
Molecular Pathogenesis in Myeloid Neoplasms with Germline Predisposition

Abstract
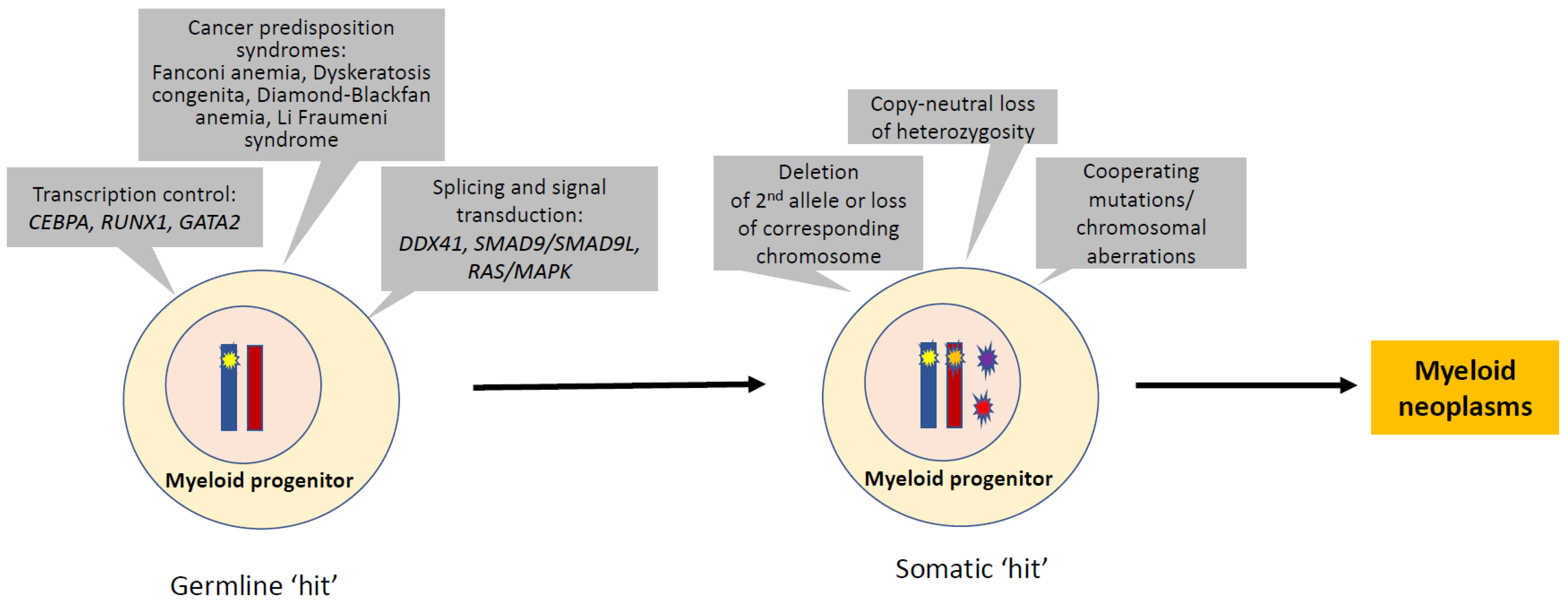
:1. Introduction
2. Transcription Control
2.1. CEBPA
2.2. RUNX1
2.3. GATA2
3. Splicing and Signal Transduction Control
3.1. DDX41
3.2. SMAD9 and SMAD9L
3.3. RAS/MAPK Pathway
4. Bone Marrow Failure Syndromes and Other Inherited Disorders
4.1. Fanconi Anemia (FA)
4.2. Dyskeratosis Congenita (DC)
4.3. Diamond–Blackfan Anemia (DBA)
4.4. Li–Fraumeni Syndrome
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Peterson, L.C.; Bloomfield, C.D.; Niemeyer, C.M.; Dohner, H.; Godley, L.A. Myeloid Neoplasms with Germline Predisposition. In WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D., Hasserjian, R.P., Le Beau, M.M., Orazi, A., Siebert, R., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 122–129. [Google Scholar]
- Zhang, J.; Nichols, K.E.; Downing, J.R. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2016, 374, 1391. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Bannon, S.A.; Routbort, M.; Franklin, A.; Mork, M.; Armanios, M.; Mace, E.M.; Orange, J.S.; Jeff-Eke, M.; Churpek, J.E.; et al. Evaluation of Patients and Families With Concern for Predispositions to Hematologic Malignancies Within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin. Lymphoma Myeloma Leuk 2016, 16, 417–428.e2. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Iwanaga, E.; Iwanaga, K.; Endo, S.; Inoue, Y.; Tokunaga, K.; Nagahata, Y.; Masuda, K.; Kawamoto, H.; Matsuoka, M. A regulatory element in the 3′-untranslated region of CEBPA is associated with myeloid/NK/T-cell leukemia. Eur. J. Haematol. 2021, 106, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.M.; Hu, J.B.; Yang, J.; Qian, W.; Yao, D.M.; Deng, Z.Q.; Zhang, Y.Y.; Zhu, X.W.; Guo, H.; Lin, J.; et al. CEBPA methylation and mutation in myelodysplastic syndrome. Med. Oncol. 2015, 32, 192. [Google Scholar] [CrossRef] [PubMed]
- Tawana, K.; Wang, J.; Renneville, A.; Bodor, C.; Hills, R.; Loveday, C.; Savic, A.; Van Delft, F.W.; Treleaven, J.; Georgiades, P.; et al. Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 2015, 126, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmson, A.S.; Porse, B.T. CCAAT enhancer binding protein alpha (CEBPA) biallelic acute myeloid leukaemia: Cooperating lesions, molecular mechanisms and clinical relevance. Br. J. Haematol. 2020, 190, 495–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, M.P.T.; Kavelaars, F.G.; Lowenberg, B.; Valk, P.J.M.; Raaijmakers, M. RUNX1 germline variants in RUNX1-mutant AML: How frequent? Blood 2021, 137, 1428–1431. [Google Scholar] [CrossRef]
- Mendler, J.H.; Maharry, K.; Radmacher, M.D.; Mrozek, K.; Becker, H.; Metzeler, K.H.; Schwind, S.; Whitman, S.P.; Khalife, J.; Kohlschmidt, J.; et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J. Clin. Oncol. 2012, 30, 3109–3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Kohne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016, 30, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Drazer, M.W.; Kadri, S.; Sukhanova, M.; Patil, S.A.; West, A.H.; Feurstein, S.; Calderon, D.A.; Jones, M.F.; Weipert, C.M.; Daugherty, C.K.; et al. Prognostic tumor sequencing panels frequently identify germ line variants associated with hereditary hematopoietic malignancies. Blood Adv. 2018, 2, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Godley, L.A. Inherited predisposition to acute myeloid leukemia. Semin. Hematol. 2014, 51, 306–321. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.L.; Churpek, J.E.; Malcovati, L.; Dohner, H.; Godley, L.A. Recognition of familial myeloid neoplasia in adults. Semin. Hematol. 2017, 54, 60–68. [Google Scholar] [CrossRef]
