2. Materials and Methods
2.1. General
The preparation and characterization of starting sulfonyl chloride
1 as well as amine
5 were described previously [
15,
16,
17]. Other reactants and solvents were obtained from commercial sources and used without purification. All reactions were performed in an open flask without any protection from CO
2 and H
2O. Reactions were monitored using analytical thin layer chromatography (TLC) Macherey-Nagel, TLC plates Polygram
® Sil G/UV254. Visualization of the developed chromatograms was performed by fluorescence quenching at 254 nm.
1H and
13C NMR spectra were measured on Bruker AVANCE DPX 400 (400 MHz for
1H and 101 MHz for
13C, respectively). All chemical shifts (δ) are given in parts per million (ppm) with reference to solvent residues in DMSO-
d6 (2.50 for proton and 39.52 for carbon), and coupling constants (
J) are reported in hertz (Hz). Multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Melting points were determined using an Electrothermal IA 9300 series Digital Melting Point Apparatus. Mass spectra were recorded on microTOF spectrometers (ESI ionization).
2.2. Synthesis and Characterization of Compounds 2a–i
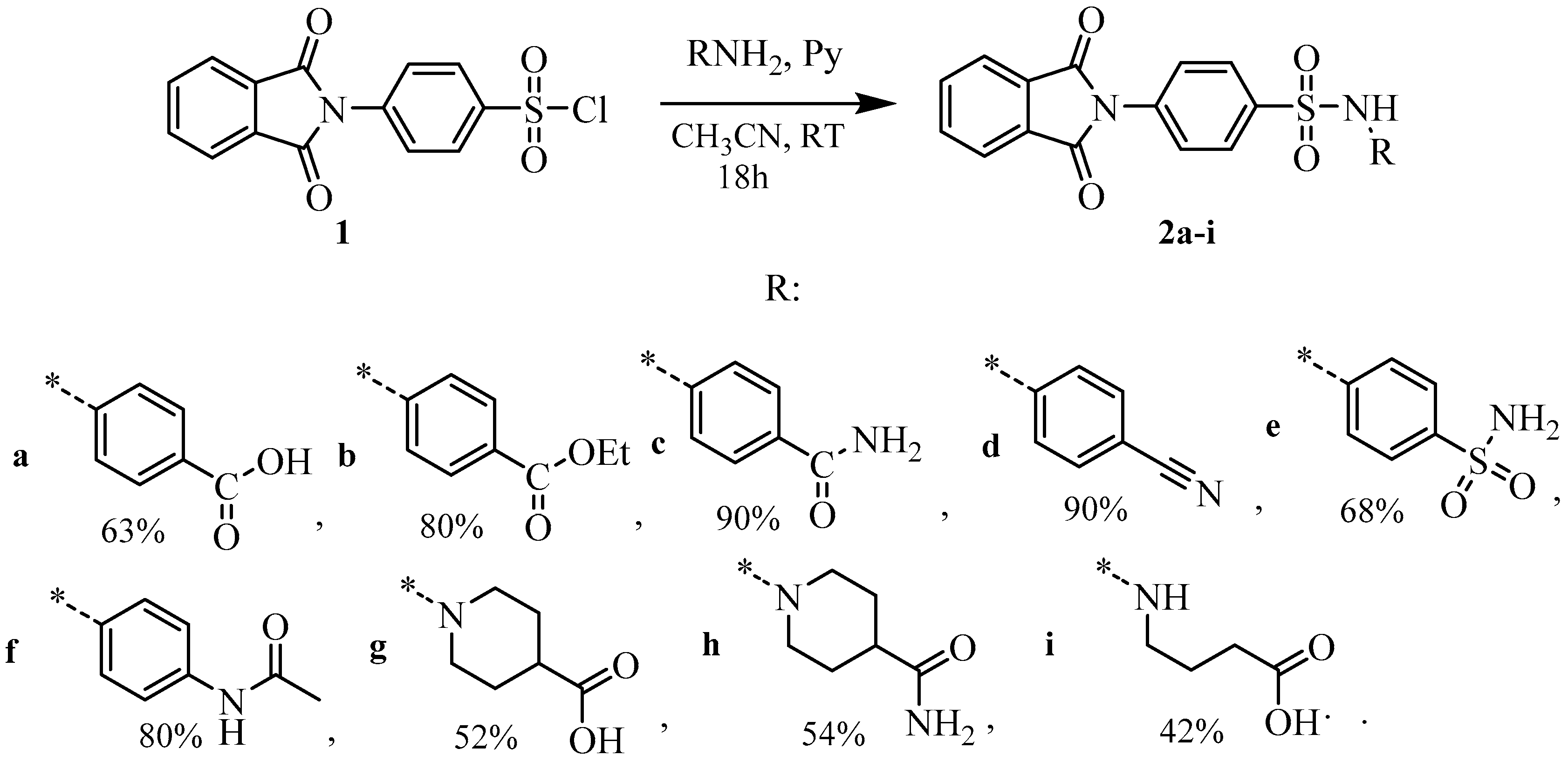
Pyridine (1.05 mmol) and 4-(1,3-dioxoisoindolin-2-yl)benzenesulfonyl chloride 1 (1 mmol) were added to a solution of the corresponding amine (1 mmol) in acetonitrile (2 mL). The mixture was stirred at room temperature for 6–12 h (TLC monitoring). Acetonitrile was removed in vacuo and the residue was treated with water (10 mL), and the precipitate was filtered off and dried. Compounds 2b, 2d, and 2i were isolated in acceptable purity, whereas other compounds (2a, 2c, 2e–h) were recrystallized from ethanol.
