
CRISPR-Based Tools for Fighting Rare Diseases
Abstract
:1. Introduction
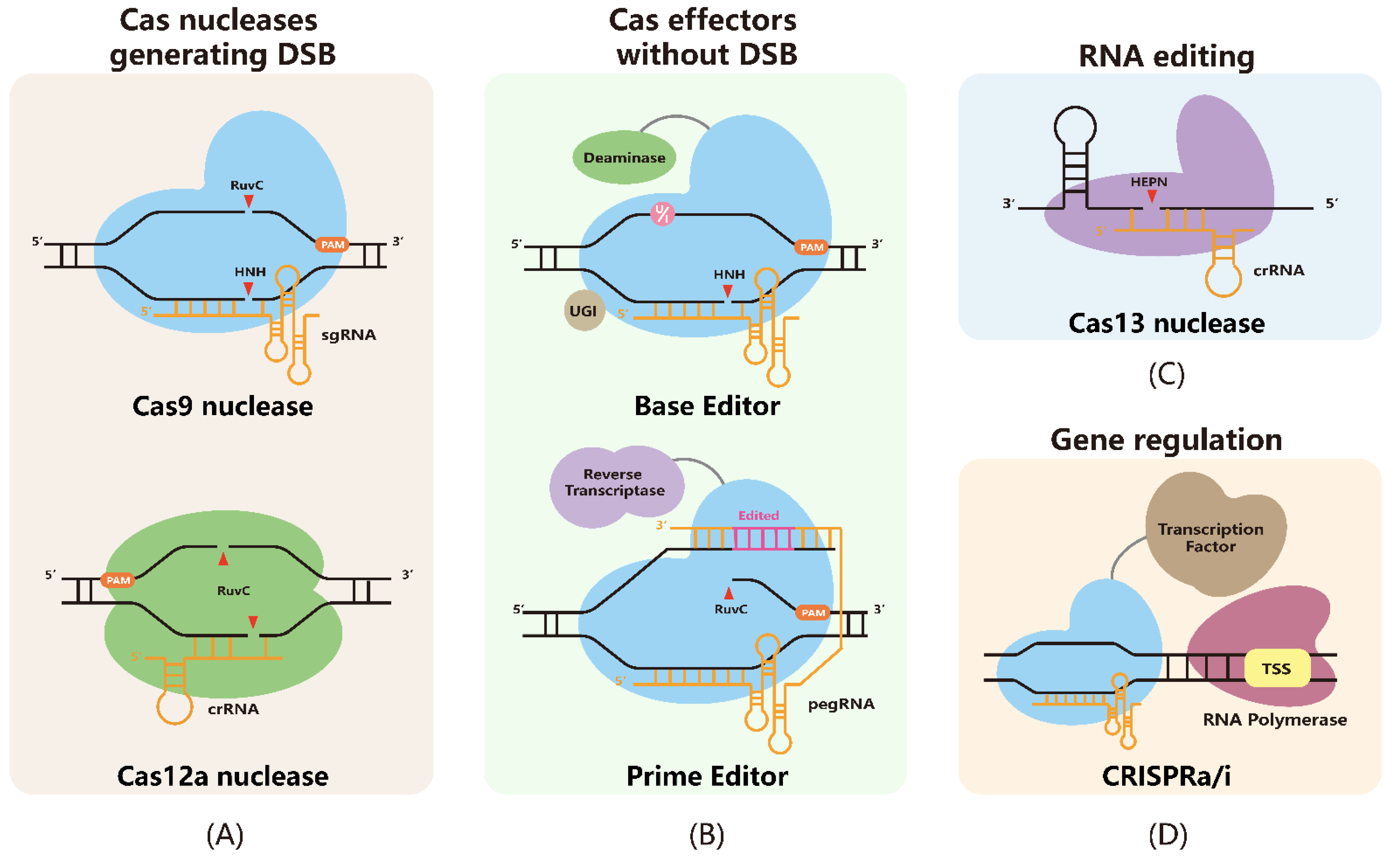
2. CRISPR-Based Tools for Genome Engineering
2.1. Cas Nucleases-Mediated Genome Editing
2.2. Base Editors-Mediated Genome Editing
2.3. Prime Editors-Mediated Genome Editing
2.4. CRISPR-Cas13-Mediated RNA Editing
2.5. CIRSPRa/i-Mediated Transcription Regulation
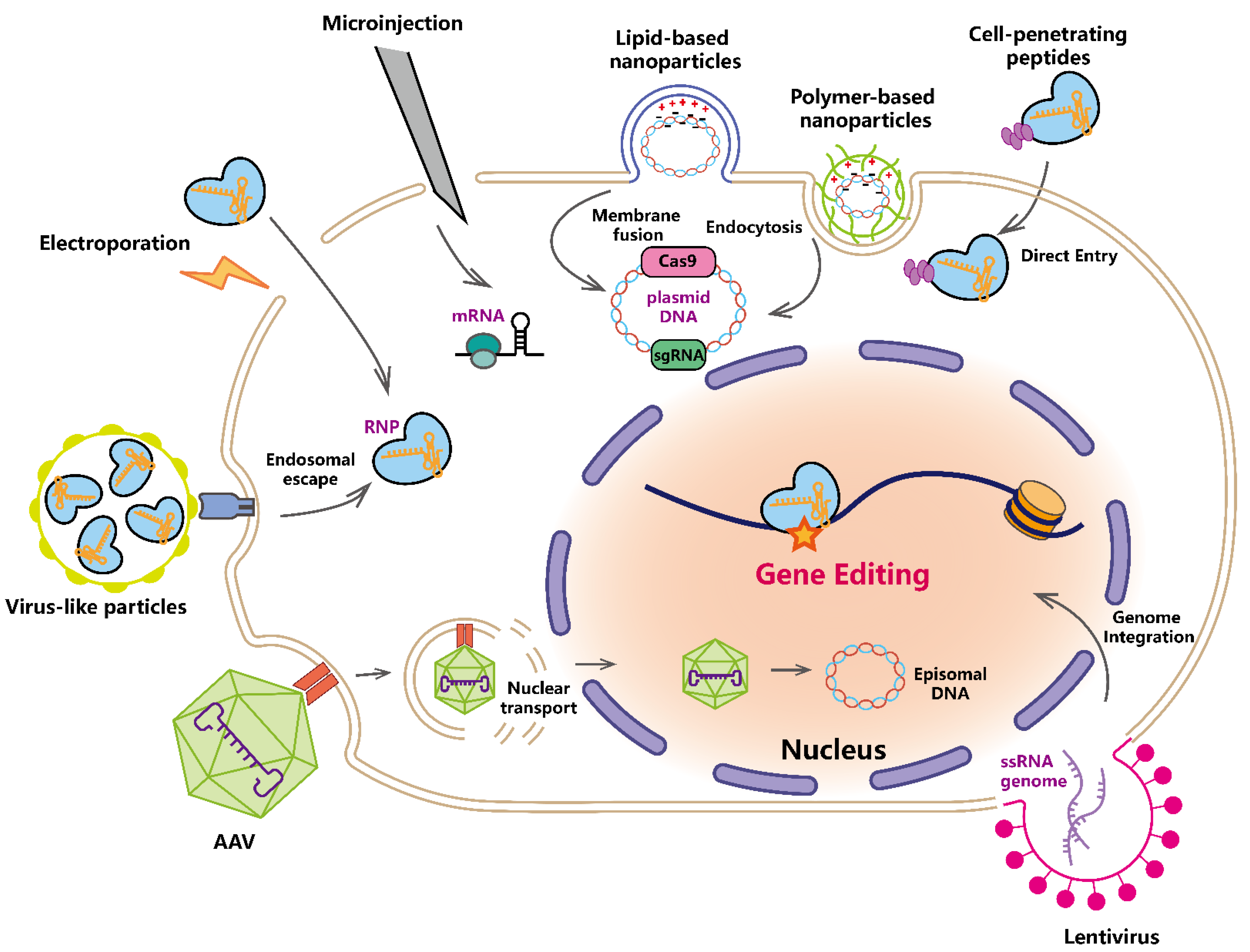
3. Delivery System of CRISPR-Based Tool
3.1. Physical Delivery
3.2. Non-Viral Chemical Delivery
3.3. Viral Delivery System
3.4. Virus-like Particles (VLPs)
4. Applications of CRISPR Tools in Rare Disease Therapy
4.1. Blood Disorders

{kind=link}
{kind=link}
| Diseases Type | Diseases | Gene | Strategy | Animal Model or Cells | Delivery | Outcome | References; Clinical Trials |
|---|---|---|---|---|---|---|---|
| Blood disorder | SCD/b-thalassemia | HBB | ABE8e-NRCH-mediated point mutation correction | HSPCs from patients with SCD | RNP; electroporation | 80% editing efficiency, 72% decrease in the pathogenic protein | [124] |
| HBB | IBE-mediated point mutation correction | CD34+ HSPCs/erythroid differentiated cells | mRNA; electroporation | 77% editing efficiency, 80% Makassar editing | [125] Clinical trials | ||
| HBG1, HBG2 | ABE8s-mediated point mutation to create “British mutation” and increase levels of γ-globin | CD34+ cells, human T cells | RNP; electroporation | 80% editing efficiency, 60% protein knockdown efficiency | [129] Clinical trials | ||
| HBB (−28) | A3A(N57Q)-BE3-mediated BCL11A enhancer disruption to reproduce γ-globin | CD34+ HSPCs | RNP; electroporation | >20% editing efficiency | [47] | ||
| BCL11A (+58) | A3A(N57Q)-BE3-mediated BCL11A erythroid-specific enhancer disruption to reproduce γ-globin | CD34+ HSPCs | RNP; electroporation | 93.3% editing efficiency, restoring >60% of γ-globin | [130] | ||
| BCL11A | NHEJ-mediated BCL11A erythroid-specific enhancer disruption | CD34+ HSPCs | RNP; electroporation | 80% editing efficiency, 30% decrease in sickle hemoglobin | [131] Clinical trials | ||
| HBB | NHEJ-mediated mRNA splicing | CD34+ HSPCs | Cas12a RNP; electroporation | >30% editing efficiency, restoring >60% of γ-globin | [132] | ||
| BCL11A | NHEJ-mediated BCL11A erythroid-specific enhancer disruption | HSPCs | AsCas12a/Cpf1 RNP; electroporation | ~80% editing efficiency | [133] | ||
| HBG | NHEJ-mediated HBG disruption | CD34+ cells | AsCas12a/Cpf1 RNP; electroporation | >80% editing efficiency | [134] Clinical trials | ||
| Eye diseases | LCA10 | CEP290 | HDR-mediated IVS26 mutation deletion | HEK293FT/Mice | Dual AAV5 | 7.5–26.4% editing efficiency | [135] |
| CEP290 | NHEJ-mediated IVS26 mutation deletion | iPSC | AAV5 | >50% editing efficiency | [136] | ||
| Cep290 | HDR-mediated IVS26 mutation deletion or inversion | Mice | AAV5 | ~30% editing efficiency | [137] Clinical trials | ||
| LCA2 | Rpe65 | HDR-mediated point mutation correction | Mice | AAV9 | >1% editing efficiency | [138] | |
| Sox2, Klf4 or Oct4 | HyperdCas12a activator-mediated endogenous transcription factor gene activation | Mice | In vivo electroporation | [139] | |||
| Metabolic disorders | FH | LDLR | HDR-mediated base insertion | iPSC | Cas9 RNP; electroporation | ~10% editing efficiency | [140] |
| Ldlr | HDR-mediated point mutation correction | Mice | Single AAV8 | 6.7% editing efficiency, restoring 18% of LDLR protein | [141] | ||
| Pcsk9 | Intein-mediated SaCBE-mediated point mutation correction | Mice | Dual AAV8 | 25% editing efficiency | [142] | ||
| PCSK9 | ABE8.8-mediated mRNA splicing | Macaca fascicularis | LNP | 90% PCSK9 reduction | [143,144] Clinical trials | ||
| Pcsk9 | SaKKH-ABE8e-mediated mRNA splicing | Mice | Single AAV8 | 66% editing efficiency, 93% PCSK9 knockdown efficiency | [145] | ||
| Pcsk9 | Split-cPE573-mediated stop codon insertion | Mice | Dual AAV8 | 13.5% editing efficiency | [146] | ||
| Pcsk9 | BE3-mediated stop codon generation | Mice (in utero) | Adenoviral (Ad) vectors | 14.5% editing efficiency | [147] | ||
| Pcsk9 | ABE-eVLPs-mediated the splice donor disruption for Pcsk9 knockdown | Mice | eVLPs | 63% editing efficiency | [119] | ||
| HT1 | Fah | HDR-mediated point mutation correction | Adult Mice | Hydrodynamic injection | 0.4% editing efficiency, restoring 8–36% of Fah mRNA | [148] | |
| Fah | HDR-mediated point mutation correction | Adult Mice | Lipid Nanoparticles (Cas9 mRNA) and AAV (sgRNA/HDR) | 6% editing efficiency, restoring 9.5% of Fah mRNA | [149] | ||
| Fah | Intein-split BE4max-mediated point mutation correction | Mice | Dual rAAV | 31% editing efficiency | [150] | ||
| Hpd | NHEJ-mediated gene deletion | Mice | Hydrodynamic tail vein injection | The editing efficiencies at 1 and 4 weeks were 8% and 68% | [151] | ||
| Hpd | BE3-mediated stop codon generation | Mice | Adenoviral (Ad) vectors | 36% editing efficiency | [147] | ||
| PKU | Pah | Intein-split nSaKKH-BE3-mediated point mutation correction | Mice | Dual AAV8 | 25.1% editing efficiency, restoring 63% of PAH mRNA | [152] | |