- Brown, A.L.; Arts, P.; Carmichael, C.L.; Babic, M.; Dobbins, J.; Chong, C.E.; Schreiber, A.W.; Feng, J.; Phillips, K.; Wang, P.P.S.; et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 2020, 4, 1131–1144. [Google Scholar] [CrossRef] [Green Version]
- Antony-Debre, I.; Duployez, N.; Bucci, M.; Geffroy, S.; Micol, J.B.; Renneville, A.; Boissel, N.; Dhedin, N.; Rea, D.; Nelken, B.; et al. Somatic mutations associated with leukemic progression of familial platelet disorder with predisposition to acute myeloid leukemia. Leukemia 2016, 30, 999–1002. [Google Scholar] [CrossRef]
- Churpek, J.E.; Pyrtel, K.; Kanchi, K.L.; Shao, J.; Koboldt, D.; Miller, C.A.; Shen, D.; Fulton, R.; O’Laughlin, M.; Fronick, C.; et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood 2015, 126, 2484–2490. [Google Scholar] [CrossRef] [Green Version]
- Collin, M.; Dickinson, R.; Bigley, V. Haematopoietic and immune defects associated with GATA2 mutation. Br. J. Haematol. 2015, 169, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Spinner, M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood 2014, 123, 809–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micol, J.B.; Abdel-Wahab, O. Collaborating constitutive and somatic genetic events in myeloid malignancies: ASXL1 mutations in patients with germline GATA2 mutations. Haematologica 2014, 99, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Muramatsu, H.; Okuno, Y.; Sakaguchi, H.; Yoshida, K.; Kawashima, N.; Xu, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; et al. GATA2 and secondary mutations in familial myelodysplastic syndromes and pediatric myeloid malignancies. Haematologica 2015, 100, e398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, R.E.; Griffin, H.; Bigley, V.; Reynard, L.N.; Hussain, R.; Haniffa, M.; Lakey, J.H.; Rahman, T.; Wang, X.N.; McGovern, N.; et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 2011, 118, 2656–2658. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.E.; Milne, P.; Jardine, L.; Zandi, S.; Swierczek, S.I.; McGovern, N.; Cookson, S.; Ferozepurwalla, Z.; Langridge, A.; Pagan, S.; et al. The evolution of cellular deficiency in GATA2 mutation. Blood 2014, 123, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Polprasert, C.; Schulze, I.; Sekeres, M.A.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.A.; Gu, X.; Phillips, J.G.; et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Lewinsohn, M.; Brown, A.L.; Weinel, L.M.; Phung, C.; Rafidi, G.; Lee, M.K.; Schreiber, A.W.; Feng, J.; Babic, M.; Chong, C.E.; et al. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood 2016, 127, 1017–1023. [Google Scholar] [CrossRef] [Green Version]
- Quesada, A.E.; Routbort, M.J.; DiNardo, C.D.; Bueso-Ramos, C.E.; Kanagal-Shamanna, R.; Khoury, J.D.; Thakral, B.; Zuo, Z.; Yin, C.C.; Loghavi, S.; et al. DDX41 mutations in myeloid neoplasms are associated with male gender, TP53 mutations and high-risk disease. Am. J. Hematol. 2019, 94, 757–766. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebert, M.; Passet, M.; Raimbault, A.; Rahme, R.; Raffoux, E.; Sicre de Fontbrune, F.; Cerrano, M.; Quentin, S.; Vasquez, N.; Da Costa, M.; et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood 2019, 134, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Nagamachi, A.; Matsui, H.; Asou, H.; Ozaki, Y.; Aki, D.; Kanai, A.; Takubo, K.; Suda, T.; Nakamura, T.; Wolff, L.; et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell 2013, 24, 305–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narumi, S.; Amano, N.; Ishii, T.; Katsumata, N.; Muroya, K.; Adachi, M.; Toyoshima, K.; Tanaka, Y.; Fukuzawa, R.; Miyako, K.; et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat. Genet. 2016, 48, 792–797. [Google Scholar] [CrossRef]
