
The Complementarity Principle—One More Step towards Analytical Docking on the Example of Dihydrofolate Reductase Complexes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Considered Complexes
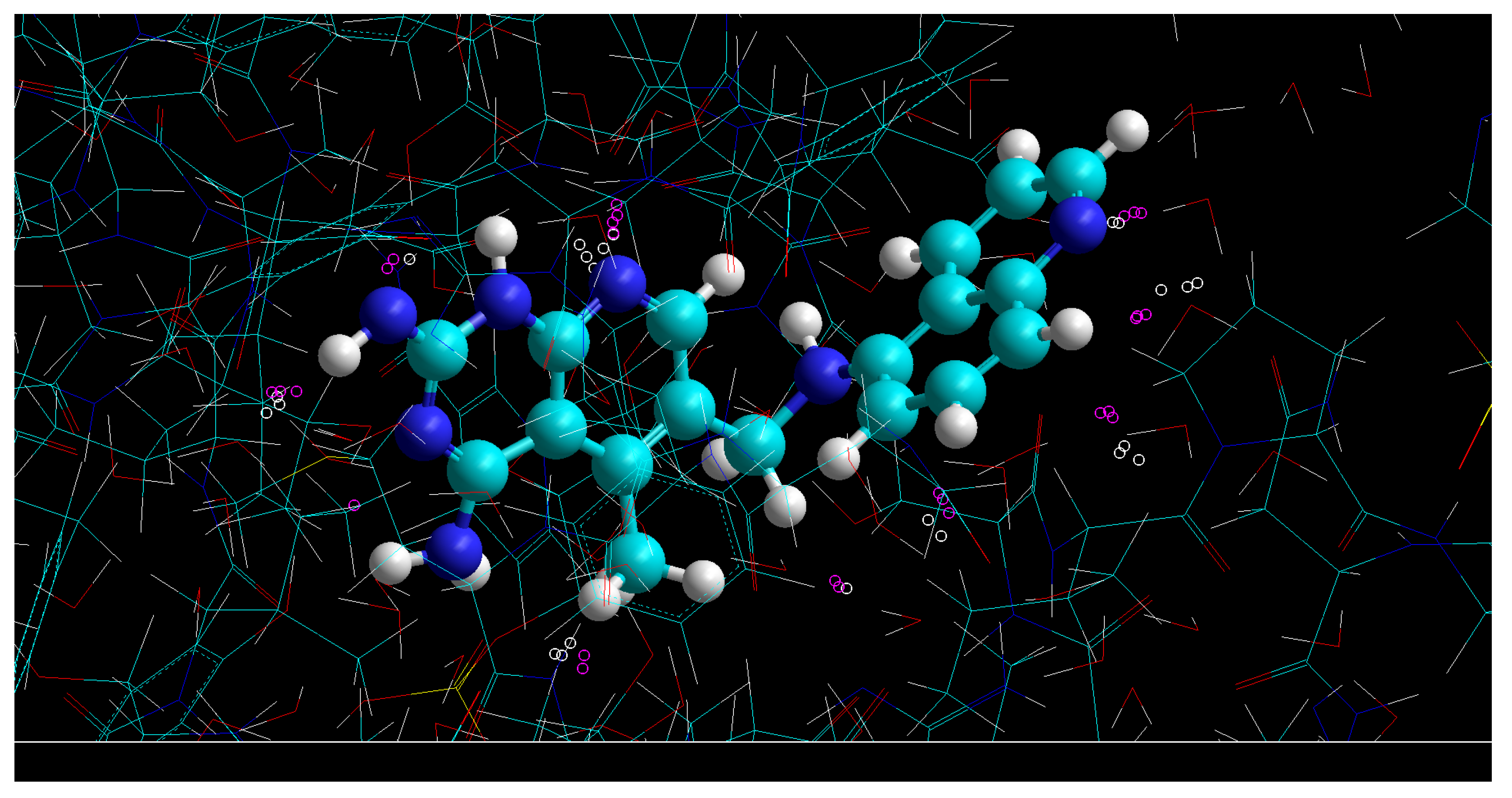
2.2. D Maps of Electron Density
