
Two Novel Homozygous HPS6 Mutations (Double Mutant) Identified by Whole-Exome Sequencing in a Saudi Consanguineous Family Suspected for Oculocutaneous Albinism

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient and Ethical Approval
2.2. DNA Isolation
2.3. Whole-Exome Sequencing
2.4. Exome Sequencing Data Analysis
2.5. Variant Filtration and Prioritization
2.6. Sanger Sequencing
2.7. Sequence and Structure Analysis
3. Results
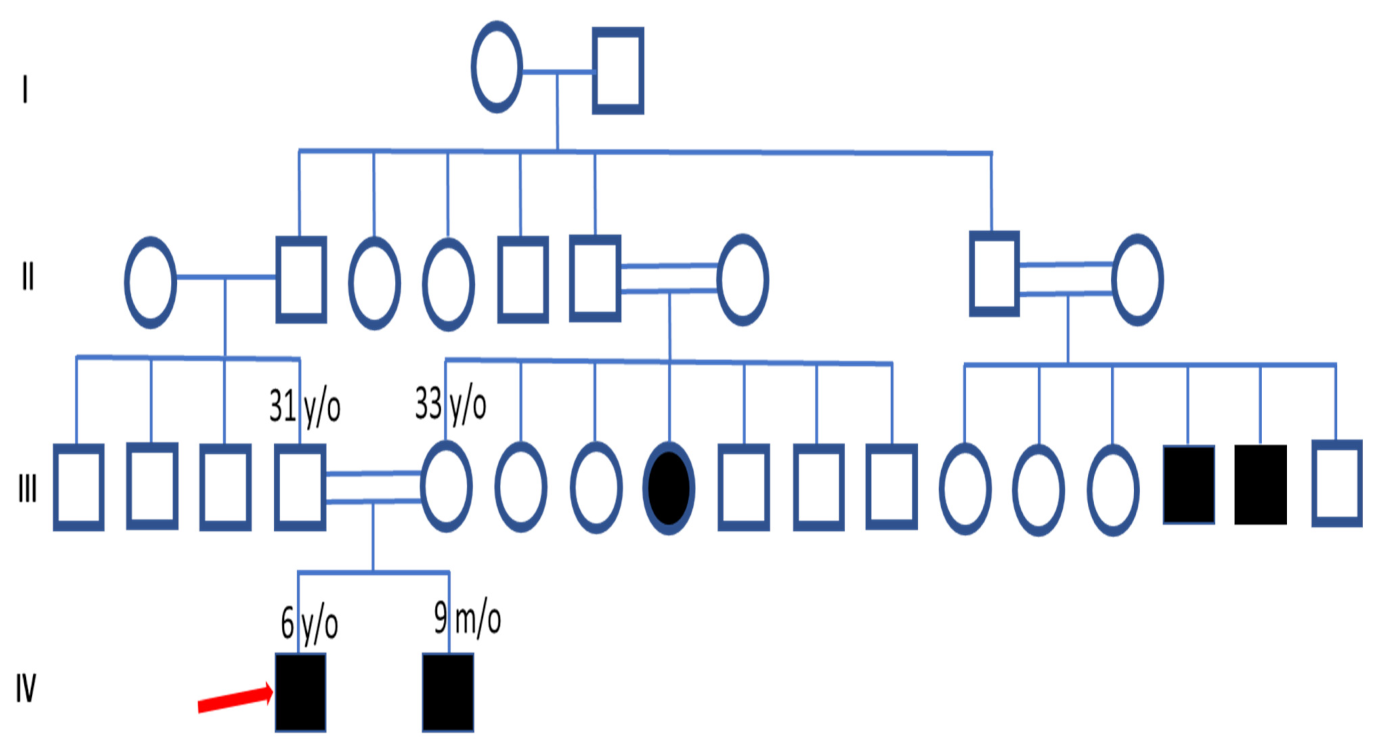
3.1. Clinical Features and Pedigree Analysis
3.2. Identification of the HPS6 Variants in OCA Patient
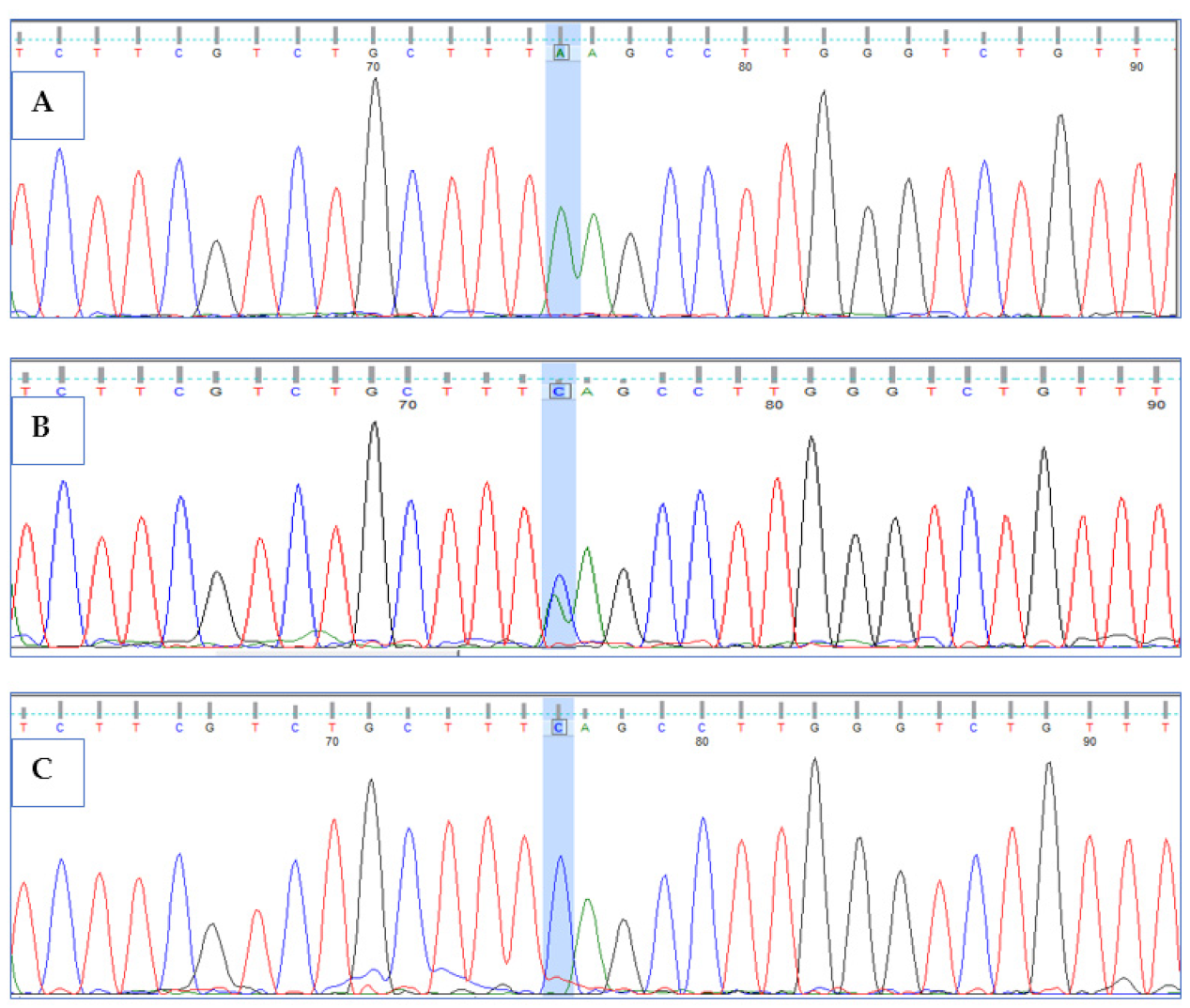
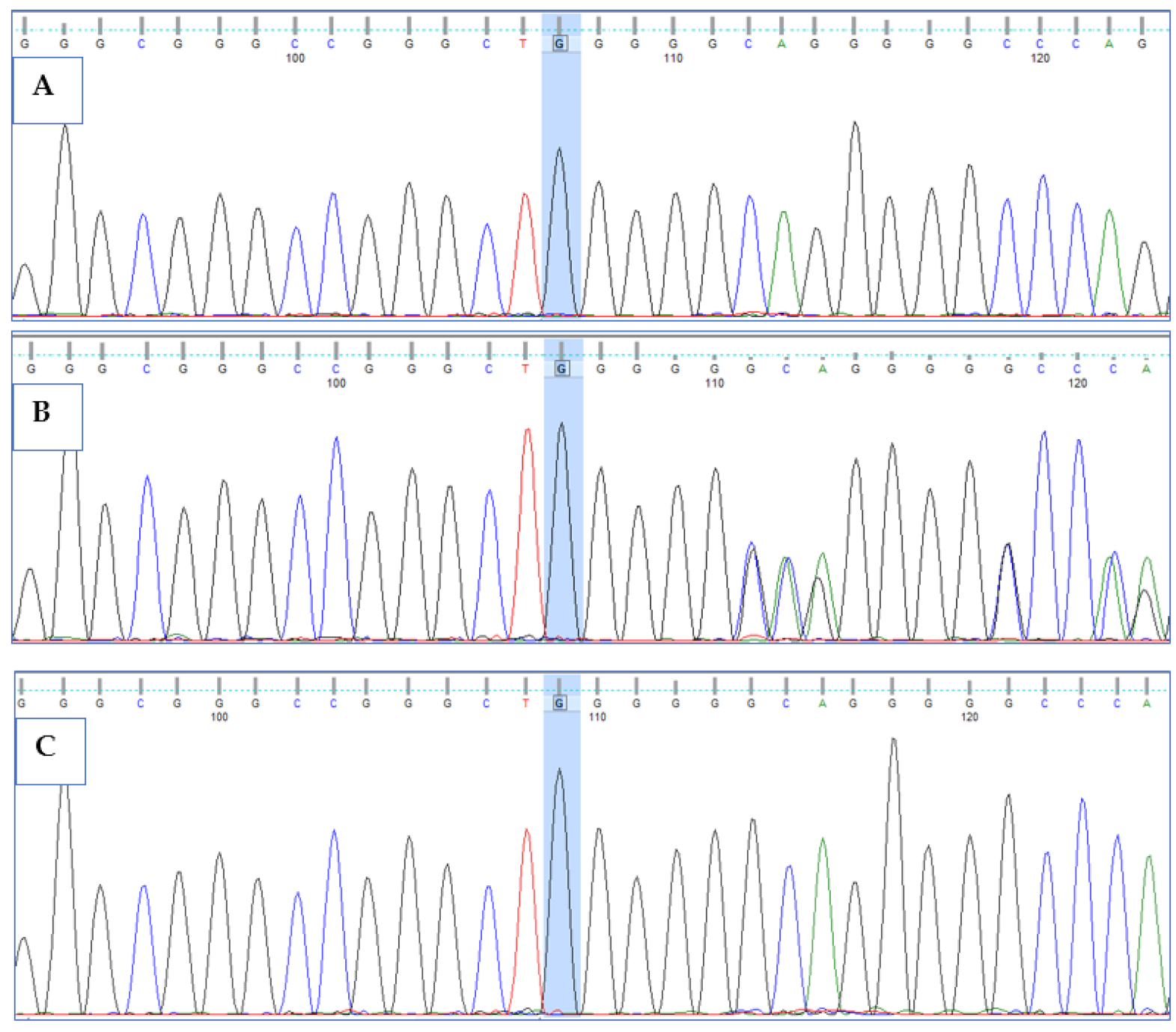
3.3. Validation of HPS6 Mutations
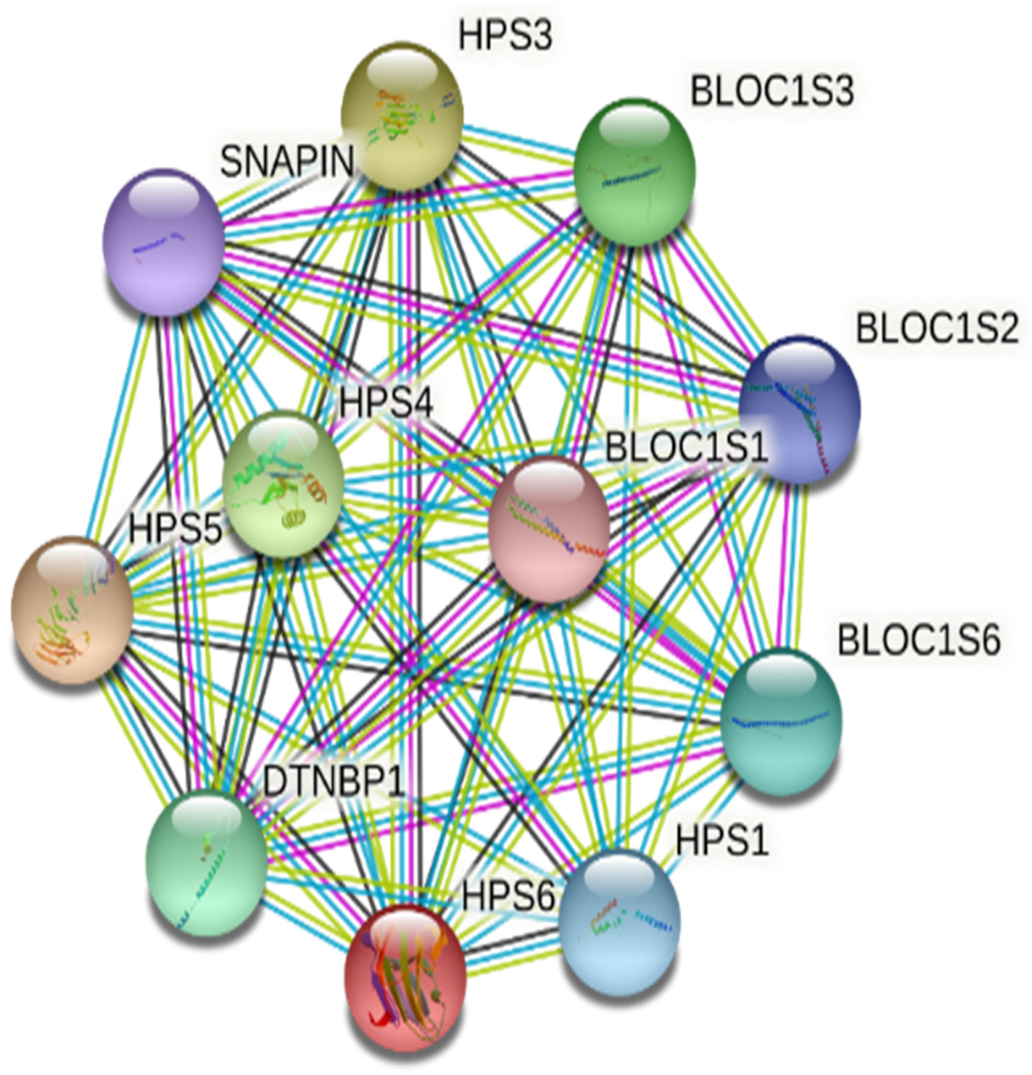
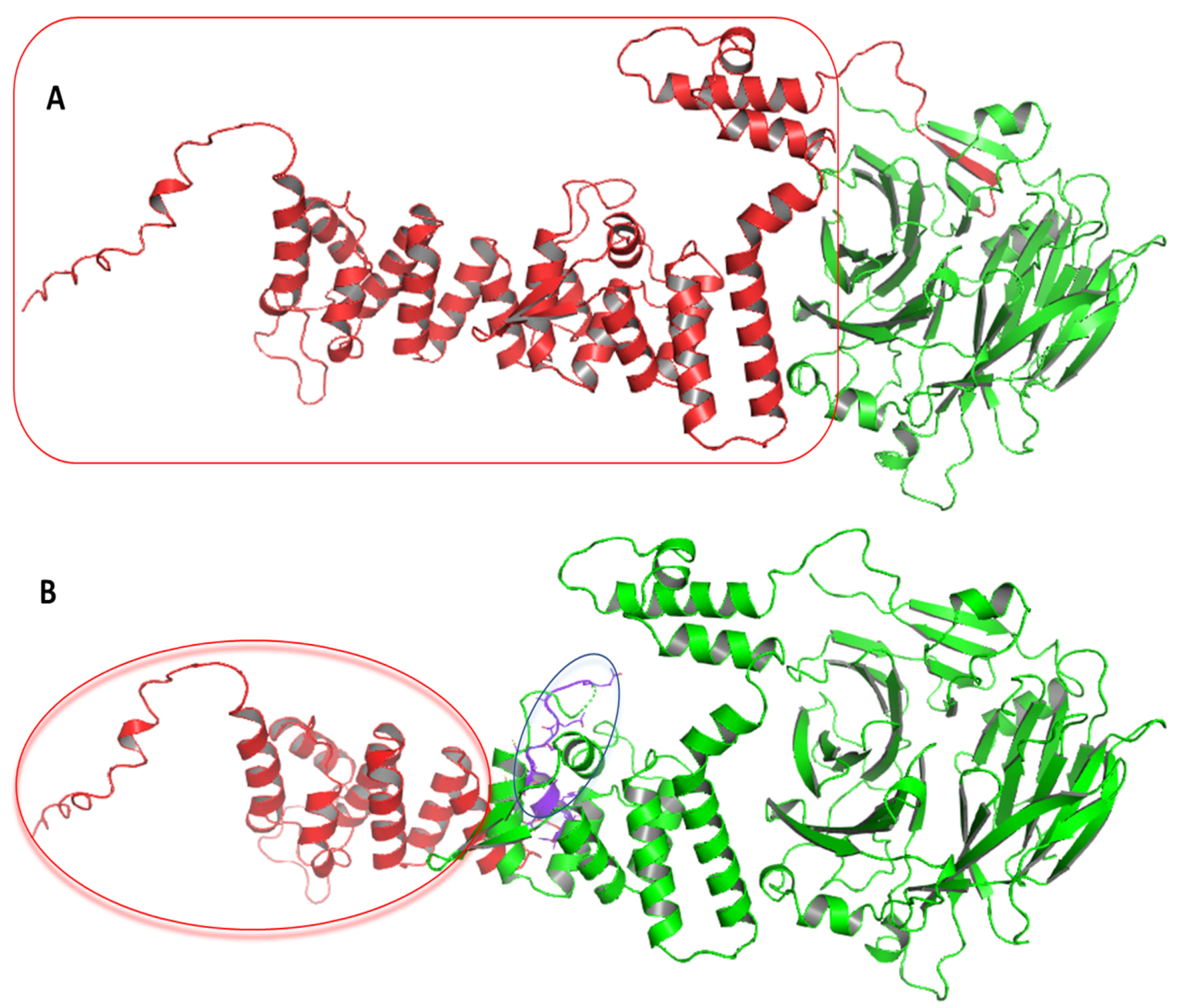
3.4. Computational Analysis of HPS6 Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OCA | Oculocutaneous albinism |
| HPS | Hermansky-Pudlak syndrome |
| WES | Whole-exome sequencing |
| ACMGG | American College of Medical Genetics and Genomics |
| HGVS | Human Genome Variation Society |
| CADD | Combined Annotation-Dependent Depletion |
References
- Yang, Q.; Yi, S.; Li, M.; Xie, B.; Luo, J.; Wang, J.; Rong, X.; Zhang, Q.; Qin, Z.; Hang, L.; et al. Genetic analyses of oculocutaneous albinism types 1 and 2 with four novel mutations. BMC Med. Genet. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Suzuki, T. Current landscape of Oculocutaneous Albinism in Japan. Pigment. Cell Melanoma Res. 2020, 34, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, B.; Purohit, R. Mutational Analysis of Oculocutaneous Albinism: A Compact Review. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagore, F.; Lund, P.M. Oculocutaneous albinism among schoolchildren in Harare, Zimbabwe. J. Med. Genet. 1995, 32, 859–861. [Google Scholar] [CrossRef] [Green Version]