- Davidsson, J.; Puschmann, A.; Tedgard, U.; Bryder, D.; Nilsson, L.; Cammenga, J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 2018, 32, 1106–1115. [Google Scholar] [CrossRef]
- Schwartz, J.R.; Ma, J.; Lamprecht, T.; Walsh, M.; Wang, S.; Bryant, V.; Song, G.; Wu, G.; Easton, J.; Kesserwan, C.; et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat. Commun. 2017, 8, 1557. [Google Scholar] [CrossRef] [Green Version]
- Drosten, M.; Dhawahir, A.; Sum, E.Y.; Urosevic, J.; Lechuga, C.G.; Esteban, L.M.; Castellano, E.; Guerra, C.; Santos, E.; Barbacid, M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010, 29, 1091–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, P. Ras-MAP kinase signaling pathways and control of cell proliferation: Relevance to cancer therapy. Crit. Rev. Clin. Lab. Sci. 2002, 39, 285–330. [Google Scholar] [CrossRef] [PubMed]
- Strullu, M.; Caye, A.; Lachenaud, J.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Pouvreau, N.; Pereira, S.; Baumann, C.; Contet, A.; et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J. Med. Genet. 2014, 51, 689–697. [Google Scholar] [CrossRef]
- Perez, B.; Mechinaud, F.; Galambrun, C.; Ben Romdhane, N.; Isidor, B.; Philip, N.; Derain-Court, J.; Cassinat, B.; Lachenaud, J.; Kaltenbach, S.; et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J. Med. Genet. 2010, 47, 686–691. [Google Scholar] [CrossRef]
- Martinelli, S.; De Luca, A.; Stellacci, E.; Rossi, C.; Checquolo, S.; Lepri, F.; Caputo, V.; Silvano, M.; Buscherini, F.; Consoli, F.; et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 2010, 87, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Baumann, I.; Niemeyer, C.M.; Thiele, J. Juvenile Myelomonocytic Leukaemia. In WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D., Hasserjian, R.P., Le Beau, M.M., Orazi, A., et al., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 89–92. [Google Scholar]
- Loh, M.L.; Sakai, D.S.; Flotho, C.; Kang, M.; Fliegauf, M.; Archambeault, S.; Mullighan, C.G.; Chen, L.; Bergstraesser, E.; Bueso-Ramos, C.E.; et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 2009, 114, 1859–1863. [Google Scholar] [CrossRef] [Green Version]
- Caye, A.; Strullu, M.; Guidez, F.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Lainey, E.; Nouri, K.; Nakhaei-Rad, S.; Dvorsky, R.; et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat. Genet. 2015, 47, 1334–1340. [Google Scholar] [CrossRef]
- Stieglitz, E.; Taylor-Weiner, A.N.; Chang, T.Y.; Gelston, L.C.; Wang, Y.D.; Mazor, T.; Esquivel, E.; Yu, A.; Seepo, S.; Olsen, S.; et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat. Genet. 2015, 47, 1326–1333. [Google Scholar] [CrossRef]