4-(4-(1,3-Dioxoisoindolin-2-yl)phenylsulfonamido)benzoic acid2a [13]: Yield 63%, white powder, mp 263–265 °C.
1H NMR (400 MHz, DMSO-
d6) δ: 12.75 (br.s, 1 H), 10.95 (s, 1 H), 8.02–7.97 (m, 4 H), 7.93–7.89 (m, 2 H), 7.83 (d,
J = 8.3 Hz, 2 H), 7.70 (d,
J = 8.4 Hz, 2 H), 7.25 (d,
J = 8.6 Hz, 2 H).
13C NMR (101 MHz, DMSO-
d6) δ: 167.36 (C=O), 167.10 (2 C=O), 142.46, 138.88, 136.71, 135.58 (2 C), 132.06 (2 C), 131.50 (2 C), 128.25 (2 C), 128.07 (2 C), 126.45, 124.28 (2 C), 118.82 (2 C).
Ethyl 4-(4-(1,3-dioxoisoindolin-2-yl)phenylsulfonamido)benzoate2b: Yield 80%, light pink powder, mp 255–257 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.99 (s, 1 H), 8.02–7.96 (m, 4 H), 7.95–7.88 (m, 2 H), 7.85 (d, J = 8.8 Hz, 2 H), 7.69 (d, J = 8.6 Hz, 2 H), 7.28 (d, J = 8.8 Hz, 2 H), 4.24 (q, J = 7.1 Hz, 2 H), 1.26 (t, J = 7.1 Hz, 3 H). 13C NMR (101 MHz, DMSO-d6) δ: 167.10 (2 C=O), 165.78 (C=O), 142.77, 138.78, 136.74, 135.58 (2 C), 132.07 (2 C), 131.34 (2 C), 128.26 (2 C), 128.09 (2 C), 125.54, 124.28 (2 C), 118.87 (2 C), 61.17 (CH2), 14.83 (CH3). HRMS (ESI), Calc. for C23H19N2O6S+: [M+H]+ 451.0958; found m/z 451.0966.
4-(4-(1,3-Dioxoisoindolin-2-yl)phenylsulfonamido)benzamide 2c: Yield 90%, white powder, mp 240–242 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.78 (s, 1 H), 8.01–7.94 (m, 4 H), 7.91 (dd, J = 5.5, 3.1 Hz, 2 H), 7.82 (s, 1 H), 7.75 (d, J = 8.4 Hz, 2 H), 7.69 (d, J = 8.3 Hz, 2 H), 7.24 (s, 1 H), 7.19 (d, J = 8.3 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 167.87 (CONH2), 167.12 (2 C=O), 140.93, 138.95, 136.62, 135.59 (2 C), 132.08 (2 C), 130.28, 129.52 (2 C), 128.24 (2 C), 128.06 (2 C), 124.29 (2 C), 118.91 (2 C). HRMS (ESI), Calc. for C21H16N3O5S+: [M+H]+ 422.0805; found m/z 422.0816.
N-(4-Cyanophenyl)-4-(1,3-dioxoisoindolin-2-yl)benzenesulfonamide 2d: Yield 90%, white powder, mp 224–226 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.20 (s, 1 H), 8.01 (d, J = 8.6 Hz, 2 H), 8.01–7.93 (m, 4 H), 7.74–7.70 (m, 4 H), 7.31 (d, J = 8.7 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 167.10 (2 C=O), 142.75, 138.63, 136.90, 135.60 (2 C), 134.47 (2 C), 132.09 (2 C), 128.37 (2 C), 128.09 (2 C), 124.30 (2 C), 119.34, 119.19 (2 C), 106.26 (C≡N). HRMS (ESI), Calc. for C21H14N3O4S+: [M+H]+ 404.0700; found m/z 404.0707.
4-(1,3-Dioxoisoindolin-2-yl)-N-(4-sulfamoylphenyl)benzenesulfonamide 2e: Yield 68%, white powder, mp 261–263 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.95 (s, 1 H), 8.03–7.95 (m, 4 H), 7.91 (dd, J = 5.5, 3.1 Hz, 2 H), 7.73–7.69 (m, 4 H), 7.32 (d, J = 8.8 Hz, 2 H), 7.20 (s, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 167.14 (2 C=O), 141.38, 139.65, 138.75, 136.76, 135.60 (2 C), 132.07(2 C), 128.32 (2 C), 128.13 (2 C), 127.97 (2 C), 124.29 (2 C), 119.10 (2 C). HRMS (ESI), Calc. for C20H16N3O6S2+: [M+H]+ 458.0475; found m/z 458.0489.