- Manga, P.; Kromberg, J.; Box, N.; Sturm, R.; Jenkins, T.; Ramsay, M. Rufous Oculocutaneous Albinism in Southern African Blacks Is Caused by Mutations in the TYRP1 Gene. Am. J. Hum. Genet. 1997, 61, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Grønskov, K.; Dooley, C.M.; Østergaard, E.; Kelsh, R.N.; Hansen, L.; Levesque, M.P.; Vilhelmsen, K.; Møllgård, K.; Stemple, D.L.; Rosenberg, T. Mutations in C10orf11, a Melanocyte-Differentiation Gene, Cause Autosomal-Recessive Albinism. Am. J. Hum. Genet. 2013, 92, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Kausar, T.; Bhatti, M.; Ali, M.; Shaikh, R.S.; Ahmed, Z.M. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin. Genet. 2012, 84, 91–93. [Google Scholar] [CrossRef]
- Liu, N.; Kong, X.D.; Shi, H.R.; Wu, Q.H.; Jiang, M. Tyrosinase gene mutations in the Chinese Han population with OCA1. Genet. Res. 2014, 96, e14. [Google Scholar] [CrossRef]
- Mauri, L.; Barone, L.; Al Oum, M.; Del Longo, A.; Piozzi, E.; Manfredini, E.; Stanzial, F.; Benedicenti, F.; Penco, S.; Patrosso, M.C. SLC45A2 mutation frequency in Oculocutaneous Albinism Italian patients doesn’t differ from other European studies. Gene 2014, 533, 398–402. [Google Scholar] [CrossRef]
- Bertolotti, A.; Lasseaux, E.; Plaisant, C.; Trimouille, A.; Morice-Picard, F.; Rooryck, C.; Lacombe, D.; Couppie, P.; Arveiler, B. Identification of a homozygous mutation of SLC24A5 (OCA6) in two patients with oculocutaneous albinism from French Guiana. Pigment. Cell Melanoma Res. 2015, 29, 104–106. [Google Scholar] [CrossRef]
- Pennamen, P.; Tingaud-Sequeira, A.; Gazova, I.; Keighren, M.; McKie, L.; Marlin, S.; Halem, S.G.; Kaplan, J.; Delevoye, C.; Lacombe, D.; et al. Dopachrome tautomerase variants in patients with oculocutaneous albinism. Genet. Med. 2020, 23, 479–487. [Google Scholar] [CrossRef]