- Steinemann, D.; Arning, L.; Praulich, I.; Stuhrmann, M.; Hasle, H.; Stary, J.; Schlegelberger, B.; Niemeyer, C.M.; Flotho, C. Mitotic recombination and compound-heterozygous mutations are predominant NF1-inactivating mechanisms in children with juvenile myelomonocytic leukemia and neurofibromatosis type 1. Haematologica 2010, 95, 320–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butturini, A.; Gale, R.P.; Verlander, P.C.; Adler-Brecher, B.; Gillio, A.P.; Auerbach, A.D. Hematologic abnormalities in Fanconi anemia: An International Fanconi Anemia Registry study. Blood 1994, 84, 1650–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lensch, M.W.; Rathbun, R.K.; Olson, S.B.; Jones, G.R.; Bagby, G.C., Jr. Selective pressure as an essential force in molecular evolution of myeloid leukemic clones: A view from the window of Fanconi anemia. Leukemia 1999, 13, 1784–1789. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, M.; Maurer, A.; Wlodarski, M.W.; Ventura Ferreira, M.S.; Bouillon, A.S.; Halfmeyer, I.; Blau, W.; Kreuter, M.; Rosewich, M.; Corbacioglu, S.; et al. Recurrent somatic mutations are rare in patients with cryptic dyskeratosis congenita. Leukemia 2018, 32, 1762–1767. [Google Scholar] [CrossRef]
- Zebisch, A.; Lal, R.; Muller, M.; Lind, K.; Kashofer, K.; Girschikofsky, M.; Fuchs, D.; Wolfler, A.; Geigl, J.B.; Sill, H. Acute myeloid leukemia with TP53 germ line mutations. Blood 2016, 128, 2270–2272. [Google Scholar] [CrossRef] [Green Version]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef]
- Geyer, J.T. Myeloid Neoplasms with Germline Predisposition. Pathobiology 2019, 86, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.; Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; De Rocco, D.; et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: Analysis of 78 patients from 21 families. Blood 2011, 117, 6673–6680. [Google Scholar] [CrossRef] [Green Version]
- Saliba, J.; Saint-Martin, C.; Di Stefano, A.; Lenglet, G.; Marty, C.; Keren, B.; Pasquier, F.; Valle, V.D.; Secardin, L.; Leroy, G.; et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat. Genet. 2015, 47, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Plo, I.; Bellanne-Chantelot, C.; Vainchenker, W. ATG2B and GSKIP: 2 new genes predisposing to myeloid malignancies. Mol. Cell Oncol. 2016, 3, e1094564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemeyer, C.M.; Kang, M.W.; Shin, D.H.; Furlan, I.; Erlacher, M.; Bunin, N.J.; Bunda, S.; Finklestein, J.Z.; Gorr, T.A.; Mehta, P.; et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat. Genet. 2010, 42, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 2013, 1833, 122–139. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.L.; Cavenagh, J.D.; Lister, T.A.; Fitzgibbon, J. Mutation of CEBPA in familial acute myeloid leukemia. N. Engl. J. Med. 2004, 351, 2403–2407. [Google Scholar] [CrossRef]
- Noetzli, L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.; Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.; et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat. Genet. 2015, 47, 535–538. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.Y.; Churpek, J.E.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat. Genet. 2015, 47, 180–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, T.; Metzger, M.L.; Wu, G.; Nishii, R.; Qian, M.; Devidas, M.; Yang, W.; Cheng, C.; Cao, X.; Quinn, E.; et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: A systematic genetic study. Lancet Oncol. 2015, 16, 1659–1666. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.P.; Sampaio, E.P.; Khan, J.; Calvo, K.R.; Lemieux, J.E.; Patel, S.Y.; Frucht, D.M.; Vinh, D.C.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef]