| Neuromuscular diseases | DMD | Dmd | Split-intein NG-ABEmax-mediated exon 50 skipping | Mice | Dual AAV9 | 35% editing efficiency, restoring 54% of dystrophin protein expression | [153] |
| DMD | PE2-mediated two bases insertion | iPSC-derived cardiomyocytes | P3 Primary Cell 4D-Nucleofector X Kit | 54% editing efficiency, restoring 39.7% of dystrophin protein expression | [153] | ||
| Dmd | ABE7.10-mediated point mutation correction | Mice | Trans-splicing AAV | 3.3% editing efficiency, restoring 17% of dystrophin protein expression | [154] | ||
| Dmd | SaCas9-mediated exon 23 deletion | Neonatal Mice | AAV9 | 39% editing efficiency, restoring 7% of dystrophin protein expression | [155] | ||
| DMD | TwinPE-mediated exon 51 deletion | HEK293T | Lipofectamine 2000 | 28% editing efficiency | [70] | ||
| Dmd | NHEJ-mediated mRNA (exon 50) splicing | Mice | AAV9 | 27.9% editing efficiency, restoring 90% of dystrophin protein expression | [156] | ||
| DMD | KKH SaCas9-based TAM-mediated exon 50 skipping | iPSC | Lipofectamine LTX Reagent | 90% editing efficiency | [157] | ||
| Dmd | CRISPR-Cas9-AID (eTAM)-mediated exon 4 skipping | Mice | AAV9 | >50% editing efficiency, restoring ~90% of dystrophin protein expression | [158] | ||
| Dmd | ADAR-mediated point mutation correction | Mice | AAV8 | 3.6% editing efficiency, restoring 1–2.5% of dystrophin protein expression | [159] | ||
| ALS | SOD1 | HDR-mediated point mutation correction | iPSCs | Plasmid; electroporation | 20% editing efficiency | [160] | |
| FUS | HDR-mediated point mutation correction | iPSCs | Plasmid; electroporation | 1% editing efficiency | [160] | ||
| C9orf72 | HDR-mediated MRE correction | iPSCs | Plasmid; electroporation | 0.6–4.5% editing efficiency | [161] | ||
| C9orf72 | HDR-mediated MRE correction | iPSCs | Plasmid; electroporation | Reducing 22.5% of C9orf72 MRE expression | [162] | ||
| DM1 | DMPK | NHEJ-mediated MRE excision | myoblasts | Nucleofection | 51% editing efficiency | [163] | |
| DMPK | NHEJ-mediated MRE excision | Fibroblast/myoblasts | RNP; electroporation | 14% editing efficiency | [164] | ||
| DMPK | NHEJ-mediated MRE excision | DM1 iPSCs | Nucleofection | >50% editing efficiency | [165] | ||
| DMPK | NHEJ-mediated MRE excision | iPSCs and myogenic cells | Lentivirus | >50% editing efficiency | [166] | ||
| DMPK | PIN-dRCas9 mediated CUG repeat RNA cleavage | myoblasts and fibroblasts | AAV9/Lentivirus | [167] | |||
| Dmpk | PIN-dRCas9 mediated CUG repeat RNA cleavage | Neonatal mice/Adult mice | AAV9 | [168] | |||
| DMPK | LshCas13a-mediated CUG repeat RNA cleavage | myoblasts | Lentivirus | 20% reduction in DMPK mRNA level | [169] | ||
| NPC | Npc | BE3-mediated point mutation correction | Mice | AAV9 | 59% editing efficiency | [142] |
4.2. Eye Diseases
4.3. Metabolic Disorders
4.4. Neuromuscular Disease
5. Discussion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Schieppati, A.; Henter, J.I.; Daina, E.; Aperia, A. Why rare diseases are an important medical and social issue. Lancet 2008, 371, 2039–2041. [Google Scholar] [CrossRef] [PubMed]
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Waldboth, V.; Patch, C.; Mahrer-Imhof, R.; Metcalfe, A. Living a normal life in an extraordinary way: A systematic review investigating experiences of families of young people’s transition into adulthood when affected by a genetic and chronic childhood condition. Int. J. Nurs. Stud. 2016, 62, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Angural, A.; Spolia, A.; Mahajan, A.; Verma, V.; Sharma, A.; Kumar, P.; Dhar, M.K.; Pandita, K.K.; Rai, E.; Sharma, S. Review: Understanding Rare Genetic Diseases in Low Resource Regions Like Jammu and Kashmir—India. Front. Genet. 2020, 11, 415. [Google Scholar] [CrossRef]
- Evans, W.R.; Rafi, I. Rare diseases in general practice: Recognising the zebras among the horses. Br. J. Gen. Pract. 2016, 66, 550–551. [Google Scholar] [CrossRef]
- Papasavva, P.; Kleanthous, M.; Lederer, C.W. Rare Opportunities: CRISPR/Cas-Based Therapy Development for Rare Genetic Diseases. Mol. Diagn. Ther. 2019, 23, 201–222. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Thierry, A.; Dujon, B. Nested chromosomal fragmentation in yeast using the meganuclease I-Sce I: A new method for physical mapping of eukaryotic genomes. Nucleic Acids. Res. 1992, 20, 5625–5631. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asmamaw, M.; Zawdie, B. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biologics 2021, 15, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 2019, 566, 218–223. [Google Scholar] [CrossRef]
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E.; Tsuchida, C.A.; Li, Z.; Cress, B.F.; Knott, G.J.; Jacobsen, S.E.; Banfield, J.F.; Doudna, J.A. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 2020, 369, 333–337. [Google Scholar] [CrossRef]
- Xu, X.; Chemparathy, A.; Zeng, L.; Kempton, H.R.; Shang, S.; Nakamura, M.; Qi, L.S. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol. Cell 2021, 81, 4333–4345.e4334. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [Green Version]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef]