- Chan, H.; Schiff, E.; Tailor, V.; Malka, S.; Neveu, M.; Theodorou, M.; Moosajee, M. Prospective Study of the Phenotypic and Mutational Spectrum of Ocular Albinism and Oculocutaneous Albinism. Genes 2021, 12, 508. [Google Scholar] [CrossRef]
- Zhang, Y.; McMahon, R.; Charles, S.; Green, J.; Moore, A.; Barton, E.D.; Yates, J. Genetic mapping of the Kallmann syndrome and X linked ocular albinism gene loci. J. Med. Genet. 1993, 30, 923–925. [Google Scholar] [CrossRef]
- Oh, J.; Ho, L.; Ala-Mello, S.; Amato, D.; Armstrong, L.; Bellucci, S.; Carakushansky, G.; Ellis, J.P.; Fong, C.-T.; Green, J.S.; et al. Mutation Analysis of Patients with Hermansky-Pudlak Syndrome: A Frameshift Hot Spot in the HPS Gene and Apparent Locus Heterogeneity. Am. J. Hum. Genet. 1998, 62, 593–598. [Google Scholar] [CrossRef] [Green Version]
- King, R.A.; Willaert, R.K.; Schmidt, R.M.; Pietsch, J.; Savage, S.; Brott, M.J.; Fryer, J.P.; Summers, C.G.; Oetting, W.S. MC1R Mutations Modify the Classic Phenotype of Oculocutaneous Albinism Type 2 (OCA2). Am. J. Hum. Genet. 2003, 73, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Shimazaki, H.; Honda, J.; Naoi, T.; Namekawa, M.; Nakano, I.; Yazaki, M.; Nakamura, K.; Yoshida, K.; Ikeda, S.-I.; Ishiura, H.; et al. Autosomal-recessive complicated spastic paraplegia with a novel lysosomal trafficking regulator gene mutation. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1024–1028. [Google Scholar] [CrossRef]
- Miyamichi, D.; Asahina, M.; Nakajima, J.; Sato, M.; Hosono, K.; Nomura, T.; Negishi, T.; Miyake, N.; Hotta, Y.; Ogata, T.; et al. Novel HPS6 mutations identified by whole-exome sequencing in two Japanese sisters with suspected ocular albinism. J. Hum. Genet. 2016, 61, 839–842. [Google Scholar] [CrossRef]