- Hahn, C.N.; Chong, C.E.; Carmichael, C.L.; Wilkins, E.J.; Brautigan, P.J.; Li, X.C.; Babic, M.; Lin, M.; Carmagnac, A.; Lee, Y.K.; et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 2011, 43, 1012–1017. [Google Scholar] [CrossRef]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Stary, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 2016, 127, 1387–1397; quiz 1518. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Kozyra, E.J.; Wlodarski, M.W. Germline predisposition in myeloid neoplasms: Unique genetic and clinical features of GATA2 deficiency and SAMD9/SAMD9L syndromes. Best Pract. Res. Clin. Haematol. 2020, 33, 101197. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.A.; Chew, E.; Flensburg, C.; Zeilemaker, A.; Miller, S.E.; Al Hinai, A.S.; Bajel, A.; Luiken, B.; Rijken, M.; McLennan, T.; et al. MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood 2018, 132, 1526–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niihori, T.; Ouchi-Uchiyama, M.; Sasahara, Y.; Kaneko, T.; Hashii, Y.; Irie, M.; Sato, A.; Saito-Nanjo, Y.; Funayama, R.; Nagashima, T.; et al. Mutations in MECOM, Encoding Oncoprotein EVI1, Cause Radioulnar Synostosis with Amegakaryocytic Thrombocytopenia. Am. J. Hum. Genet. 2015, 97, 848–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripperger, T.; Hofmann, W.; Koch, J.C.; Shirneshan, K.; Haase, D.; Wulf, G.; Issing, P.R.; Karnebogen, M.; Schmidt, G.; Auber, B.; et al. MDS1 and EVI1 complex locus (MECOM): A novel candidate gene for hereditary hematological malignancies. Haematologica 2018, 103, e55–e58. [Google Scholar] [CrossRef] [Green Version]
- Germeshausen, M.; Ancliff, P.; Estrada, J.; Metzler, M.; Ponstingl, E.; Rutschle, H.; Schwabe, D.; Scott, R.H.; Unal, S.; Wawer, A.; et al. MECOM-associated syndrome: A heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood Adv. 2018, 2, 586–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, N.V.; Daly, M.E. Gene of the issue: RUNX1 mutations and inherited bleeding. Platelets 2017, 28, 208–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonocore, F.; Kuhnen, P.; Suntharalingham, J.P.; Del Valle, I.; Digweed, M.; Stachelscheid, H.; Khajavi, N.; Didi, M.; Brady, A.F.; Blankenstein, O.; et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J. Clin. Investig. 2017, 127, 1700–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Rundberg Nilsson, A.; Ripperger, T.; et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirwan, M.; Walne, A.J.; Plagnol, V.; Velangi, M.; Ho, A.; Hossain, U.; Vulliamy, T.; Dokal, I. Exome sequencing identifies autosomal-dominant SRP72 mutations associated with familial aplasia and myelodysplasia. Am. J. Hum. Genet. 2012, 90, 888–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| MN with germline predisposition without a preexisting disorder or organ dysfunction | AML with germline CEBPA mutation |
| MN with germline DDX41 mutation | |
| MN with germline predisposition and preexisting platelet disorder | MN with germline RUNX1 mutation |
| MN with germline ANKRD26 mutation | |
| MN with germline ETV6 mutation | |
| MN with germline predisposition and other rgan dysfunction | MN with germline GATA2 mutation |
| MN associated with bone marrow failure syndromes | |
| MN associated with telomere biology disorders | |
| Juvenile myelomonocytic leukemia associated with neurofibromatosis, Noonan syndrome, or Noonan-syndrome-like disorders | |
| MN associated with Down syndrome |
| Gene (chr. Band) | Syndrome Name | Key Clinical and Pathologic Features | Mechanism | References |
|---|---|---|---|---|
