
Hematopoietic Prostaglandin D Synthase Inhibitor PK007 Decreases Muscle Necrosis in DMD mdx Model Mice


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Muscle Grip Strength Assessments
2.3. Muscle Dissection and Histochemical Staining
2.4. Blood Collection and Muscle Creatine Kinase (CK-MM) Assay
2.5. Statistics
3. Results
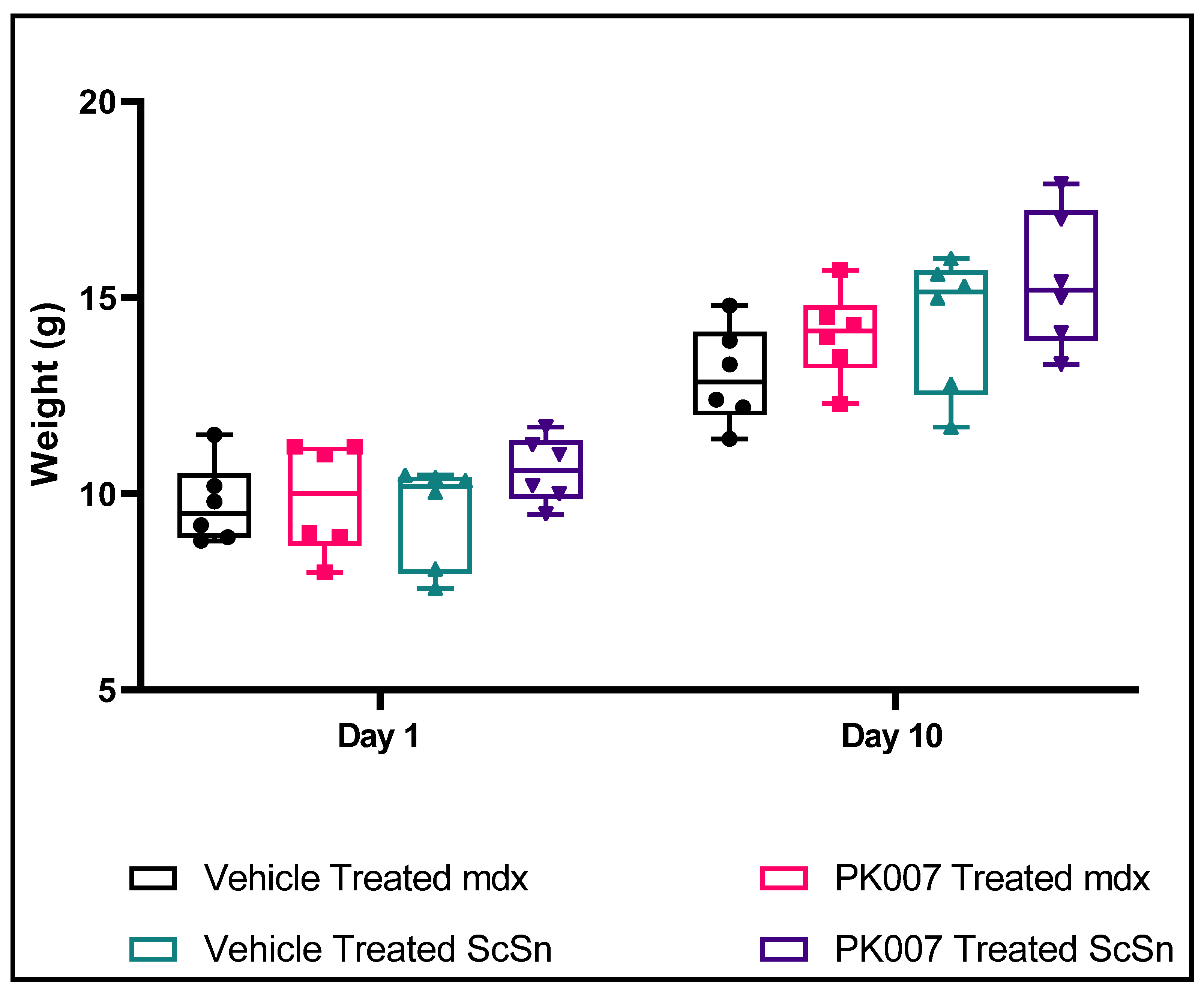
3.1. No Change in Mean Body Weight over the 10-Day Treatment Period for Both Treated and Untreated Groups and WT Groups
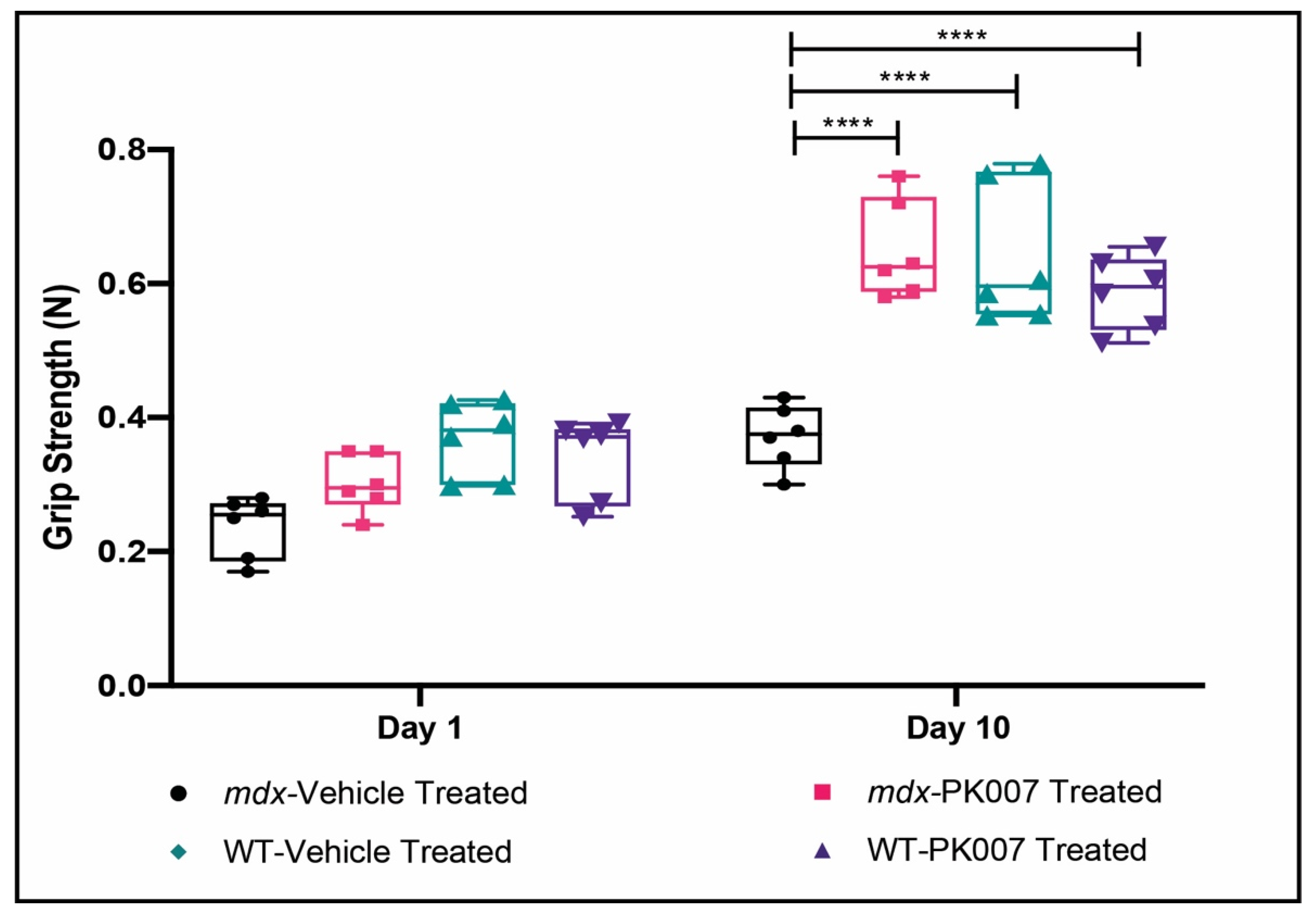
3.2. Mdx Mice Display Improved Muscle Grip by the End of Treatment with PK007
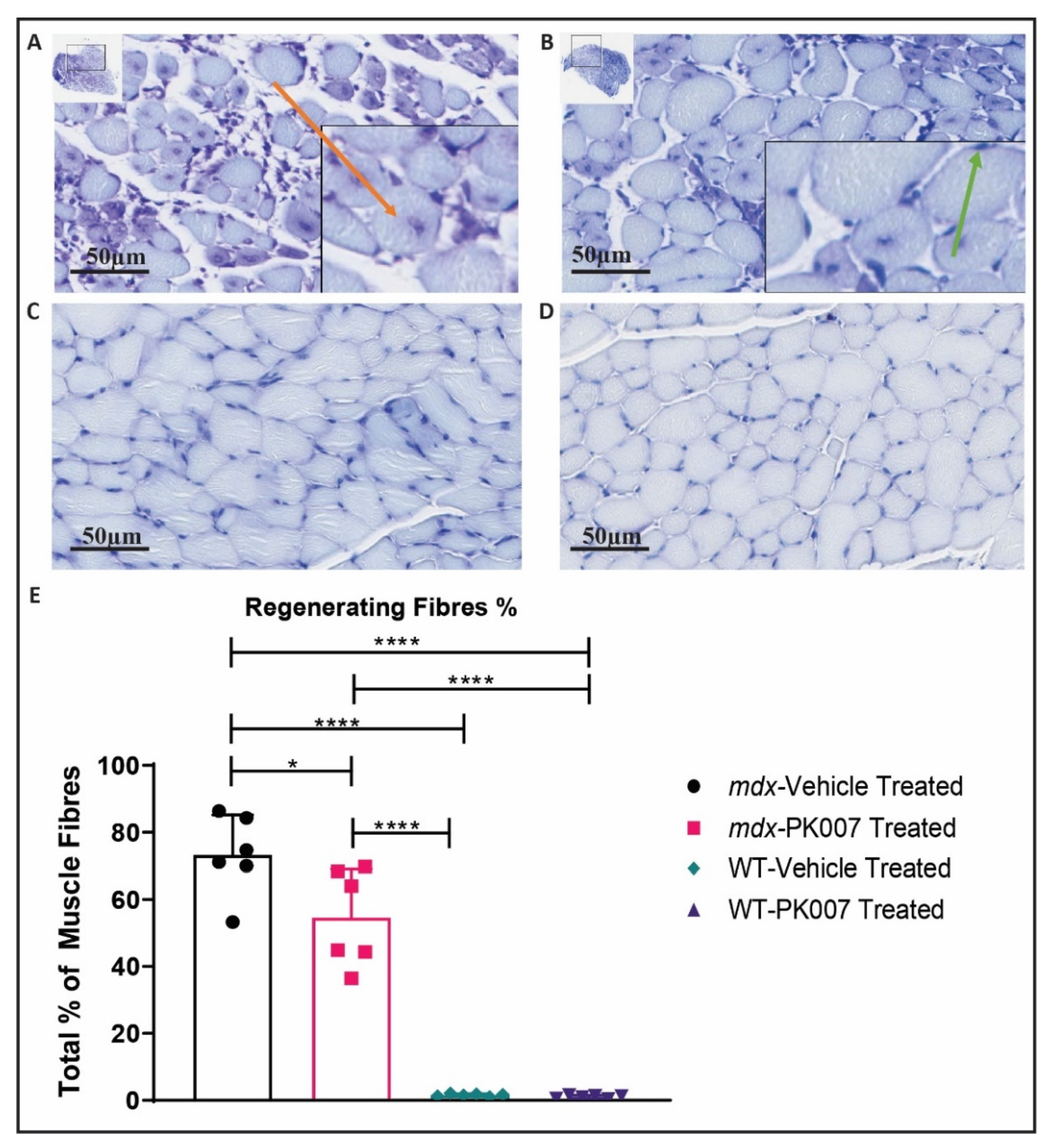
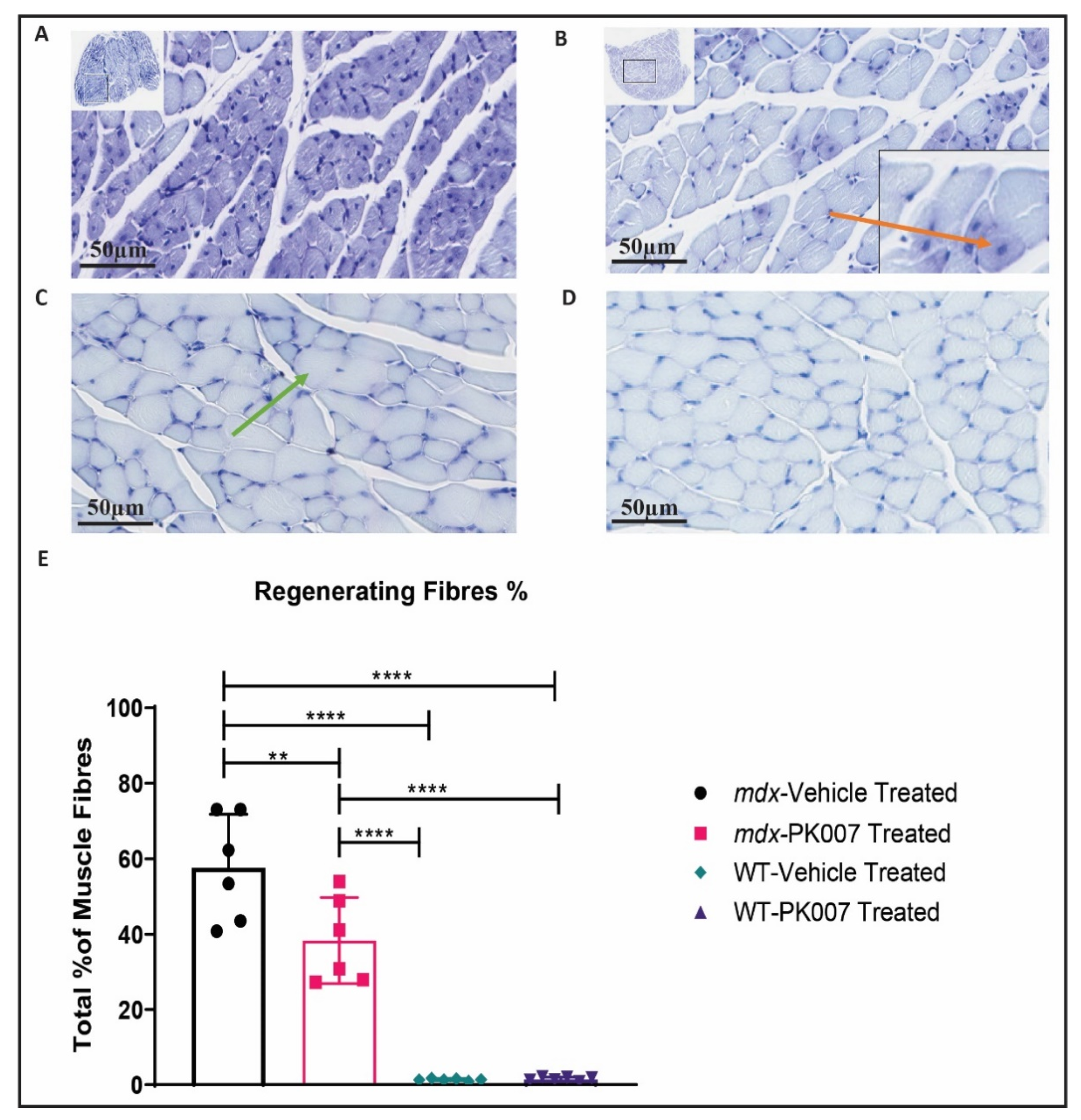
3.3. PK007-Treated mdx Group Exhibits Fewer Regenerating Muscle Fibers in the TA and GA
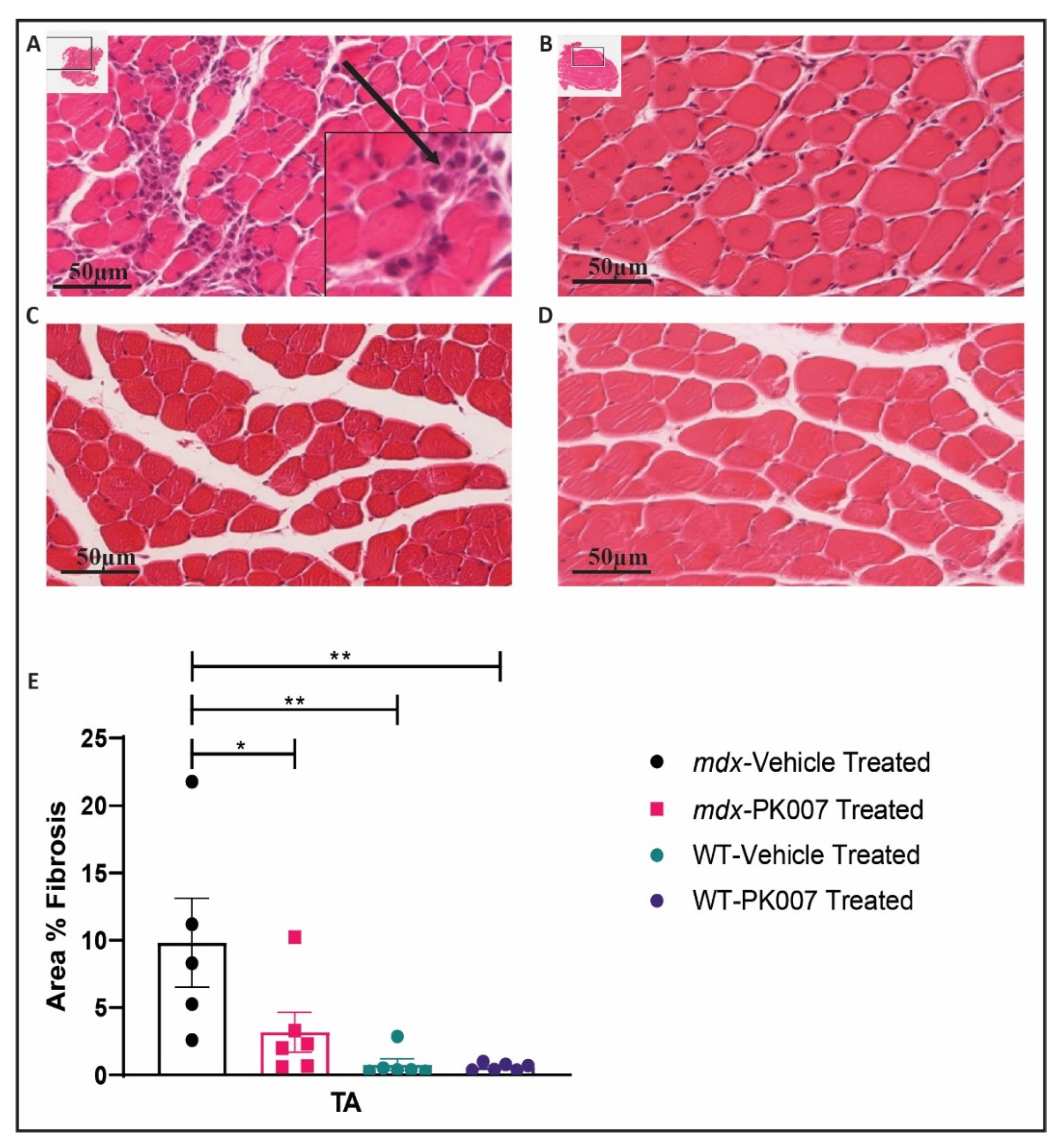
3.4. TA Muscles from PK007-Treated Mice Display Lower Levels of Muscle Fibrosis Compared to TA Muscles from mdx Vehicle-Treated Mice
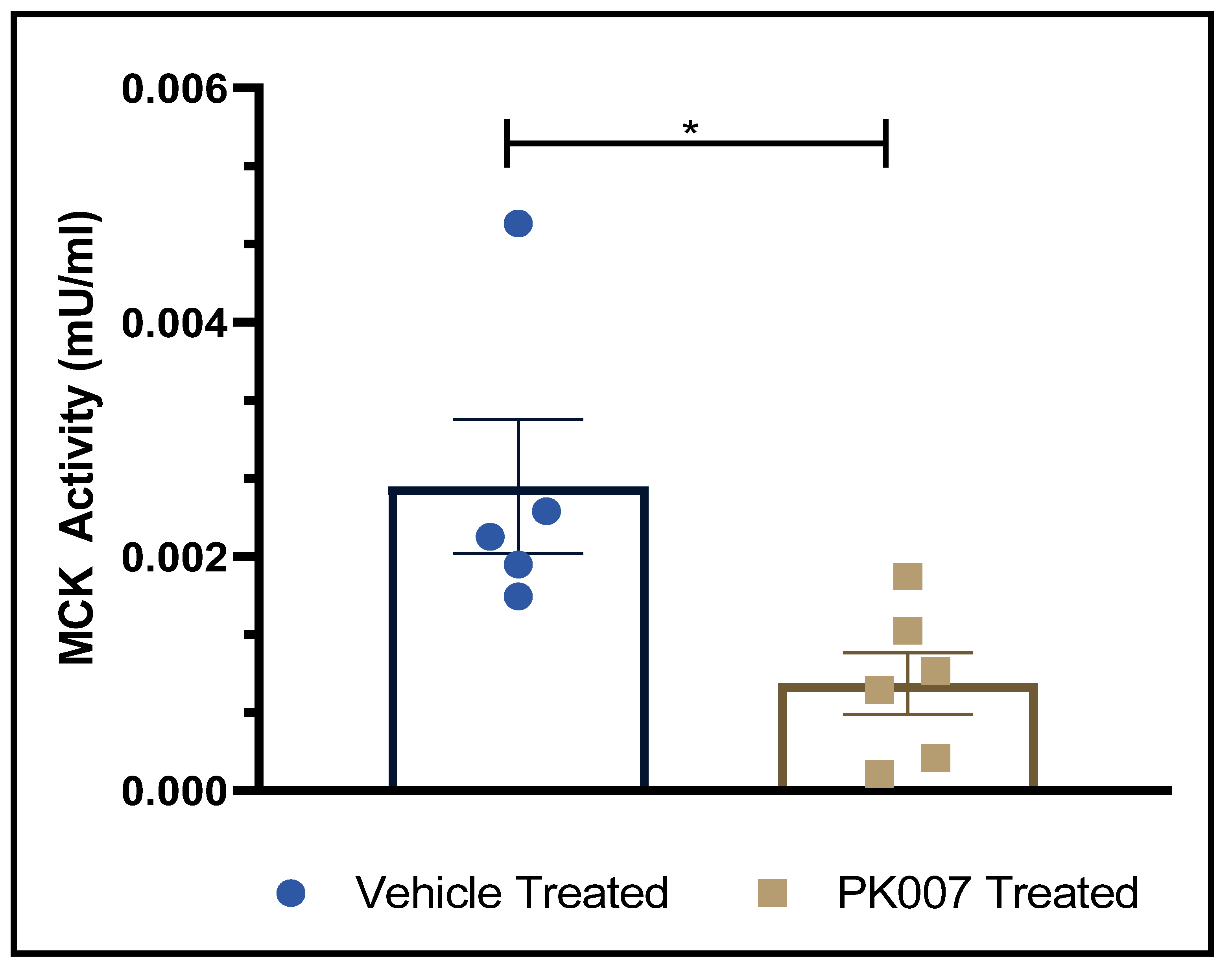
3.5. PK007 Treatment Reduces Serum Muscle Creatine Kinase (CK-MM) in mdx Mice
4. Discussion
4.1. PK007 Improves Muscle Grip in mdx Mice
4.2. Mdx Mice Treated with PK007 Results in a Greater Amount of Mature Muscle Fibers, Fewer Regenerating Muscle Fibers, and Decreased Percentage of Fibrosis in the TA Muscle
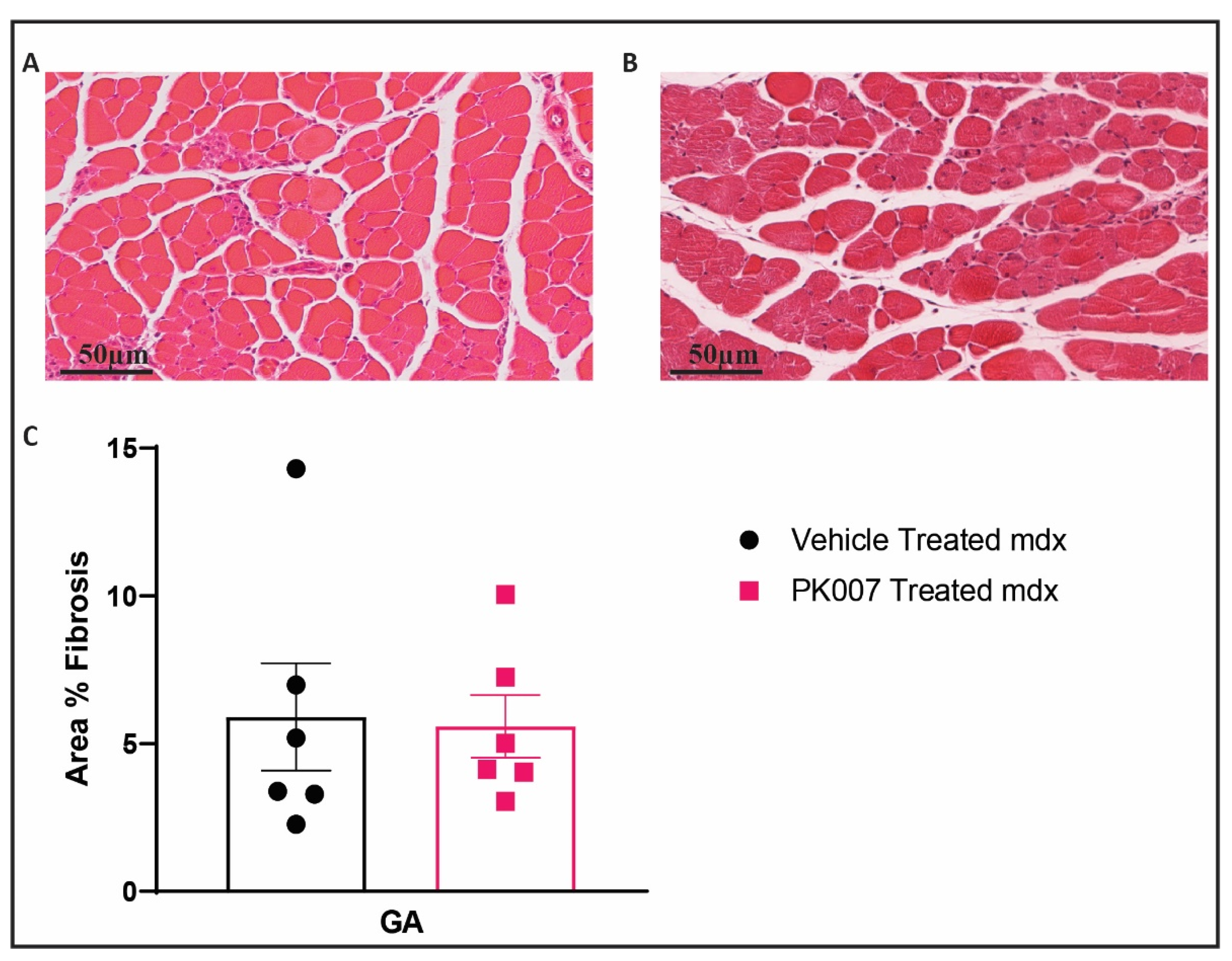
4.3. PK007 Treatment of mdx Mice Results in Decreased Fibrosis in TA Muscles but Not GA Muscles
4.4. Decreased CK-MM Levels in PK007-Treated mdx Mice Indicate Lower Levels of Muscle Damage
4.5. Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Annexstad, E.J.; Lund-Petersen, I.; Rasmussen, M. Duchenne muscular dystrophy. Tidsskr. Nor. Laegeforen. 2014, 134, 1361–1364. [Google Scholar] [CrossRef]