- Paul, B.; Montoya, G. CRISPR-Cas12a: Functional overview and applications. Biomed. J. 2020, 43, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Fonfara, I.; Richter, H.; Bratovič, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Toth, E.; Varga, E.; Kulcsar, P.I.; Kocsis-Jutka, V.; Krausz, S.L.; Nyeste, A.; Welker, Z.; Huszar, K.; Ligeti, Z.; Talas, A.; et al. Improved LbCas12a variants with altered PAM specificities further broaden the genome targeting range of Cas12a nucleases. Nucleic Acids. Res. 2020, 48, 3722–3733. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef]
- Cai, Y.; Cheng, T.; Yao, Y.; Li, X.; Ma, Y.; Li, L.; Zhao, H.; Bao, J.; Zhang, M.; Qiu, Z.; et al. In vivo genome editing rescues photoreceptor degeneration via a Cas9/RecA-mediated homology-directed repair pathway. Sci. Adv. 2019, 5, eaav3335. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, R.; Offen, D. Single-Base Resolution: Increasing the Specificity of the CRISPR-Cas System in Gene Editing. Mol. Ther. 2021, 29, 937–948. [Google Scholar] [CrossRef]
- Ryan, D.E.; Taussig, D.; Steinfeld, I.; Phadnis, S.M.; Lunstad, B.D.; Singh, M.; Vuong, X.; Okochi, K.D.; McCaffrey, R.; Olesiak, M.; et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids. Res. 2018, 46, 792–803. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Luk, K.; Wolfe, S.A.; Kim, J.S. Evaluating and Enhancing Target Specificity of Gene-Editing Nucleases and Deaminases. Annu. Rev. Biochem. 2019, 88, 191–220. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, J.; Yin, W.; Zhang, Z.; Song, Y.; Chang, X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods 2016, 13, 1029–1035. [Google Scholar] [CrossRef]
- Yu, Y.; Leete, T.C.; Born, D.A.; Young, L.; Barrera, L.A.; Lee, S.J.; Rees, H.A.; Ciaramella, G.; Gaudelli, N.M. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 2020, 11, 2052. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, J.M.; Cervantes, O.; Clement, M.K.; Wu, Y.; Zeng, J.; Bauer, D.E.; Pinello, L.; Joung, J.K. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol. 2018, 36, 977–982. [Google Scholar] [CrossRef]
- Thuronyi, B.W.; Koblan, L.W.; Levy, J.M.; Yeh, W.H.; Zheng, C.; Newby, G.A.; Wilson, C.; Bhaumik, M.; Shubina-Oleinik, O.; Holt, J.R.; et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 2019, 37, 1070–1079. [Google Scholar] [CrossRef]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [Green Version]
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grunewald, J.; Joung, J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021, 39, 41–46. [Google Scholar] [CrossRef]
- Chen, L.; Park, J.E.; Paa, P.; Rajakumar, P.D.; Prekop, H.T.; Chew, Y.T.; Manivannan, S.N.; Chew, W.L. Programmable C:G to G:C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat. Commun. 2021, 12, 1384. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, B.; Ru, G.; Meng, H.; Yan, Y.; Hong, M.; Zhang, D.; Luan, C.; Zhang, S.; Wu, H.; et al. Re-engineering the adenine deaminase TadA-8e for efficient and specific CRISPR-based cytosine base editing. Nat. Biotechnol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.H.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 2018, 9, 3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Kim, D.E.; Lee, G.; Cho, S.I.; Kim, J.S. Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat. Biotechnol. 2019, 37, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Doman, J.L.; Raguram, A.; Newby, G.A.; Liu, D.R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 2020, 38, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, M.E.; Hsu, A.; Arbab, M.; Krasnow, N.A.; McElroy, A.N.; Pandey, S.; Doman, J.L.; Huang, T.P.; Raguram, A.; Banskota, S.; et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nat. Biotechnol. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Sun, Y.; Yan, R.; Liu, Y.; Zuo, E.; Gu, C.; Han, L.; Wei, Y.; Hu, X.; Zeng, R.; et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 2019, 571, 275–278. [Google Scholar] [CrossRef]
- Grünewald, J.; Zhou, R.; Garcia, S.P.; Iyer, S.; Lareau, C.A.; Aryee, M.J.; Joung, J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 2019, 569, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Wilson, C.; Doman, J.L.; Liu, D.R. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv. 2019, 5, eaax5717. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, J.; Zhou, R.; Iyer, S.; Lareau, C.A.; Garcia, S.P.; Aryee, M.J.; Joung, J.K. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 2019, 37, 1041–1048. [Google Scholar] [CrossRef]
- Li, J.; Yu, W.; Huang, S.; Wu, S.; Li, L.; Zhou, J.; Cao, Y.; Huang, X.; Qiao, Y. Structure-guided engineering of adenine base editor with minimized RNA off-targeting activity. Nat. Commun. 2021, 12, 2287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tao, W.; Huang, S.; Sun, W.; Wang, Y.; Jiang, W.; Huang, X.; Lin, C.P. Engineering an adenine base editor in human embryonic stem cells with minimal DNA and RNA off-target activities. Mol. Ther. Nucleic Acids 2022, 29, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Cirincione, A.; Adamson, B. Prime Editing: Precision Genome Editing by Reverse Transcription. Mol. Cell 2020, 77, 210–212. [Google Scholar] [CrossRef]