| ANKRD26 (10p12.1) | Thrombocytopenia 2 | Thrombocytopenia/platelet dysfunction No organ dysfunction MDS, AML, rarely CML, CMML | ANKRD26 germline missense mutations result in increased MPL signaling pathway and impaired pro-platelet formation by megakaryocytes; ANKRD26 promoter mutations prevent RUNX1 binding to the promoter, resulting in consecutive activation of MAPK signaling and abnormal platelet function. | WHO 2017 NCCN 2021 [47,48] |
| ATG2B (14q32.2) GSKIP (14q32.2) | MNs with germline predisposition due to duplications of ATG2B and GSKIP | ET or PMF progress to AML, CMML, CML, aCML Penetrance > 80% | An approximately 700 kb tandem duplication results in overexpression of ATG2B and GSKIP, enhancing sensitivity to thrombopoietin; cooperates with acquired JAK2, MPL, and CALR mutations during MPN development. | NCCN 2021 [49,50] |
| CBL (11q23.3) | CBL syndrome | JMML, clonal macrophage/monocyte proliferation | CBL is a proto-oncogene that encodes a RING finger E3 ubiquitin ligase, which plays a role in tyrosine kinase signaling; the mutational spectrum is mainly missense variants or splice-site variants in the linker region and zinc-coordinating amino acids of the RING finger domain. Heterozygous germline variants are typically accompanied by copy-neutral loss of heterozygosity (CN-LOH) of the 11q23 region with the consecutive homozygous state of the CBL variant; thus, a high allele frequency of the identified CBL variant or CN-LOH at 11q23 is suggestive of CBL syndrome. | [35,51,52] |
| CEBPA (19q13.11) | Familial AML with mutated CEBPA | AML (normal karyotype, blasts have aberrant CD7 expression) No organ dysfunction Median age at diagnosis: 25 years ~100% penetrance | The gene encodes granulocyte differentiation factor. Germline mutations are typically N-terminal and frame-shift variants. C-terminal mutations are typically somatic and in-frame insertions, deletions, frameshift, or missense variants; they are typically unstable, and a novel clone during recurrence is not unusual. Acquired mutations in GATA2 and WT1 are common and mutually exclusive. | WHO 2017 NCCN 2021 [6,47,53] |
| DDX41 (5q35.3) | Familial AML with mutated DDX41 | MDS, AML, CMML, CML Normal karyotype in 80% of patients No organ dysfunction Median age at diagnosis is 62 years | DEAD/H-box helicase gene encodes an RNA helicase protein with a function in RNA splicing. Many patients have biallelic mutations (frameshift, missense, splicing). Although rare in MNs (1.5%), if detected, DDX41 mutation is germline in 50% of cases. | WHO 2017 NCCN 2021 [23,24,47] |
| ETV6 (12p13.2) | MNs with germline ETV6 mutation/Thrombocytopenia 5 | Thrombocytopenia/platelet dysfunction and no organ dysfunction ALL, MDS, AML, CMML, PV, aplastic anemia | The gene encodes a transcription factor. Mutations abrogate DNA binding, alter subcellular localization, decrease transcriptional repression in a dominant-negative effect, and impair expression of platelet-associated genes with defective maturation of megakaryocytes. Germline variants are associated with lymphoid and myeloid neoplasms. | WHO 2017 NCCN 2021 [54,55,56] |
| GATA2 (3q21.3) | Familial MDS/AML with mutated GATA2 (known as GATA2 deficiency syndromes) | Organ dysfunction (lymphedema, hydrocele, congenital deafness, vulnerability to viral infections) 70–75% penetrance MDS, AML, common in children with MDS and monosomy 7 Dysplasia is frequent, particularly dysmegakaryopoiesis | Gata2 is a zinc-finger transcription factor that is important in the control of hematopoiesis and autoimmunity; germline mutations are loss-of-function variants. Mutations are frequent in MDS and AML cases together with ASXL1 mutations, indicative of a possible collaborative mechanism. | WHO 2017 NCCN 2021 [18,21,57,58,59,60] |