(N-(4-(4-(1,3-Dioxoisoindolin-2-yl)phenylsulfonamido)phenyl)acetamide 2f: Yield 81%, white powder, mp 220–222 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.18 (s, 1 H), 9.86 (s, 1 H), 8.00–7.93 (m, 4 H), 7.86 (d, J = 8.7 Hz, 2 H), 7.65 (d, J = 8.7 Hz, 2 H), 7.44 (d, J = 8.9 Hz, 2 H), 7.04 (d, J = 8.9 Hz, 2 H), 1.98 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ: 168.73 (C=O), 167.15 (2 C=O), 139.10, 136.86, 136.30, 135.57 (2 C), 132.88, 132.09 (2 C), 128.03 (4 C), 124.26 (2 C), 122.17 (2 C), 120.41 (2 C), 24.53 (CH3). HRMS (ESI), Calc. for C22H18N3O5S+: [M+H]+ 436.0962; found m/z 436.0969.
1-((4-(1,3-Dioxoisoindolin-2-yl)phenyl)sulfonyl)piperidine-4-carboxylic acid 2g: Yield 52%, white powder, mp 207–210 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.27 (br.s, 1 H), 8.02–7.98 (m, 2 H), 7.95–7.92 (m, 2 H), 7.91 (d, J = 8.3 Hz, 2 H), 7.79 (d, J = 8.3 Hz, 2 H), 3.53 (d, J = 11.7 Hz, 2 H), 2.53–2.47 (m, 2 H), 2.34 (t, J = 11.1 Hz, 1 H), 1.91 (dd, J = 13.2, 3.8 Hz, 2 H), 1.58 (td, J = 14.4, 13.6, 6.6 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 175.86 (C=O), 167.20 (2 C=O), 136.67, 135.60 (2 C), 135.24, 132.13 (2 C), 128.80 (2 C), 128.18 (2 C), 124.30 (2 C), 45.90 (2 CH2), 39.36 (CH), 27.90 (2 CH2). HRMS (ESI), Calc. for C20H19N2O6S+: [M+H]+ 415.0958; found m/z 415.0972.
1-((4-(1,3-Dioxoisoindolin-2-yl)phenyl)sulfonyl)piperidine-4-carboxamide 2h: Yield 54%, white powder, mp 217–220 °C. 1H NMR (400 MHz, DMSO-d6) δ: 8.03–7.98 (m, 2 H), 7.96–7.93 (m, 2 H), 7.91 (d, J = 8.3 Hz, 2 H), 7.77 (d, J = 8.3 Hz, 2 H), 7.20 (s, 1 H), 6.82 (s, 1 H), 3.62 (d, J = 11.6 Hz, 2 H), 2.36 (t, J = 11.7 Hz, 2 H), 2.10 (dd, J = 13.5, 8.8 Hz, 1 H), 1.80 (d, J = 11.9 Hz, 2 H), 1.57 (qd, J = 12.8, 4.4 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 175.48 (C=O), 166.52 (2 C=O), 135.97, 134.91 (2 C), 134.44, 131.45 (2 C), 128.13 (2 C), 127.47 (2 C), 123.62 (2 C), 45.47 (2 C), 40.08 (CH), 27.72 (2 C). HRMS (ESI), Calc. for C20H19N3O5S+: [M+H]+ 414.1118; found m/z 414.1122.
4-(4-(1,3-Dioxoisoindolin-2-yl)phenylsulfonamido)butanoic acid2i: Yield 42%, white powder, mp 160–162 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.09 (s, 1 H), 8.02–7.97 (m, 2 H), 7.96–7.91 (m, 4 H), 7.78–7.67 (m, 3 H), 2.81 (q, J = 6.9, 6.3 Hz, 2 H), 2.25 (t, J = 7.6 Hz, 2 H), 1.63 (p, J = 8.0, 7.5 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 174.61 (2 C=O), 167.25 (C=O), 140.24, 135.95, 135.56 (2 C), 132.15 (2 C), 128.25 (2 C), 127.78 (2 C), 124.27 (2 C), 42.66, 31.29, 25.36. HRMS (ESI), Calc. for C18H17N2O6S+: [M+H]+ 389.0802; found m/z 389.0813.