- Gao, R.; Fu, Z.C.; Li, X.; Wang, Y.; Wei, J.; Li, G.; Wang, L.; Wu, J.; Huang, X.; Yang, L.; et al. Genomic and Transcriptomic Analyses of Prime Editing Guide RNA-Independent Off-Target Effects by Prime Editors. CRISPR J. 2022, 5, 276–293. [Google Scholar] [CrossRef]
- Nelson, J.W.; Randolph, P.B.; Shen, S.P.; Everette, K.A.; Chen, P.J.; Anzalone, A.V.; An, M.; Newby, G.A.; Chen, J.C.; Hsu, A.; et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022, 40, 402–410. [Google Scholar] [CrossRef]
- Choi, J.; Chen, W.; Suiter, C.C.; Lee, C.; Chardon, F.M.; Yang, W.; Leith, A.; Daza, R.M.; Martin, B.; Shendure, J. Precise genomic deletions using paired prime editing. Nat. Biotechnol. 2022, 40, 218–226. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Gao, X.D.; Podracky, C.J.; Nelson, A.T.; Koblan, L.W.; Raguram, A.; Levy, J.M.; Mercer, J.A.M.; Liu, D.R. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 2022, 40, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Liu, J.; Wu, H.; Zhu, Q.; Yan, Y.; Meng, H.; Chen, P.R.; Yi, C. Increasing the efficiency and precision of prime editing with guide RNA pairs. Nat. Chem. Biol. 2022, 18, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Han, Y.; Wang, Y.; Huang, H.; Qian, P. Programmable System of Cas13-Mediated RNA Modification and Its Biological and Biomedical Applications. Front. Cell Dev. Biol. 2021, 9, 677587. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [Green Version]
- Kellner, M.J.; Koob, J.G.; Gootenberg, J.S.; Abudayyeh, O.O.; Zhang, F. SHERLOCK: Nucleic acid detection with CRISPR nucleases. Nat. Protoc. 2019, 14, 2986–3012. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Zhou, J.; Yang, C.; Wang, G.; Tan, Y.; Wu, Y.; Zhang, S.; Yi, K.; Kang, C. The CRISPR-Cas13a Gene-Editing System Induces Collateral Cleavage of RNA in Glioma Cells. Adv. Sci. 2019, 6, 1901299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, H.; Huang, J.; Xiao, Q.; He, B.; Dong, X.; Liu, Y.; Yang, X.; Han, D.; Wang, Z.; Wang, X.; et al. High-fidelity Cas13 variants for targeted RNA degradation with minimal collateral effects. Nat. Biotechnol. 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A cytosine deaminase for programmable single-base RNA editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Altae-Tran, H.; Jin, X.; Madigan, V.J.; Oshiro, R.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Compact RNA editors with small Cas13 proteins. Nat. Biotechnol. 2022, 40, 194–197. [Google Scholar] [CrossRef]
- Vogel, P.; Stafforst, T. Critical review on engineering deaminases for site-directed RNA editing. Curr. Opin. Biotechnol. 2019, 55, 74–80. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.R.P.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Behr, M.; Zhou, J.; Xu, B.; Zhang, H. In vivo delivery of CRISPR-Cas9 therapeutics: Progress and challenges. Acta Pharm. Sin. B 2021, 11, 2150–2171. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, F.; Ding, Y. CRISPR/Cas9 Delivery System Engineering for Genome Editing in Therapeutic Applications. Pharmaceutics 2021, 13, 1649. [Google Scholar] [CrossRef]
- Li, L.; Hu, S.; Chen, X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials 2018, 171, 207–218. [Google Scholar] [CrossRef]
- Xu, W. Microinjection and Micromanipulation: A Historical Perspective. Methods Mol. Biol. 2019, 1874, 1–16. [Google Scholar] [CrossRef]
- Pensado, A.; Seijo, B.; Sanchez, A. Current strategies for DNA therapy based on lipid nanocarriers. Expert Opin. Drug Deliv. 2014, 11, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Nishikawa, A.; Kume, S.; Chayama, K.; Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014, 4, 5400. [Google Scholar] [CrossRef] [Green Version]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials 2020, 10, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X. Gold Nanoparticles: Recent Advances in the Biomedical Applications. Cell Biochem. Biophys. 2015, 72, 771–775. [Google Scholar] [CrossRef]
- Suresh, B.; Ramakrishna, S.; Kim, H. Cell-Penetrating Peptide-Mediated Delivery of Cas9 Protein and Guide RNA for Genome Editing. Methods Mol. Biol. 2017, 1507, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Campbell, S.; Vogt, V.M. In vitro assembly of virus-like particles with Rous sarcoma virus Gag deletion mutants: Identification of the p10 domain as a morphological determinant in the formation of spherical particles. J. Virol. 1997, 71, 4425–4435. [Google Scholar] [CrossRef] [Green Version]
- Urbinati, F.; Arumugam, P.; Higashimoto, T.; Perumbeti, A.; Mitts, K.; Xia, P.; Malik, P. Mechanism of reduction in titers from lentivirus vectors carrying large inserts in the 3′LTR. Mol. Ther. 2009, 17, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J., Jr.; Gusella, G.L.; Najfeld, V.; Klotman, M.E.; Cara, A. Novel integrase-defective lentiviral episomal vectors for gene transfer. Hum. Gene. Ther. 2004, 15, 361–372. [Google Scholar] [CrossRef]