- Nicholson, L.V.; Johnson, M.A.; Bushby, K.M.; Gardner-Medwin, D. Functional significance of dystrophin positive fibres in Duchenne muscular dystrophy. Arch. Dis. Child. 1993, 68, 632–636. [Google Scholar] [CrossRef] [Green Version]
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. Neurol. India 2008, 56, 236. [Google Scholar] [CrossRef]
- Hoogerwaard, E.; Bakker, E.; Ippel, P.; Oosterwijk, J.; Majoor-Krakauer, D.; Leschot, N.; Van Essen, A.; Brunner, H.; van der Wouw, P.; Wilde, A.; et al. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in the Netherlands: A cohort study. Lancet 1999, 353, 2116–2119. [Google Scholar] [CrossRef]
- Iwańczak, F.; Stawarski, A.; Potyrała, M.; Siedlecka-Dawidko, J.; Agrawal, G.S. Early symptoms of Duchenne muscular dystrophy—description of cases of an 18-month-old and an 8-year-old patient. Med. Sci. Monit. 2001, 6, 592–595. [Google Scholar]
- Pasternak, C.; Wong, S.; Elson, E.L. Mechanical function of dystrophin in muscle cells. J. Cell Biol. 1995, 128, 355–361. [Google Scholar] [CrossRef]
- Horsley, V.; Pavlath, G.K. Prostaglandin F2α stimulates growth of skeletal muscle cells via an NFATC2-dependent pathway. J. Cell Biol. 2003, 161, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Kachaeva, E.V.; Shenkman, B.S. Various Jobs of Proteolytic Enzymes in Skeletal Muscle during Unloading: Facts and Speculations. J. Biomed. Biotechnol. 2012, 2012, 493618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Westering, T.; Betts, C.A.; Wood, M.J.A. Current Understanding of Molecular Pathology and Treatment of Cardiomyopathy in Duchenne Muscular Dystrophy. Molecules 2015, 20, 8823–8855. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Viña, J.; Ji, L.L. Role of Redox Signaling and Inflammation in Skeletal Muscle Adaptations to Training. Antioxidants 2016, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- van Furth, A.M.; Roord, J.J.; van Furth, R. Roles of proinflammatory and anti-inflammatory cytokines in pathophysiology of bacterial meningitis and effect of adjunctive therapy. Infect. Immun. 1996, 64, 4883–4890. [Google Scholar] [CrossRef] [Green Version]