2.3. Synthesis and Characterization of Compounds 3 and 4
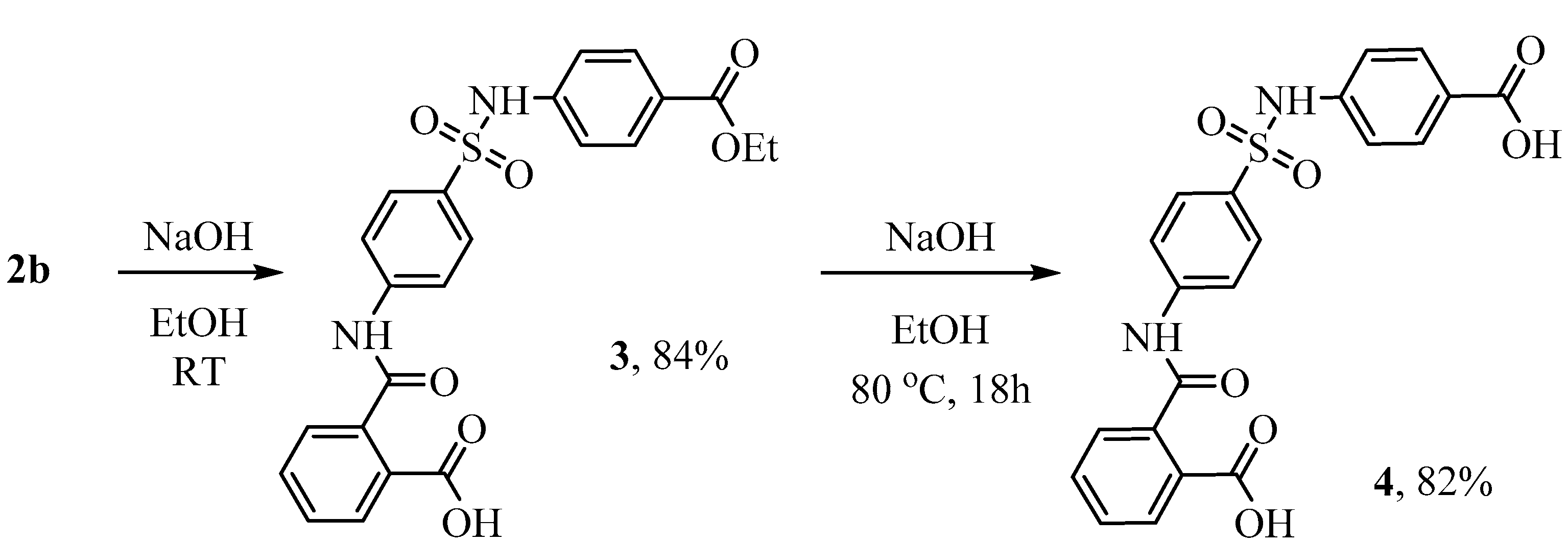
2-((4-(N-(4-(Ethoxycarbonyl)phenyl)sulfamoyl)phenyl)carbamoyl)benzoic acid 3 was obtained by heating ethyl 4-(4-(1,3-dioxoisoindolin-2-yl)phenylsulfonamido)benzoate 2b (1 mmol) at 50 °C in 50% aqueous ethanol with sodium hydroxide (2 mmol) for 2 h. The reaction mixture was then cooled to room temperature and acidified with 1 N hydrochloric acid to pH 2. The precipitate was collected by filtration, washed with water and dried. Yield 84%, white powder, mp 176–178 °C. 1H NMR (400 MHz, DMSO-d6) δ: 13.03 (br.s, 1 H), 10.75 (s, 2 H), 7.89 (d, J = 6.3 Hz, 1 H), 7.85–7.76 (m, 6 H), 7.66 (t, J = 8.2 Hz, 1 H), 7.58 (t, J = 8.2 Hz, 1 H), 7.53 (d, J = 8.0 Hz, 1 H), 7.23 (d, J = 8.8 Hz, 2 H), 4.24 (q, J = 7.1 Hz, 2 H), 1.27 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ: 168.67 (C=O), 167.80 (C=O), 165.83 (C=O), 144.29, 143.09, 139.03, 133.59, 132.57, 131.39, 131.24 (2 C), 130.35 (2 C), 128.67 (2 C), 128.41, 125.25, 119.83 (2 C), 118.77 (2 C), 61.14 (CH2), 14.84 (CH3). HRMS (ESI), Calc. for C23H21N2O7S+: [M+H]+ 469.1064; found m/z 469.1070.
2-((4-(N-(4-Carboxyphenyl)sulfamoyl)phenyl)carbamoyl)benzoic acid 4: The mixture (2b+3) or 2b (1 mmol) in 50% aqueous ethanol with sodium hydroxide (45 mmol) was refluxed for 12 h, cooled to room temperature and acidified with 1 N hydrochloric acid to pH 2. The solid was collected by filtration, washed with water and dried to give (2-((4-(N-(4-carboxyphenyl)sulfamoyl)phenyl)carbamoyl)benzoic acid 4 as a white solid. Yield 82%, white powder, mp 203–205 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.90 (s, 2 H), 10.73 (s, 2 H), 7.89 (d, J = 7.7 Hz, 1 H), 7.86–7.77 (m, 6 H), 7.66 (t, J = 7.5 Hz, 1 H), 7.58 (t, J = 7.6 Hz, 1 H), 7.53 (d, J = 7.6 Hz, 1 H), 7.20 (d, J = 8.8 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 168.68 (C=O), 167.80 (C=O), 167.42 (C=O), 144.26, 142.78, 139.06, 133.68, 132.57, 131.40 (2 C), 130.34, 130.32, 130.29, 128.65 (2 C), 128.41, 126.13, 119.84 (2 C), 118.70 (2 C). HRMS (ESI), Calc. for C21H17N2O7S+: [M+H]+ 441.0751; found m/z 441.0764.