- Sánchez-Hernández, S.; Gutierrez-Guerrero, A.; Martín-Guerra, R.; Cortijo-Gutierrez, M.; Tristán-Manzano, M.; Rodriguez-Perales, S.; Sanchez, L.; Garcia-Perez, J.L.; Chato-Astrain, J.; Fernandez-Valades, R.; et al. The IS2 Element Improves Transcription Efficiency of Integration-Deficient Lentiviral Vector Episomes. Mol. Ther. Nucleic Acids 2018, 13, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [Green Version]
- Wiethoff, C.M.; Nemerow, G.R. Adenovirus membrane penetration: Tickling the tail of a sleeping dragon. Virology 2015, 479–480, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Granelli-Piperno, A.; Choi, Y.; Steinman, R.M. Recombinant adenovirus is an efficient and non-perturbing genetic vector for human dendritic cells. Eur. J. Immunol. 1999, 29, 964–972. [Google Scholar] [CrossRef]
- Sonntag, F.; Bleker, S.; Leuchs, B.; Fischer, R.; Kleinschmidt, J.A. Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 2006, 80, 11040–11054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [Green Version]
- Cearley, C.N.; Vandenberghe, L.H.; Parente, M.K.; Carnish, E.R.; Wilson, J.M.; Wolfe, J.H. Expanded repertoire of AAV vector serotypes mediate unique patterns of transduction in mouse brain. Mol. Ther. 2008, 16, 1710–1718. [Google Scholar] [CrossRef]
- Gray, S.J.; Nagabhushan Kalburgi, S.; McCown, T.J.; Jude Samulski, R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene. Ther. 2013, 20, 450–459. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Stanford, W.; de Solis, C.; Aradhana; Abraham, N.D.; Dao, T.J.; Thaseen, S.; Sairavi, A.; Gonzalez, C.U.; Ploski, J.E. The Development of an AAV-Based CRISPR SaCas9 Genome Editing System That Can Be Delivered to Neurons in vivo and Regulated via Doxycycline and Cre-Recombinase. Front. Mol. Neurosci. 2018, 11, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, D.; Yue, Y.; Yan, Z.; Engelhardt, J.F. A new dual-vector approach to enhance recombinant adeno-associated virus-mediated gene expression through intermolecular cis activation. Nat. Med. 2000, 6, 595–598. [Google Scholar] [CrossRef]
- Ghosh, A.; Yue, Y.; Lai, Y.; Duan, D. A hybrid vector system expands adeno-associated viral vector packaging capacity in a transgene-independent manner. Mol. Ther. 2008, 16, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, W.; Wang, B.; Xiao, X.; Liu, X.Q. Protein trans-splicing as a means for viral vector-mediated in vivo gene therapy. Hum. Gene. Ther. 2008, 19, 958–964. [Google Scholar] [CrossRef] [Green Version]
- Zhi, S.; Chen, Y.; Wu, G.; Wen, J.; Wu, J.; Liu, Q.; Li, Y.; Kang, R.; Hu, S.; Wang, J.; et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 2022, 30, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: Preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnol. 2021, 19, 59. [Google Scholar] [CrossRef]
- Campbell, L.A.; Coke, L.M.; Richie, C.T.; Fortuno, L.V.; Park, A.Y.; Harvey, B.K. Gesicle-Mediated Delivery of CRISPR/Cas9 Ribonucleoprotein Complex for Inactivating the HIV Provirus. Mol. Ther. 2019, 27, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Bell, N.M.; Lever, A.M. HIV Gag polyprotein: Processing and early viral particle assembly. Trends Microbiol. 2013, 21, 136–144. [Google Scholar] [CrossRef]
- Aoki, T.; Miyauchi, K.; Urano, E.; Ichikawa, R.; Komano, J. Protein transduction by pseudotyped lentivirus-like nanoparticles. Gene. Ther. 2011, 18, 936–941. [Google Scholar] [CrossRef] [Green Version]
- Gee, P.; Lung, M.S.Y.; Okuzaki, Y.; Sasakawa, N.; Iguchi, T.; Makita, Y.; Hozumi, H.; Miura, Y.; Yang, L.F.; Iwasaki, M.; et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat. Commun. 2020, 11, 1334. [Google Scholar] [CrossRef] [Green Version]
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 2022, 185, 250–265.e216. [Google Scholar] [CrossRef]
- Raguram, A.; Banskota, S.; Liu, D.R. Therapeutic in vivo delivery of gene editing agents. Cell 2022, 185, 2806–2827. [Google Scholar] [CrossRef] [PubMed]
- Roessler, H.I.; Knoers, N.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef]
- Blin, O.; Lefebvre, M.N.; Rascol, O.; Micallef, J. Orphan drug clinical development. Therapie 2020, 75, 141–147. [Google Scholar] [CrossRef]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Prim. 2018, 4, 18010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newby, G.A.; Yen, J.S.; Woodard, K.J.; Mayuranathan, T.; Lazzarotto, C.R.; Li, Y.; Sheppard-Tillman, H.; Porter, S.N.; Yao, Y.; Mayberry, K.; et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 2021, 595, 295–302. [Google Scholar] [CrossRef]