- Grounds, M.D.; Radley, H.G.; Gebski, B.L.; Bogoyevitch, M.A.; Shavlakadze, T. Implications of Cross-Talk between Tumour Necrosis Factor and Insulin-Like Growth Factor-1 Signalling in Skeletal Muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 846–851. [Google Scholar] [CrossRef]
- Hoxha, M. Duchenne muscular dystrophy: Focus on arachidonic acid metabolites. Biomed. Pharmacother. 2019, 110, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Narumiya, S. Prostaglandins and chronic inflammation. Trends Pharmacol. Sci. 2012, 33, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Mohri, I.; Aritake, K.; Taniguchi, H.; Sato, Y.; Kamauchi, S.; Nagata, N.; Maruyama, T.; Taniike, M.; Urade, Y. Inhibition of Prostaglandin D Synthase Suppresses Muscular Necrosis. Am. J. Pathol. 2009, 174, 1735–1744. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.T.V.; Palla, A.R.; Blake, M.R.; Yucel, N.D.; Wang, Y.X.; Magnusson, K.E.G.; Holbrook, C.A.; Kraft, P.E.; Delp, S.L.; Blau, H.M. Prostaglandin E2 is essential for efficacious skeletal muscle stem-cell function, augmenting regeneration and strength. Proc. Natl. Acad. Sci. USA 2017, 114, 6675–6684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorris, S.L.; Peebles, R.S. PGI2as a Regulator of Inflammatory Diseases. Mediat. Inflamm. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Okinaga, T.; Mohri, I.; Fujimura, H.; Imai, K.; Ono, J.; Urade, Y.; Taniike, M. Induction of hematopoietic prostaglandin D synthase in hyalinated necrotic muscle fibers: Its implication in grouped necrosis. Acta Neuropathol. 