- Huizing, M.; Malicdan, M.C.V.; Wang, J.A.; Pri-Chen, H.; Hess, R.A.; Fischer, R.; O’Brien, K.J.; Merideth, M.A.; Gahl, W.A.; Gochuico, B.R. Hermansky-Pudlak syndrome: Mutation update. Hum. Mutat. 2019, 41, 543–580. [Google Scholar] [CrossRef]
- Huizing, M.; Malicdan, M.C.V.; Gochuico, B.R.; Gahl, W.A. Hermansky-Pudlak Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zhang, Q.; Zhao, B.; Li, W.; Oiso, N.; Novak, E.K.; Rusiniak, M.E.; Gautam, R.; Chintala, S.; O’Brien, E.P.; Zhang, Y.; et al. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat. Genet. 2003, 33, 145–153. [Google Scholar] [CrossRef]
- Wei, A.; Yuan, Y.; Bai, D.; Ma, J.; Hao, Z.; Zhang, Y.; Yu, J.; Zhou, Z.; Yang, L.; Yang, X.; et al. NGS-based 100-gene panel of hypopigmentation identifies mutations in Chinese Hermansky-Pudlak syndrome patients. Pigment. Cell Melanoma Res. 2016, 29, 702–706. [Google Scholar] [CrossRef]
- Bastida, J.M.; Morais, S.; Palma-Barqueros, V.; Benito, R.; Bermejo, N.; Karkucak, M.; Trapero-Marugan, M.; Bohdan, N.; Pereira, M.; Marin-Quilez, A.; et al. Identification of novel variants in ten patients with Hermansky-Pudlak syndrome by high-throughput sequencing. Ann. Med. 2019, 51, 141–148. [Google Scholar] [CrossRef]
- Wang, C.; Shi, P.; Li, Q.; Chen, C.; Zhao, X.; Zhang, R.; Kong, X. Hermansky-Pudlak syndrome: Five Chinese patients with novel variants in HPS1 and HPS6. Eur. J. Med. Genet. 2021, 64, 1–5. [Google Scholar] [CrossRef]
- Huang, X.-F.; Huang, F.; Wu, K.-C.; Wu, J.; Chen, J.; Pang, C.-P.; Lu, F.; Qu, J.; Jin, Z.-B. Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Absar, M.; Mahmood, A.; Akhtar, T.; Basit, S.; Ramzan, K.; Jameel, A.; Afzal, S.; Ullah, A.; Qureshi, K.; Alanazi, N.; et al. Whole exome sequencing identifies a novel FANCD2 gene splice site mutation associated with disease progression in chronic myeloid leukemia: Implication in targeted therapy of advanced phase CML. Pak. J. Pharm. Sci. 2020, 33, 1419–1426. [Google Scholar]