- Chu, S.H.; Packer, M.; Rees, H.; Lam, D.; Yu, Y.; Marshall, J.; Cheng, L.I.; Lam, D.; Olins, J.; Ran, F.A.; et al. Rationally Designed Base Editors for Precise Editing of the Sickle Cell Disease Mutation. CRISPR J. 2021, 4, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Uda, M.; Galanello, R.; Sanna, S.; Lettre, G.; Sankaran, V.G.; Chen, W.; Usala, G.; Busonero, F.; Maschio, A.; Albai, G.; et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 1620–1625. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.E.; Kamran, S.C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M.C.; Smith, E.C.; Pinello, L.; et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013, 342, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forget, B.G. Molecular basis of hereditary persistence of fetal hemoglobin. Ann. N. Y. Acad. Sci. 1998, 850, 38–44. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Sola-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.J.; Liquori, A.J.; et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Zeng, J.; Wu, Y.; Ren, C.; Bonanno, J.; Shen, A.H.; Shea, D.; Gehrke, J.M.; Clement, K.; Luk, K.; Yao, Q.; et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 2020, 26, 535–541. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Luk, K.; Yao, Q.; Shen, A.H.; Zeng, J.; Wu, Y.; Luo, H.Y.; Brendel, C.; Pinello, L.; Chui, D.H.K.; et al. Editing aberrant splice sites efficiently restores beta-globin expression in beta-thalassemia. Blood 2019, 133, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zuris, J.A.; Viswanathan, R.; Edelstein, J.N.; Turk, R.; Thommandru, B.; Rube, H.T.; Glenn, S.E.; Collingwood, M.A.; Bode, N.M.; et al. AsCas12a ultra nuclease facilitates the rapid generation of therapeutic cell medicines. Nat. Commun. 2021, 12, 3908. [Google Scholar] [CrossRef] [PubMed]
- Sousa, P.; Janoudi, T.; deDreuzy, E.; Shearman, M.S.; Zhang, K.; Chang, K.-H. Preclinical Development of EDIT301, an Autologous Cell Therapy Comprising AsCas12a-RNP Modified Mobilized Peripheral Blood-CD34 + Cells for the Potential Treatment of Transfusion Dependent Beta Thalassemia. Blood 2021, 138, 1858. [Google Scholar] [CrossRef]
- Ruan, G.X.; Barry, E.; Yu, D.; Lukason, M.; Cheng, S.H.; Scaria, A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol. Ther. 2017, 25, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Burnight, E.R.; Gupta, M.; Wiley, L.A.; Anfinson, K.R.; Tran, A.; Triboulet, R.; Hoffmann, J.M.; Klaahsen, D.L.; Andorf, J.L.; Jiao, C.; et al. Using CRISPR-Cas9 to Generate Gene-Corrected Autologous iPSCs for the Treatment of Inherited Retinal Degeneration. Mol. Ther. 2017, 25, 1999–2013. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef]
- Jo, D.H.; Song, D.W.; Cho, C.S.; Kim, U.G.; Lee, K.J.; Lee, K.; Park, S.W.; Kim, D.; Kim, J.H.; Kim, J.S.; et al. CRISPR-Cas9-mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Sci. Adv. 2019, 5, eaax1210. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.Y.; Bian, J.; Davis, A.E.; Liu, P.; Kempton, H.R.; Zhang, X.; Chemparathy, A.; Gu, B.; Lin, X.; Rane, D.A.; et al. Multiplexed genome regulation in vivo with hyper-efficient Cas12a. Nat. Cell Biol. 2022, 24, 590–600. [Google Scholar] [CrossRef]
- Omer, L.; Hudson, E.A.; Zheng, S.; Hoying, J.B.; Shan, Y.; Boyd, N.L. CRISPR Correction of a Homozygous Low-Density Lipoprotein Receptor Mutation in Familial Hypercholesterolemia Induced Pluripotent Stem Cells. Hepatol. Commun. 2017, 1, 886–898. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.; Yeh, W.H.; Pendse, N.; Davis, J.R.; Hennessey, E.; Butcher, R.; Koblan, L.W.; Comander, J.; Liu, Q.; Liu, D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020, 4, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-course CRISPR Base Editing Therapy Targeting PCSK9 in Non-human Primate and Mouse Models. Circulation 2022. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Davis, J.R.; Wang, X.; Witte, I.P.; Huang, T.P.; Levy, J.M.; Raguram, A.; Banskota, S.; Seidah, N.G.; Musunuru, K.; Liu, D.R. Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors. Nat. Biomed. Eng. 2022, 6, 1272–1283. [Google Scholar] [CrossRef]
- Zheng, C.; Liang, S.Q.; Liu, B.; Liu, P.; Kwan, S.Y.; Wolfe, S.A.; Xue, W. A flexible split prime editor using truncated reverse transcriptase improves dual-AAV delivery in mouse liver. Mol. Ther. 2022, 30, 1343–1351. [Google Scholar] [CrossRef]
- Rossidis, A.C.; Stratigis, J.D.; Chadwick, A.C.; Hartman, H.A.; Ahn, N.J.; Li, H.; Singh, K.; Coons, B.E.; Li, L.; Lv, W.; et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med. 2018, 24, 1513–1518. [Google Scholar] [CrossRef]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Song, C.Q.; Dorkin, J.R.; Zhu, L.J.; Li, Y.; Wu, Q.; Park, A.; Yang, J.; Suresh, S.; Bizhanova, A.; et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016, 34, 328–333. [Google Scholar] [CrossRef]
- Yang, L.; Wang, L.; Huo, Y.; Chen, X.; Yin, S.; Hu, Y.; Zhang, X.; Zheng, R.; Geng, H.; Han, H.; et al. Amelioration of an Inherited Metabolic Liver Disease through Creation of a De Novo Start Codon by Cytidine Base Editing. Mol. Ther. 2020, 28, 1673–1683. [Google Scholar] [CrossRef]