2002, 104, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Aritake, K.; Kado, Y.; Inoue, T.; Miyano, M.; Urade, Y. Structural and Functional Characterization of HQL-79, an Orally Selective Inhibitor of Human Hematopoietic Prostaglandin D Synthase. J. Biol. Chem. 2006, 281, 15277–15286. [Google Scholar] [CrossRef] [Green Version]
- Moniot, B.; Farhat, A.; Aritake, K.; Declosmenil, F.; Nef, S.; Eguchi, N.; Urade, Y.; Poulat, F.; Boizet-Bonhoure, B. Hematopoietic prostaglandin D synthase (H-Pgds) is expressed in the early embryonic gonad and participates to the initial nuclear translocation of the SOX9 protein. Dev. Dyn. 2011, 240, 2335–2343. [Google Scholar] [CrossRef]
- Mohri, I.; Taniike, M.; Taniguchi, H.; Kanekiyo, T.; Aritake, K.; Inui, T.; Fukumoto, N.; Eguchi, N.; Kushi, A.; Sasai, H.; et al. Prostaglandin D2-Mediated Microglia/Astrocyte Interaction Enhances Astrogliosis and Demyelination in twitcher. J. Neurosci. 2006, 26, 4383–4393. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, E.; Komaki, H.; Shimizu-Motohashi, Y.; Ishiyama, A.; Sasaki, M.; Takeda, S. A phase I study of TAS-205 in patients with Duchenne muscular dystrophy. Ann. Clin. Transl. Neurol. 2018, 5, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Aritake, K. Novel inhibitor of hematopoietic prostaglandin D synthase improves the muscle disorder in an experimental model of Duchenne muscular dystrophy. Neuromuscul. Disord. 2014, 24, 821. [Google Scholar] [CrossRef]
- Korotkova, M.; Lundberg, I. The skeletal muscle arachidonic acid cascade in health and inflammatory disease. Nat. Rev. Rheumatol. 2014, 10, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Radley, H.G.; Lynch, G.; Nagaraju, K.; De Luca, A. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol. Dis. 2008, 31, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.; Goemans, N.; Takeda, S.I. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 1–19. [Google Scholar] [CrossRef]