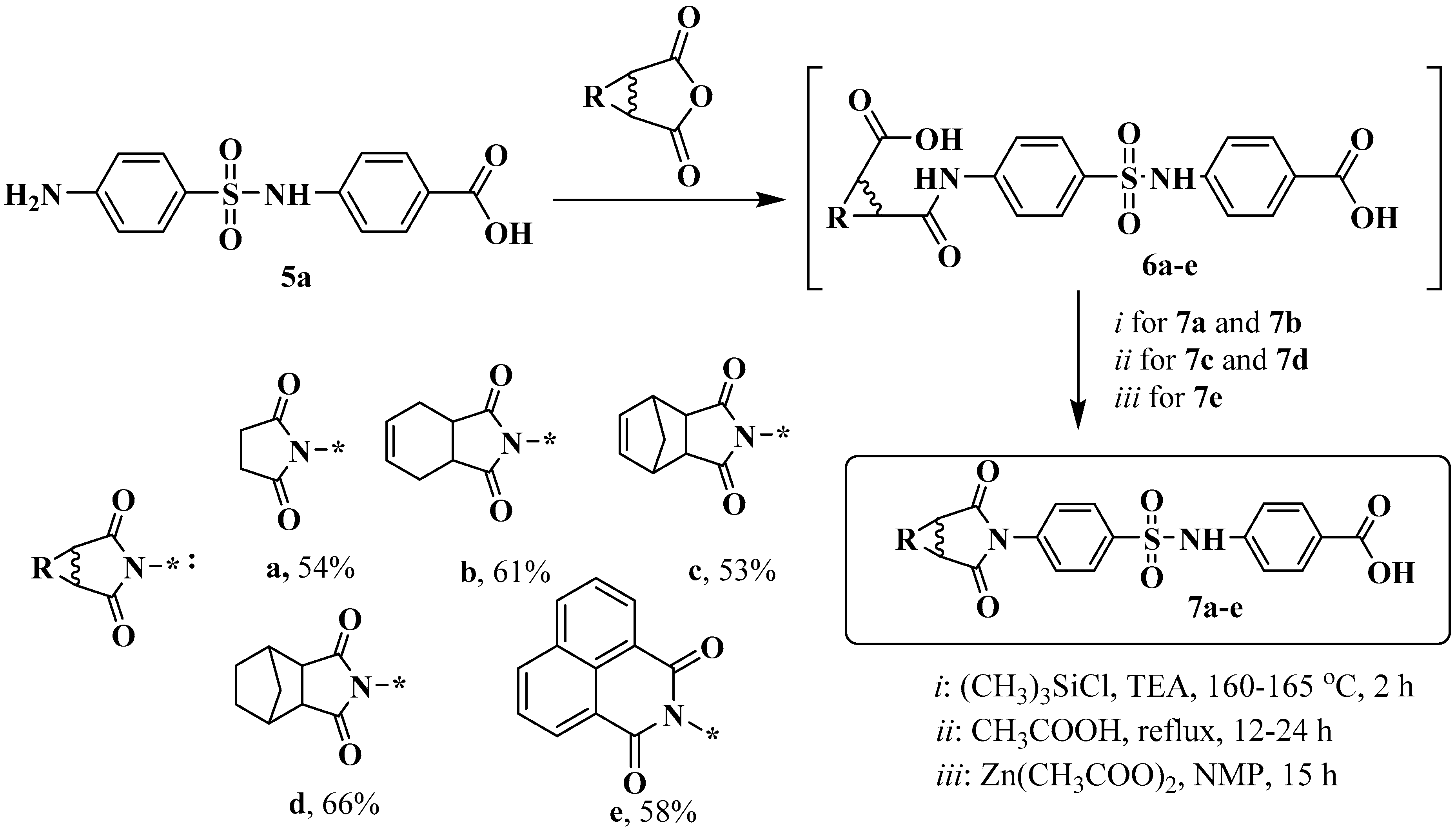
2.4. Synthesis and Characterization of Compounds 7a and 7b
The corresponding anhydride (1 mmol) was added to a solution of 4-((4-aminophenyl)sulfonamido)benzoic acid 6 (1 mmol) in glacial acetic acid (2 mL). The reaction mixture was stirred for 24 h (TLC monitoring). It was diluted with water (1 mL), filtered off, and washed with water. The resulting crude product was dried at 50 °C. A mixture of 4-(4-(3-carboxypropanamido)phenylsulfonamido)benzoic acid 6 (0.2 g, 0.5 mmol), TMSCl (0.07 g), and NMP (0.6 mL) was then heated for 2 h in a bath at 160–165 °C, cooled to room temperature, and diluted with water. The precipitate was separated by filtration, washed with water, and dried in air to obtain the target compound. These compounds were purified by flash column chromatography using CH2Cl2:MeOH (97:3) mixture as an eluent.
