGE levels in gingival tissues of smokers | [64] | |
| Peri-implant tissues | Increased RAGE levels | [65] | |
| Peri-apical tissues | Increased RAGE levels | [66] | |
| Saliva and apical-coronal tissues | Increased AGEs levels and oxidative stress | [71] | |
| Saliva, peri-implant tissues, and periodontal tissues | Increased AGEs levels and oxidative stress | [72] | |
| PISF | Increased AGEs levels in T2DM patients | [73] | |
| PISF | Increased AGEs levels in T2DM patients | [74] | |
| PISF | Increased AGEs levels in smokers | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plemmenos, G.; Piperi, C. Pathogenic Molecular Mechanisms in Periodontitis and Peri-Implantitis: Role of Advanced Glycation End Products. Life 2022, 12, 218. https://doi.org/10.3390/life12020218
Plemmenos G, Piperi C. Pathogenic Molecular Mechanisms in Periodontitis and Peri-Implantitis: Role of Advanced Glycation End Products. Life. 2022; 12(2):218. https://doi.org/10.3390/life12020218
Chicago/Turabian StylePlemmenos, Grigorios, and Christina Piperi. 2022. "Pathogenic Molecular Mechanisms in Periodontitis and Peri-Implantitis: Role of Advanced Glycation End Products" Life 12, no. 2: 218. https://doi.org/10.3390/life12020218