- Sampath, S.C.; Sampath, S.C.; Millay, D.P. Myoblast fusion confusion: The resolution begins. Skelet. Muscle 2018, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef]
- Collins, C.A.; Morgan, J.E. Duchenne’s muscular dystrophy: Animal models used to investigate pathogenesis and develop therapeutic strategies. Int. J. Exp. Pathol. 2003, 84, 165–172. [Google Scholar] [CrossRef] [PubMed]
- TREAT-NMD. Experimental Protocols for DMD Animal Models. 2020. Available online: https://treat-nmd.org/research-overview/preclinical-research/experimental-protocols-for-dmd-animal-models/ (accessed on 13 July 2021).
- Wang, C.; Yue, F.; Kuang, S. Muscle Histology Characterization Using H&E Staining and Muscle Fiber Type Classification Using Immunofluorescence Staining. Bio-Protocol 2017, 7, e2279. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Zelikovich, A.S.; Salamone, I.M.; Fischer, J.A.; McNally, E.M. Mechanisms and Clinical Applications of Glucocorticoid Steroids in Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, 39–52. [Google Scholar] [CrossRef]
- Keeling, R.M.; Golumbek, P.T. Weekly oral prednisolone improves survival and strength in male mdx mice. Muscle Nerve 2007, 35, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Golumbek, P.T.; Keeling, R.M.; Connolly, A.M. Strength and corticosteroid responsiveness of mdx mice is unchanged by RAG2 gene knockout. Neuromuscul. Disord. 2007, 17, 376–384. [Google Scholar] [CrossRef]
- Anderson, J.E.; McIntosh, L.M.; Poettcker, R. Deflazacort but not prednisone improves both muscle repair and fiber growth in diaphragm and limb muscle in vivo in the mdx dystrophic mouse. Muscle Nerve 1996, 19, 1576–1585. [Google Scholar] [CrossRef]