4-(4-(2,5-Dioxopyrrolidin-1-yl)phenylsulfonamido)benzoic acid 7a: Yield 54%, beige powder, mp 243–245 °C (with decomposition). 1H NMR (400 MHz, DMSO-d6) δ: 12.61 (br.s, 1 H), 10.90 (s, 1 H), 7.94 (d, J = 8.4 Hz, 2 H), 7.81 (d, J = 8.5 Hz, 2 H), 7.50 (d, J = 8.3 Hz, 2 H), 7.22 (d, J = 8.5 Hz, 2 H), 2.77 (s, 4 H). 13C NMR (101 MHz, DMSO-d6) δ: 177.12 (2 C=O), 167.34 (C=O), 142.40, 139.21, 137.32, 131.46 (2 C), 128.36 (2 C), 128.02 (2 C), 126.47, 118.86 (2 C), 29.18 (CH2CH2). HRMS (ESI), Calc. for C17H15N2O6S+: [M+H]+ 375.0645; found m/z 375.0649.
4-(4-(1,3-Dioxo-3a,4,7,7a-tetrahydro-1H-isoindol-2(3H)-yl)phenylsulfonamido)benzoic acid 7b: Yield 61%, beige powder, mp 250–252 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.68 (br.s, 1 H), 10.89 (br.s, 1 H), 7.94 (d, J = 8.4 Hz, 2 H), 7.81 (d, J = 8.5 Hz, 2 H), 7.46 (d, J = 8.5 Hz, 2 H), 7.22 (d, J = 8.4 Hz, 2 H), 5.91 (s, 2 H), 3.32–3.27 (m, 2 H), 2.44 (d, J = 14.8 Hz, 2 H), 2.26 (d, J = 15.0 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 179.45 (2 C=O), 167.34 (C=O), 142.36, 139.39, 136.92, 131.46 (2 C), 128.32 (2 C), 128.15 (2 C), 126.53, 118.94 (2 C), 39.56 (2 C), 23.86 (2 C). HRMS (ESI), Calc. for C21H19N2O6S+: [M+H]+ 427.0958; found m/z 427.0971.
2.5. Synthesis and Characterization of Compounds 7c and 7d
The corresponding anhydride (1 mmol) was added to a solution of 4-((4-aminophenyl)sulfonamido)benzoic acid 5 (1 mmol) in glacial acetic acid (2 mL). The reaction mass was warmed to reflux and stirred for 12–24 h under reflux (TLC monitoring). It was cooled to room temperature, diluted with water (1 mL), filtered and the filter cake was washed with water. The resulting crude product was recrystallized from ethanol.
4-(4-(1,3-Dioxo-3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindol-2(3H)-yl)phenylsulfonamido)benzoic acid 7c: Yield 53%, beige powder, mp 241–243 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.63 (br.s, 1 H), 10.91 (s, 1H, NH), 7.90 (d, J = 8.4 Hz, 2 H), 7.81 (d, J = 8.5 Hz, 2 H), 7.36 (d, J = 8.5 Hz, 2 H), 7.22 (d, J = 8.6 Hz, 2 H), 6.20 (s, 2 H), 3.50 (s, 2 H), 1.59 (s, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 176.91 (2 C=O), 167.33 (C=O), 142.37, 139.40, 136.69, 135.22 (2 C), 131.49 (2 C), 128.43 (2 C), 128.07 (2 C), 126.44, 118.75 (HC=CH), 52.41, 46.22 (2 C), 45.55 (2 C). HRMS (ESI), Calc. for C22H19N2O6S+: [M+H]+ 439.0958; found m/z 439.0967.
4-(4-(1,3-Dioxohexahydro-1H-4,7-methanoisoindol-2(3H)-yl)phenylsulfonamido)benzoic acid 7d: Yield 66%, beige powder, mp 265–267 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.68 (br.s, 1 H), 10.92 (br.s, 1 H), 7.95 (d, J = 8.6 Hz, 2 H), 7.82 (d, J = 8.7 Hz, 2 H), 7.48 (d, J = 8.6 Hz, 2 H), 7.24 (d, J = 8.6 Hz, 2 H), 3.26 (s, J = 2.5 Hz, 2 H), 2.66 (s, 2 H), 1.66 (d, J = 9.9 Hz, 1 H), 1.60–1.50 (m, 3 H), 1.25 (d, J = 8.5 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 177.56 (2 C=O), 167.33 (C=O), 142.38, 139.59, 136.80, 131.50 (2 C), 128.61 (2 C), 128.18 (2 C), 126.47, 118.80 (2 C), 49.15, 42.14, 41.11, 39.61, 25.08. HRMS (ESI), Calc. for C22H21N2O6S+: [M+H]+ 441.1115; found m/z 441.1124.
2.6. Synthesis and Characterization of Compound 7e
Anhydrous zinc acetate (1 mmol) and compound 5 (1 mmol) were added under nitrogen to a mixture of naphthalene-1,8-dicarboxylic anhydride (1 mmol) and 10 mL of anhydrous N-methylpyrrolidone. Next, the mixture was heated to 160 °C and stirred at this temperature for 15 h. After cooling to room temperature, the product was precipitated by adding 5 mL of 5% hydrochloric acid, filtered off, washed first with 5% hydrochloric acid and then with hot water, and dried. The product was obtained as a brown solid, which corresponds to a yield of 94%. The resulting crude product was purified by short-path chromatography on silica gel using ethyl acetate as an eluent.