- Pankowicz, F.P.; Barzi, M.; Legras, X.; Hubert, L.; Mi, T.; Tomolonis, J.A.; Ravishankar, M.; Sun, Q.; Yang, D.; Borowiak, M.; et al. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat. Commun. 2016, 7, 12642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villiger, L.; Grisch-Chan, H.M.; Lindsay, H.; Ringnalda, F.; Pogliano, C.B.; Allegri, G.; Fingerhut, R.; Haberle, J.; Matos, J.; Robinson, M.D.; et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018, 24, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Chemello, F.; Chai, A.C.; Li, H.; Rodriguez-Caycedo, C.; Sanchez-Ortiz, E.; Atmanli, A.; Mireault, A.A.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv. 2021, 7, eabg4910. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.T.; Kim, H.S.; Kim, D.E.; Lee, H.; Chung, E.; et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018, 36, 536–539. [Google Scholar] [CrossRef]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, eaan8081. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Ma, Y.; Huang, T.; Chen, Y.; Peng, Y.; Li, B.; Li, J.; Zhang, Y.; Song, B.; Sun, X.; et al. Genetic Modulation of RNA Splicing with a CRISPR-Guided Cytidine Deaminase. Mol. Cell 2018, 72, 380–394.e387. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, K.; Zhang, Y.; Qi, T.; Yuan, J.; Zhang, L.; Qiu, H.; Wang, J.; Yang, H.T.; Dai, Y.; et al. Therapeutic Exon Skipping Through a CRISPR-Guided Cytidine Deaminase Rescues Dystrophic Cardiomyopathy in Vivo. Circulation 2021, 144, 1760–1776. [Google Scholar] [CrossRef]
- Katrekar, D.; Chen, G.; Meluzzi, D.; Ganesh, A.; Worlikar, A.; Shih, Y.R.; Varghese, S.; Mali, P. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 2019, 16, 239–242. [Google Scholar] [CrossRef]
- Wang, L.; Yi, F.; Fu, L.; Yang, J.; Wang, S.; Wang, Z.; Suzuki, K.; Sun, L.; Xu, X.; Yu, Y.; et al. CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell 2017, 8, 365–378. [Google Scholar] [CrossRef]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca(2+)-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ababneh, N.A.; Scaber, J.; Flynn, R.; Douglas, A.; Barbagallo, P.; Candalija, A.; Turner, M.R.; Sims, D.; Dafinca, R.; Cowley, S.A.; et al. Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell-derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology-directed repair. Hum. Mol. Genet. 2020, 29, 2200–2217. [Google Scholar] [CrossRef] [PubMed]
- van Agtmaal, E.L.; Andre, L.M.; Willemse, M.; Cumming, S.A.; van Kessel, I.D.G.; van den Broek, W.; Gourdon, G.; Furling, D.; Mouly, V.; Monckton, D.G.; et al. CRISPR/Cas9-Induced (CTGCAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol. Ther. 2017, 25, 24–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, C.; Cappella, M.; Valaperta, R.; Cardani, R.; Meola, G.; Martelli, F.; Cardinali, B.; Falcone, G. CRISPR/Cas9-Mediated Deletion of CTG Expansions Recovers Normal Phenotype in Myogenic Cells Derived from Myotonic Dystrophy 1 Patients. Mol. Ther. Nucleic. Acids. 2017, 9, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hao, L.; Wang, H.; Santostefano, K.; Thapa, A.; Cleary, J.; Li, H.; Guo, X.; Terada, N.; Ashizawa, T.; et al. Therapeutic Genome Editing for Myotonic Dystrophy Type 1 Using CRISPR/Cas9. Mol. Ther. 2018, 26, 2617–2630. [Google Scholar] [CrossRef] [Green Version]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids. Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef] [Green Version]
- Batra, R.; Nelles, D.A.; Pirie, E.; Blue, S.M.; Marina, R.J.; Wang, H.; Chaim, I.A.; Thomas, J.D.; Zhang, N.; Nguyen, V.; et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 2017, 170, 899–912.e810. [Google Scholar] [CrossRef]
- Batra, R.; Nelles, D.A.; Roth, D.M.; Krach, F.; Nutter, C.A.; Tadokoro, T.; Thomas, J.D.; Sznajder, L.J.; Blue, S.M.; Gutierrez, H.L.; et al. The sustained expression of Cas9 targeting toxic RNAs reverses disease phenotypes in mouse models of myotonic dystrophy type 1. Nat. Biomed. Eng. 2021, 5, 157–168. [Google Scholar] [CrossRef]
- Zhang, N.; Bewick, B.; Xia, G.; Furling, D.; Ashizawa, T. A CRISPR-Cas13a Based Strategy That Tracks and Degrades Toxic RNA in Myotonic Dystrophy Type 1. Front. Genet. 2020, 11, 594576. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Cideciyan, A.V. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog. Retin. Eye Res. 2010, 29, 398–427. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.H.; Sharma, T. Leber Congenital Amaurosis. Adv. Exp. Med. Biol. 2018, 1085, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M. Leber congenital amaurosis—A model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 2007, 144, 791–811. [Google Scholar] [CrossRef] [PubMed]
- den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrault, I.; Delphin, N.; Hanein, S.; Gerber, S.; Dufier, J.L.; Roche, O.; Defoort-Dhellemmes, S.; Dollfus, H.; Fazzi, E.; Munnich, A.; et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum. Mutat. 2007, 28, 416. [Google Scholar] [CrossRef]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Mary, B.; Khan, N.; Arumugam, S.; Saxena, H.; Kumar, M.; Manimaran, P.; Chattopadhyay, S.; Jayandharan, G.R. Adeno-associated Virus Vectors in Gene Therapy. In Gene and Cell Therapy: Biology and Applications; Springer: Berlin/Heidelberg, Germany, 2018; pp. 29–56. [Google Scholar] [CrossRef]