- Komaki, H.; Maegaki, Y.; Matsumura, T.; Shiraishi, K.; Awano, H.; Nakamura, A.; Kinoshita, S.; Ogata, K.; Ishigaki, K.; Saitoh, S.; et al. Early phase 2 trial of TAS-205 in patients with Duchenne muscular dystrophy. Ann. Clin. Transl. Neurol. 2020, 7, 181–190. [Google Scholar] [CrossRef]
- Mâncio, R.D.; Hermes, T.D.A.; Macedo, A.B.; Mizobuti, D.S.; Rupcic, I.F.; Minatel, E. Dystrophic phenotype improvement in the diaphragm muscle of mdx mice by diacerhein. PLoS ONE 2017, 12, e0182449. [Google Scholar] [CrossRef] [Green Version]
- Krahn, M.; Anderson, J. Anabolic steroid treatment increases myofiber damage in mdx mouse muscular dystrophy. J. Neurol. Sci. 1994, 125, 138–146. [Google Scholar] [CrossRef]
- DelloRusso, C.; Crawford, R.W.; Chamberlain, J.S.; Brooks, S. Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J. Muscle Res. Cell Motil. 2001, 22, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittchen, S.; Heinemann, A. Therapeutic Potential of Hematopoietic Prostaglandin D2 Synthase in Allergic Inflammation. Cells 2019, 8, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luz, M.; Marques, M.; Neto, H.S. Impaired regeneration of dystrophin-deficient muscle fibers is caused by exhaustion of myogenic cells. Braz. J. Med. Biol. Res. 2002, 35, 691–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the Process of Fibrosis in Duchenne Muscular Dystrophy. BioMed Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef]
- Szigyarto, C.A.-K.; Spitali, P. Biomarkers of Duchenne muscular dystrophy: Current findings. Degener. Neurol. Neuromuscul. Dis. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, E.; Komaki, H.; Tachimori, H.; Miyoshi, K.; Yamamiya, I.; Shimizu-Motohashi, Y.; Ishiyama, A.; Saito, T.; Nakagawa, E.; Sugai, K.; et al. Urinary prostaglandin metabolites as Duchenne muscular dystrophy progression markers. Brain Dev. 2018, 40, 918–925. [Google Scholar] [CrossRef]