4-(4-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)phenylsulfonamido)benzoic acid 7e: Yield 58%, beige powder, mp 284–286 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.74 (br.s, 1 H), 10.98 (s, 1 H), 8.53–8.48 (m, 4 H), 7.99 (d, J = 8.4 Hz, 2 H), 7.93–7.82 (m, 4 H), 7.64 (d, J = 8.4 Hz, 2 H), 7.29 (d, J = 8.5 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ: 167.37 (C=O), 164.13 (2 C=O), 142.50, 141.01, 139.89, 135.32 (2 C), 132.12, 131.49 (2 C), 131.19 (2 C), 130.85, 130.00, 128.54, 127.99, 127.93 (2 C), 126.45, 123.10 (2 C), 120.01, 118.84 (2 C). HRMS (ESI), Calc. for C25H17N2O6S+: [M+H]+ 473.0802; found m/z 473.0815.
2.7. Viruses and Cells
Coxsackievirus B3 (CVB3, strain Nancy) was obtained from the collection of viruses of the Pasteur Institute (St. Petersburg, Russia). The permissive cell line Vero (ATCC #CCL-81) was used in the study. Infectious titers (in a 50%-tissue culture infection dose, TCID50) were determined in Vero cells for CVB3 by endpoint dilution assay using the following procedure. Permissive cells were seeded into 96-wells plates in Eagles minimal essential medium (MEM) supplemented with 5% fetal bovine serum (FBS). After 24 h, the medium was aspirated, the wells were washed with saline, fresh MEM without FBS was added to the wells and the cells were infected with serial tenfold dilutions of virus stocks (100 µL per well, 4 wells for each dilution). The plates were incubated at +37 °C in 5% CO2 and observed daily for cytopathic effect (CPE). After 72 h the viral titer was calculated in TCID50 using the method of Spearmen-Carber.
2.8. Cytoprotection Assay
Vero cells were seeded in 96-well plates in 24 h before the assay. On the assay day, the cells were washed with saline and tested compounds in appropriate concentrations (300–3 μg/mL) in MEM without FBS were added to cells. No compounds were added to virus control wells. The plates were incubated for 1 h at 37 °C at 5% CO2. The cells were then infected with CVB3 Nancy (m.o.i range of 1–0.001) and incubated for 72 h at 37 °C at 5% CO2. No virus was added to the cytotoxicity control wells. Cell viability was then assessed using an MTT test with the following procedure. The cells were washed with saline, and a solution of 3-(4,5-dimethylthiazolyl-2) 2,5-diphenyltetrazolium bromide (ICN Biochemicals Inc., Aurora, OH, USA) (0.5 µg/mL) in MEM was added to the wells (100 µL per well). After 2 h of incubation at 37 °C in 5% CO2, the supernatant from wells was discarded, and the formazan residue was dissolved in DMSO (100 µL per well). The optical density of cells was then measured on a Multiskan multifunctional reader (Thermo Fisher Scientific, Shanghai, China) at a wavelength of 540 nm.
The cytoprotective activity of compounds was considered to be their ability to increase the values of OD compared to control wells (using only a virus, without drugs). Based on the results obtained, the values of IC50, i.e., the concentration of compounds that result in 50% cells protection were calculated for m.o.i. 0.001 using GraphPad Prism 6.01 software.
2.9. Cytotoxicity Assay
The microtetrazolium test (MTT) was used to study the cytotoxicity of the compounds. The permissive cell line Vero was seeded in 96-well plates in Eagles minimal essential medium (MEM) supplemented with 10% FBS. After 24 h, the media were removed, and the wells were washed with saline. Compounds were dissolved in DMSO, and a series of two-fold dilutions of each compound in MEM without FBS were prepared and added to the cells in triplicates. Cells were incubated for 24 h at 37 °C in 5% CO2. The MTT was then performed as described above. Next, the optical density of cells was measured on a Multiskan multifunctional reader (Thermo Fisher Scientific, Shanghai, China) at a wavelength of 540 nm and plotted against the concentration of the compounds to generate the dose–response curve. The 50% cytotoxic dose (CC50) of each compound (i.e., the compound concentration that causes the death of 50% of cells in a culture, or decreases the optical density twice, as compared to the control wells) was calculated using a four-parameter logistic nonlinear regression model (GraphPad Prism 6.01).
2.10. Antiviral Activity Determination
The antiviral activity of the compounds was evaluated using viral yield reduction assay. The respective permissive cell line Vero was seeded in MEM supplemented with 5% FBS in 24-well plates. When the cell confluence reached 100%, the compounds tested were dissolved in DMSO, and a series of three-fold dilutions of each compound in MEM without FBS was added to the cells, and incubated at 37 °C in 5% CO2. After 1 h, the media were discarded; equal volumes of fresh serial dilutions of each compound and viral suspension in MEM without FBS at MOI 0.0 were added. In control wells, only MEM without FBS was added. No compounds were added in the virus control wells. The plates were incubated at 4 °C for 1 h. The unbound virus was then washed away and, again, three-fold dilutions of each compound (the final concentration 50–0.6 µg/mL) in MEM without FBS were added to the wells. After 24 h of incubation at 37 °C in 5% CO2, the infectious titer of viral progeny (in TCID50) for each compound concentration, cell control, and virus control wells were determined in permissive cell lines by endpoint dilution assay as described above.