- Rasool, M.; Pushparaj, P.N.; Buhmeida, A.; Karim, S. Mutational spectrum of BRAF gene in colorectal cancer patients in Saudi Arabia. Saudi J. Biol. Sci. 2021, 28, 5906–5912. [Google Scholar] [CrossRef]
- Abdulkareem, A.A.; Abulnaja, K.O.; Jan, M.M.; Karim, S.; Rasool, M.; Ansari, S.A.; Chaudhary, A.G.; Al-Qahtani, M.H.; Naseer, M.I. A novel homozygous nonsense mutation in CCDC88A gene cause PEHO-like syndrome in consanguineous Saudi family. Neurol. Sci. 2018, 40, 299–303. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Klein, H.-G.; Bauer, P.; Hambuch, T. Whole genome sequencing (WGS), whole exome sequencing (WES) and clinical exome sequencing (CES) in patient care. Lab. Med. 2014, 38, 221–230. [Google Scholar] [CrossRef]
- Al Asiri, S.; Basit, S.; Wood-Trageser, M.; Yatsenko, S.A.; Jeffries, E.P.; Surti, U.; Ketterer, D.M.; Afzal, S.; Ramzan, K.; Haque, M.F.-U.; et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J. Clin. Investig. 2014, 125, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Osanai, K. Rab38 Mutation and the Lung Phenotype. Int. J. Mol. Sci. 2018, 19, 2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.G.; O’Brien, K.J.; Coon, L.M.; Majerus, J.A.; Huryn, L.A.; Haroutunian, S.G.; Moka, N.; Introne, W.J.; Macnamara, E.; Gahl, W.A.; et al. Severe bleeding with subclinical oculocutaneous albinism in a patient with a novel HPS6 missense variant. Am. J. Med. Genet. Part. A 2018, 176, 2819–2823. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing–Based Oncology Panels. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, K.; Hayashi, M.; Abe, Y.; Kono, M.; Nakajima, K.; Aoyama, Y.; Nishigori, C.; Ishimoto, H.; Ishimatsu, Y.; Nakajima, M.; et al. NGS-based targeted resequencing identified rare subtypes of albinism: Providing accurate molecular diagnosis for Japanese patients with albinism. Pigment. Cell Melanoma Res. 2019, 32, 848–853. [Google Scholar] [CrossRef]
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front. Oncol. 2019, 9, 851. [Google Scholar] [CrossRef]
- Liu, T.; Yuan, Y.; Bai, D.; Qi, Z.; Yang, L.; Zhang, T.; Yang, X.; Li, W.; Wei, A. Genetic variants and mutational spectrum of Chinese Hermansky-Pudlak syndrome patients. Pigment. Cell Melanoma Res. 2020, 34, 111–121. [Google Scholar] [CrossRef]
- Khan, A.O.; Tamimi, M.; Lenzner, S.; Bolz, H.J. Hermansky-Pudlak syndrome genes are frequently mutated in patients with albinism from the Arabian Peninsula. Clin. Genet. 2016, 90, 96–98. [Google Scholar] [CrossRef]
- O’Brien, K.J.; Lozier, J.; Cullinane, A.R.; Osorio, B.; Nghiem, K.; Speransky, V.; Zein, W.M.; Mullikin, J.C.; Neff, A.T.; Simon, K.L.; et al. Identification of a novel mutation in HPS6 in a patient with hemophilia B and oculocutaneous albinism. Mol. Genet. Metab. 2016, 119, 284–287. [Google Scholar] [CrossRef] [Green Version]
- Andres, O.; Wiegering, V.; König, E.-M.; Schneider, A.L.; Semeniak, D.; Stritt, S.; Klopocki, E.; Schulze, H. A novel two-nucleotide deletion in HPS6 affects mepacrine uptake and platelet dense granule secretion in a family with Hermansky-Pudlak syndrome. Pediatr. Blood Cancer 2016, 64, e26320. [Google Scholar] [CrossRef]
- Schreyer-Shafir, N.; Huizing, M.; Anikster, Y.; Nusinker, Z.; Bejarano-Achache, I.; Maftzir, G.; Resnik, L.; Helip-Wooley, A.; Westbroek, W.; Gradstein, L.; et al. A new genetic isolate with a unique phenotype of syndromic oculocutaneous albinism: Clinical, molecular, and cellular characteristics. Hum. Mutat. 2006, 27, 1158. [Google Scholar] [CrossRef]