- Ozawa, E.; Hagiwara, Y.; Yoshida, M. Creatine kinase, cell membrane and Duchenne muscular dystrophy. Mol. Cell. Biochem. 1999, 190, 143–151. [Google Scholar] [CrossRef]
- Gutpell, K.M.; Hrinivich, W.T.; Hoffman, L.M. Skeletal muscle fibrosis in the mdx/utrn+/- mouse validates its suitability as a murine model of Duchenne muscular dystrophy. PLoS ONE 2015, 10, e0117306. [Google Scholar] [CrossRef]
- Koskinen, S.O.; Lehti, M. Molecular and Cellular Markers in Skeletal Muscle Damage after Acute Voluntary Exercise Containing Eccentric Muscle Contractions. In Muscle Cell and Tissue; Sakuma, K., Ed.; IntechOpen: Tokyo, Japan, 2018; Available online: https://www.intechopen.com/chapters/61489. [CrossRef] [Green Version]
- McDonald, A.A.; Hebert, S.L.; Kunz, M.D. Disease course in mdx:utrophin+/- mice: Comparison of three mouse models of Duchenne muscular dystrophy. Physiol. Rep. 2015, 3, e12391. [Google Scholar] [CrossRef] [Green Version]
- Squire, S.; Raymackers, J.; Vandebrouck, C.; Potter, A.; Tinsley, J.; Fisher, R.; Gillis, J.; Davies, K. Prevention of pathology in mdx mice by expression of utrophin: Analysis using an inducible transgenic expression system. Hum. Mol. Genet. 2002, 11, 3333–3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammers, D.W.; Hart, C.C.; Matheny, M.K.; Wright, L.A.; Armellini, M.; Barton, E.R.; Sweeney, H.L. The D2.mdx mouse as a preclinical model of the skeletal muscle pathology associated with Duchenne muscular dystrophy. Sci. Rep. 2020, 10, 14070. [Google Scholar] [CrossRef]
- Duddy, W.; Duguez, S.; Johnston, H.; Cohen, T.V.; Phadke, A.; Gordish-Dressman, H.; Nagaraju, K.; Gnocchi, V.; Low, S.; Partridge, T. Muscular dystrophy in the mdx mouse is a severe myopathy compounded by hypotrophy, hypertrophy and hyperplasia. Skelet. Muscle 2015, 5, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (Days) | Mean Body Weight (g) | |||