The titer of virus progeny was plotted against log concentration of the compounds tested to generate the dose–response curve. The 50% inhibition concentration (IC50) of each compound tested (i.e., the compound concentration that decreases the infectious viral progeny titer twice, as compared to the control wells) was calculated using a four-parameter logistic nonlinear regression model (GraphPad Prism 6.01).
Selectivity index (SI) was calculated for each compound tested as a ratio of CC50 to IC50 values.
2.11. Thermostability Assay
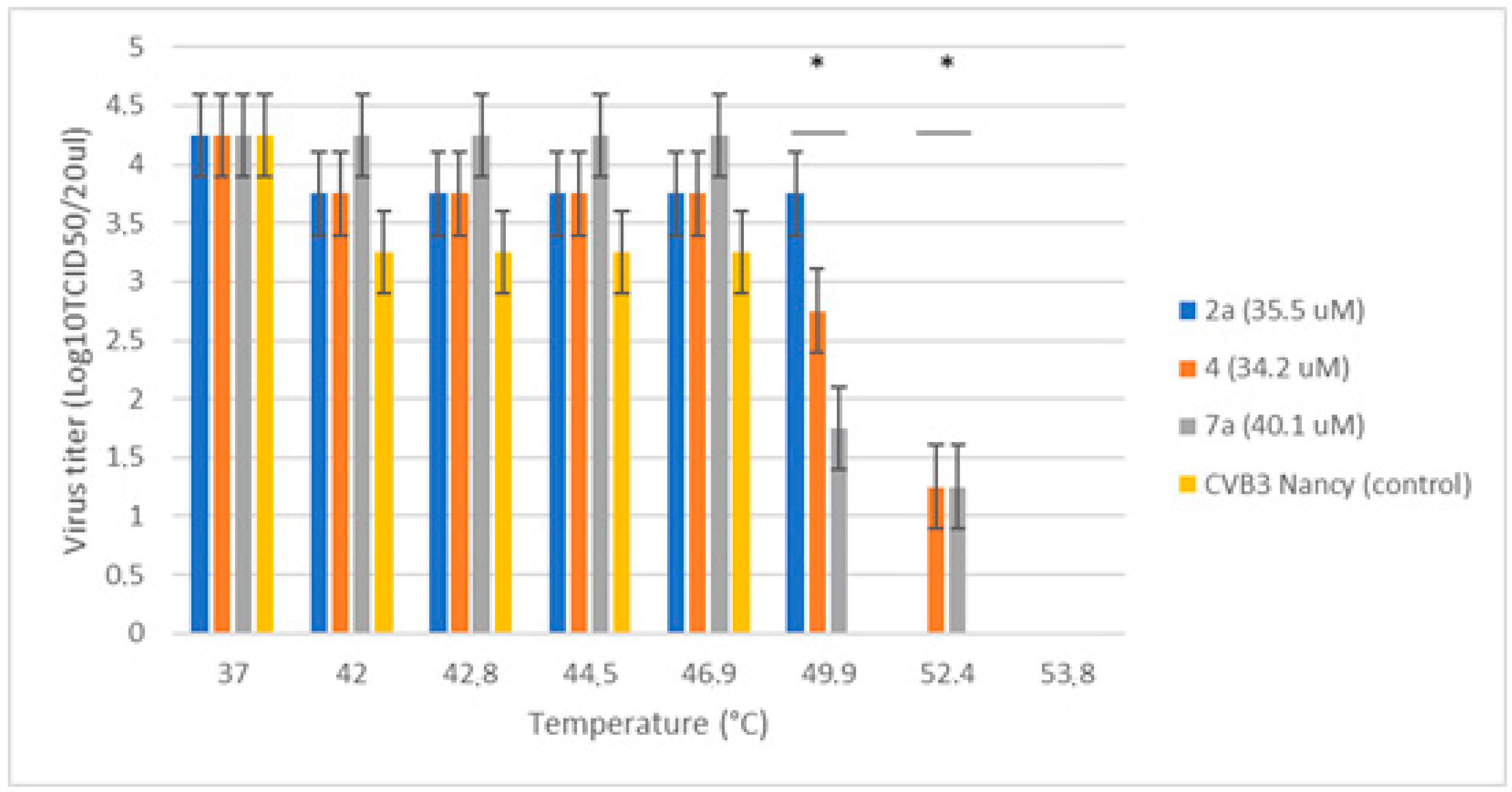
CVB3 Nancy was pre-incubated with the compounds tested or an equal volume of MEM (virus control) for 30 min at +37 °C in sterile thin-walled 200 μL PCR-tubes (7 tubes per each treatment condition) in BioRad CFX PCR-machine. The thermal gradient of 37–55 °C for 2 min was then applied, followed by rapid cooling to 4 °C. Subsequently, the infectious virus load at each thermal condition for each compound tested was quantified by end-point dilution assay.
2.12. Statistics
All in vitro experiments were repeated three times. The results were analyzed using GraphPad Prism 6.01.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}