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. Int. J. Mol. Sci. 2018, 19, 3426. [Google Scholar] [CrossRef] [Green Version]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Prim. 2017, 3, 17093. [Google Scholar] [CrossRef]
- Hobbs, H.H.; Brown, M.S.; Goldstein, J.L. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1992, 1, 445–466. [Google Scholar] [CrossRef]
- Stoekenbroek, R.M.; Kastelein, J.J.P. Proprotein convertase subtilisin/kexin type 9: From genetics to clinical trials. Curr. Opin. Cardiol. 2018, 33, 269–275. [Google Scholar] [CrossRef]
- Stein, E.A.; Gipe, D.; Bergeron, J.; Gaudet, D.; Weiss, R.; Dufour, R.; Wu, R.; Pordy, R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: A phase 2 randomised controlled trial. Lancet 2012, 380, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, A.C.; Wang, X.; Musunuru, K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arter. Thromb. Vasc. Biol. 2017, 37, 1741–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Xue, W.; Zhang, H.; Gao, R.; Qiu, H.; Wei, J.; Zhou, L.; Lei, Y.N.; Wu, X.; Li, X.; et al. Eliminating base-editor-induced genome-wide and transcriptome-wide off-target mutations. Nat. Cell Biol. 2021, 23, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Tanguay, R.M.; Bergeron, A.; Jorquera, R. Hepatorenal Tyrosinemia. Genet. Dis. Kidney 2009, 681–691. [Google Scholar] [CrossRef]
- Holme, E.; Lindstedt, S. Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione). J. Inherit. Metab. Dis. 1998, 21, 507–517. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Martinez, A.; Soblechero-Martin, P.; de-la-Puente-Ovejero, L.; Nogales-Gadea, G.; Arechavala-Gomeza, V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes 2020, 11, 1109. [Google Scholar] [CrossRef]
- Wheeler, S.; Sillence, D.J. Niemann-Pick type C disease: Cellular pathology and pharmacotherapy. J. Neurochem. 2020, 153, 674–692. [Google Scholar] [CrossRef]
- Chamberlain, J.R.; Chamberlain, J.S. Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2017, 25, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef] [Green Version]
- Farrokhi, V.; Walsh, J.; Palandra, J.; Brodfuehrer, J.; Caiazzo, T.; Owens, J.; Binks, M.; Neelakantan, S.; Yong, F.; Dua, P.; et al. Dystrophin and mini-dystrophin quantification by mass spectrometry in skeletal muscle for gene therapy development in Duchenne muscular dystrophy. Gene. Ther. 2021. [Google Scholar] [CrossRef]
- Komaki, H.; Nagata, T.; Saito, T.; Masuda, S.; Takeshita, E.; Sasaki, M.; Tachimori, H.; Nakamura, H.; Aoki, Y.; Takeda, S. Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Putten, M.; Hulsker, M.; Young, C.; Nadarajah, V.D.; Heemskerk, H.; van der Weerd, L.; Hoen, P.A.; van Ommen, G.J.; Aartsma-Rus, A.M. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB J. 2013, 27, 2484–2495. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Lyu, P.; Yoo, K.; Yadav, M.K.; Singh, R.; Atala, A.; Lu, B. Engineered extracellular vesicles as versatile ribonucleoprotein delivery vehicles for efficient and safe CRISPR genome editing. J. Extracell Vesicles 2021, 10, e12076. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.M.; Hartley, B.J.; Flaherty, E.; Rajarajan, P.; Abdelaal, R.; Obiorah, I.; Barretto, N.; Muhammad, H.; Phatnani, H.P.; Akbarian, S.; et al. Evaluating Synthetic Activation and Repression of Neuropsychiatric-Related Genes in hiPSC-Derived NPCs, Neurons, and Astrocytes. Stem Cell Rep. 2017, 9, 615–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.L.; Pickett-Leonard, M.; Riddle, M.J.; Chen, W.; Albert, F.W.; Tolar, J. Genes and compounds that increase type VII collagen expression as potential treatments for dystrophic epidermolysis bullosa. Exp. Dermatol. 2022, 31, 1065–1075. [Google Scholar] [CrossRef]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef]
- Wang, H.; Guo, R.; Du, Z.; Bai, L.; Li, L.; Cui, J.; Li, W.; Hoffman, A.R.; Hu, J.F. Epigenetic Targeting of Granulin in Hepatoma Cells by Synthetic CRISPR dCas9 Epi-suppressors. Mol. Ther. Nucleic Acids 2018, 11, 23–33. [Google Scholar] [CrossRef]
- Jin, P.; Warren, S.T. Understanding the molecular basis of fragile X syndrome. Hum. Mol. Genet. 2000, 9, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Klann, T.S.; Black, J.B.; Chellappan, M.; Safi, A.; Song, L.; Hilton, I.B.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017, 35, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Gao, X. Multiplex base- and prime-editing with drive-and-process CRISPR arrays. Nat. Commun. 2022, 13, 2771. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Gao, Y.; Wang, H. CRISPR-Based Tools for Fighting Rare Diseases. Life 2022, 12, 1968. https://doi.org/10.3390/life12121968
Li Q, Gao Y, Wang H. CRISPR-Based Tools for Fighting Rare Diseases. Life. 2022; 12(12):1968. https://doi.org/10.3390/life12121968
Chicago/Turabian StyleLi, Qingyang, Yanmin Gao, and Haifeng Wang. 2022. "CRISPR-Based Tools for Fighting Rare Diseases" Life 12, no. 12: 1968. https://doi.org/10.3390/life12121968