|---|---|---|---|---|
| Mdx + Vehicle | Mdx + PK007 | Control + Vehicle | Control + PK007 | |
| 1 | 9.73 ± 0.42 | 9.88 ± 0.58 | 9.49 ± 0.53 | 10.61 ± 0.34 |
| 2 | 9.37 ± 0.48 | 9.78 ± 0.48 | 9.78 ± 0.60 | 11.23 ± 0.43 |
| 3 | 9.33 ± 0.50 | 9.77 ± 0.42 | 10.17 ± 0.59 | 11.68 ± 0.46 |
| 4 | 9.67 ± 0.35 | 10.00 ± 0.37 | 10.83 ± 0.57 | 12.10 ± 0.63 |
| 5 | 9.93 ± 0.35 | 10.35 ± 0.32 | 11.38 ± 0.58 | 12.68 ± 0.52 |
| 6 | 10.50 ± 0.50 | 10.80 ± 0.35 | 11.87 ± 0.65 | 12.82 ± 0.74 |
| 7 | 10.52 ± 0.89 | 11.77 ± 0.33 | 12.48 ± 0.63 | 13.43 ± 0.65 |
| 8 | 11.97 ± 0.62 | 12.63 ± 0.37 | 12.98 ± 0.65 | 13.77 ± 0.68 |
| 9 | 12.60 ± 0.59 | 13.63 ± 0.55 | 13.73 ± 0.69 | 14.58 ± 0.70 |
| 10 | 13.00 ± 0.51 | 14.05 ± 0.46 | 14.40 ± 0.71 | 15.45 ± 0.71 |
| Time (Days) | Grip Strength in Newtons (N) | |||
|---|---|---|---|---|
| Mdx + Vehicle | Mdx + PK007 | Control + Vehicle | Control + PK007 | |
| 1 | 0.237 ± 0.019 | 0.302 ± 0.017 | 0.368 ± 0.023 | 0.340 ± 0.025 |
| 10 | 0.372 ± 0.019 | 0.650 ± 0.030 | 0.640 ± 0.042 | 0.587 ± 0.022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yarlagadda, S.; Kulis, C.; Noakes, P.G.; Smythe, M.L. Hematopoietic Prostaglandin D Synthase Inhibitor PK007 Decreases Muscle Necrosis in DMD mdx Model Mice. Life 2021, 11, 994. https://doi.org/10.3390/life11090994
Yarlagadda S, Kulis C, Noakes PG, Smythe ML. Hematopoietic Prostaglandin D Synthase Inhibitor PK007 Decreases Muscle Necrosis in DMD mdx Model Mice. Life. 2021; 11(9):994. https://doi.org/10.3390/life11090994
Chicago/Turabian StyleYarlagadda, Sai, Christina Kulis, Peter G. Noakes, and Mark L. Smythe. 2021. "Hematopoietic Prostaglandin D Synthase Inhibitor PK007 Decreases Muscle Necrosis in DMD mdx Model Mice" Life 11, no. 9: 994. https://doi.org/10.3390/life11090994