- Huizing, M.; Pederson, B.A.; Hess, R.; Griffin, A.; Helip-Wooley, A.; Westbroek, W.; Dorward, H.; O’Brien, K.J.; Golas, G.; Tsilou, E.; et al. Clinical and cellular characterisation of Hermansky-Pudlak syndrome type 6. J. Med. Genet. 2009, 46, 803–810. [Google Scholar] [CrossRef] [Green Version]
- Huizing, M.; Helip-Wooley, A.; Westbroek, W.; Gunay-Aygun, M.; Gahl, W.A. Disorders of Lysosome-Related Organelle Biogenesis: Clinical and Molecular Genetics. Annu. Rev. Genom. Hum. Genet. 2008, 9, 359–386. [Google Scholar] [CrossRef] [Green Version]
- Sitaram, A.; Marks, M.S. Mechanisms of Protein Delivery to Melanosomes in Pigment Cells. Physiology 2012, 27, 85–99. [Google Scholar] [CrossRef] [Green Version]
- Marks, M.S. Organelle Biogenesis: En BLOC Exchange for RAB32 and RAB38. Curr. Biol. 2012, 22, R963–R965. [Google Scholar] [CrossRef] [Green Version]
- Fourey, D.; Care, M.; Siminovitch, K.A.; Weissler-Snir, A.; Hindieh, W.; Chan, R.H.; Gollob, M.; Rakowski, H.; Adler, A. Prevalence and Clinical Implication of Double Mutations in Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Fatkin, D.; Johnson, R. Are Double Mutations Double Trouble? Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: A potential link to sudden death in the absence of conventional risk factors. Heart Rhythm. 2012, 9, 57–63. [Google Scholar] [CrossRef]
- Savov, A.; Angelicheva, D.; Balassopoulou, A.; Noussia-Arvanltakis, S.; Kalaydjieva, L.; Jordanova, A. Double mutant alleles: Are they rare? Hum. Mol. Genet. 1995, 4, 1169–1171. [Google Scholar] [CrossRef]