| MBD4 (3q21.3) | Familial AML with mutated MBD4 | AML | MBD4, methyl-CpG binding domain 4, encodes a DNA glycosylase that removes base lesions and initiates DNA repair. MBD4-deficiency AML displays a 33-fold higher mutation burden, with mostly C>T in CG dinucleotide; has a propensity for DNMT3A-mutated clonal hematopoiesis. | [61] |
| MECOM (3q26.2) | MECOM-associated syndrome (also known as congenital amegakaryocytic thrombocytopenia and radioulnar synostosis) | Amegakaryocytic thrombocytopenia, bone marrow failure, MDS Organ dysfunction (radiosynostosis, clinodactyly, presenile hearing loss, cardiac/renal malformations) | A range of genetic variants have been observed, including gene deletions and point mutations. Mutations are clustered within the 8th zinc-finger motif of the C-terminal zinc-finger domain of EVI1. | [62,63,64] |
| RUNX1 (21q22) | Familial platelet disorder with propensity for myeloid malignancies | MDS, AML, thrombocytopenia, platelet dysfunction, CMML No organ dysfunction Median age at diagnosis is 39 years old; lifetime risk of myeloid neoplasm is ~20~65%. | The gene encodes a transcription factor with a major role in megakaryocyte maturation, differentiation, ploidization, and pro-platelet formation. Germline mutations are typically frameshift or missense all over the gene, but predominantly in the RUNT domain and transactivation domain. | WHO 2017 NCCN 2021 [15,16,47,65] |
| SAMD9 (7q21.2) SAMD9L (7q21.2) | Congenital SAMD9/SAMD9L mutations MIRAGE syndrome/ Monosomy 7 myelodysplasia and leukemia syndrome 2/Ataxia pancytopenia syndrome | MDS, AML, pancytopenia Organ dysfunction (hematopoietic, immunologic, endocrine, genital, neurologic) Together with GATA2-mutated myeloid neoplasms are the most frequent oncogenic drivers in pediatric MDS (half of pediatric MDS with monosomy 7) Penetrance is not complete | SAMD9 encodes sterile alpha motif domain-containing protein 9, which is involved in endosome fusion and plays a role in signal transduction and proliferation. SAMD9L is a paralog (L stands for -like) and a negative controller of proliferation. Germline mutations cause gain of function of SAMD9 and SAMD9L, which normally results in suppression of myeloid proliferation and inhibits cell cycle progression, leading to pancytopenias, as well as other organ hypoplasias and growth restrictions. | [29,31,60,66,67] |
| SRP72 (4q12) | Familial aplastic anemia/MDS with SRP72 mutation | MDS, aplastic anemia, familial leukemia, bone marrow failure | SRP72 is a component of the signal recognition particle, a ribonucleoprotein complex responsible for the translocation of nascent membrane-bound and excreted proteins to the endoplasmic reticulum. | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Chen, Y.; Sukhanova, M. Molecular Pathogenesis in Myeloid Neoplasms with Germline Predisposition. Life 2022, 12, 46. https://doi.org/10.3390/life12010046
Gao J, Chen Y, Sukhanova M. Molecular Pathogenesis in Myeloid Neoplasms with Germline Predisposition. Life. 2022; 12(1):46. https://doi.org/10.3390/life12010046
Chicago/Turabian StyleGao, Juehua, Yihua Chen, and Madina Sukhanova. 2022. "Molecular Pathogenesis in Myeloid Neoplasms with Germline Predisposition" Life 12, no. 1: 46. https://doi.org/10.3390/life12010046