- Ogawa, T.; Tomatsu, S.; Fukuda, S.; Yamagishi, A.; Rezvi, G.M.M.; Sukegawa, K.; Kondo, N.; Suzuki, Y.; Shimozawa, N.; Orii, T. Mucopolysaccharidosis IVA: Screening and identification of mutations of the N-acetylgalactosamine-6-sulfate sulfatase gene. Hum. Mol. Genet. 1995, 4, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, A.; Ikonen, E.; Sormunen, R.; Lehto, V.-P.; Peltonen, L.; Jalanko, A. Dissection of the Molecular Consequences of a Double Mutation Causing a Human Lysosomal Disease. DNA Cell Biol. 1994, 13, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Latham, T.; Grabowski, G.; Theophilus, B.D.; Smith, I.F. Complex alleles of the acid beta-glucosidase gene in Gaucher disease. Am. J. Hum. Genet. 1990, 47, 79–86. [Google Scholar] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karim, S.; Saharti, S.; Alganmi, N.; Mirza, Z.; Alfares, A.; Turkistany, S.; Al-Attas, M.; Noureldin, H.; Al Sakkaf, K.; Abusamra, H.; et al. Two Novel Homozygous HPS6 Mutations (Double Mutant) Identified by Whole-Exome Sequencing in a Saudi Consanguineous Family Suspected for Oculocutaneous Albinism. Life 2022, 12, 14. https://doi.org/10.3390/life12010014
Karim S, Saharti S, Alganmi N, Mirza Z, Alfares A, Turkistany S, Al-Attas M, Noureldin H, Al Sakkaf K, Abusamra H, et al. Two Novel Homozygous HPS6 Mutations (Double Mutant) Identified by Whole-Exome Sequencing in a Saudi Consanguineous Family Suspected for Oculocutaneous Albinism. Life. 2022; 12(1):14. https://doi.org/10.3390/life12010014
Chicago/Turabian StyleKarim, Sajjad, Samah Saharti, Nofe Alganmi, Zeenat Mirza, Ahmed Alfares, Shereen Turkistany, Manal Al-Attas, Hend Noureldin, Khadega Al Sakkaf, Heba Abusamra, and et al. 2022. "Two Novel Homozygous HPS6 Mutations (Double Mutant) Identified by Whole-Exome Sequencing in a Saudi Consanguineous Family Suspected for Oculocutaneous Albinism" Life 12, no. 1: 14. https://doi.org/10.3390/life12010014