2.3. Complementarity Factors
2.4. Overlaps
3. Results and Discussion
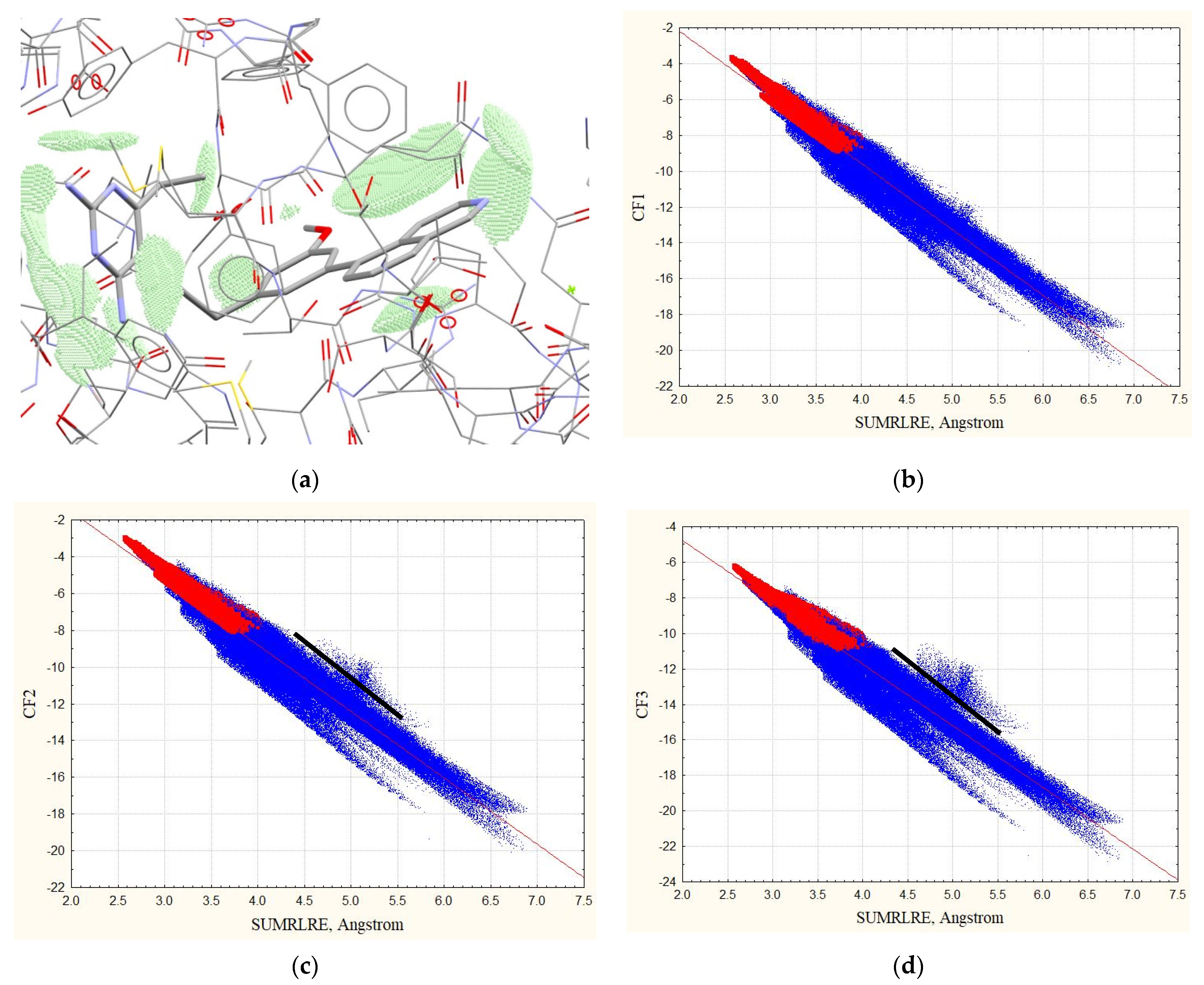
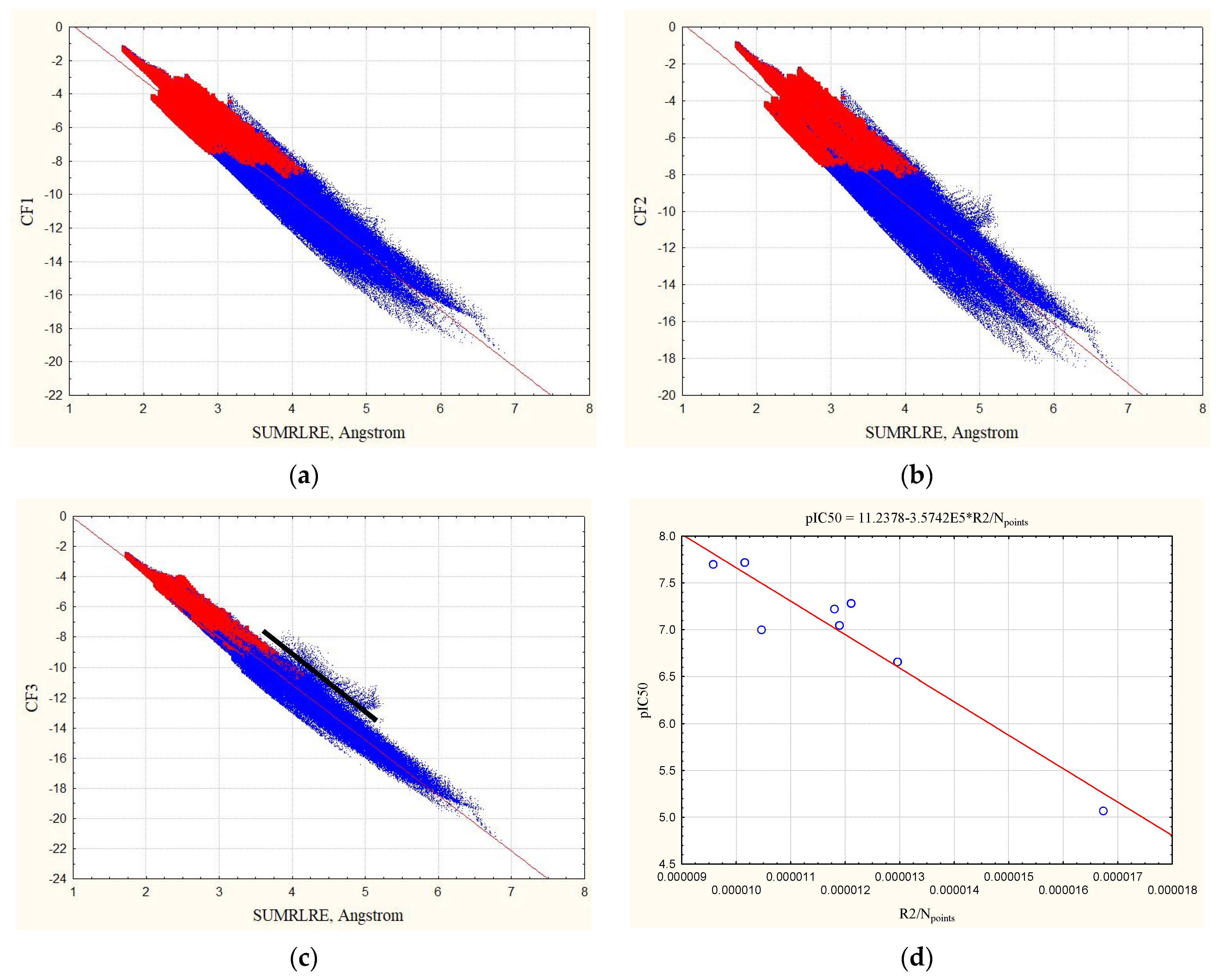
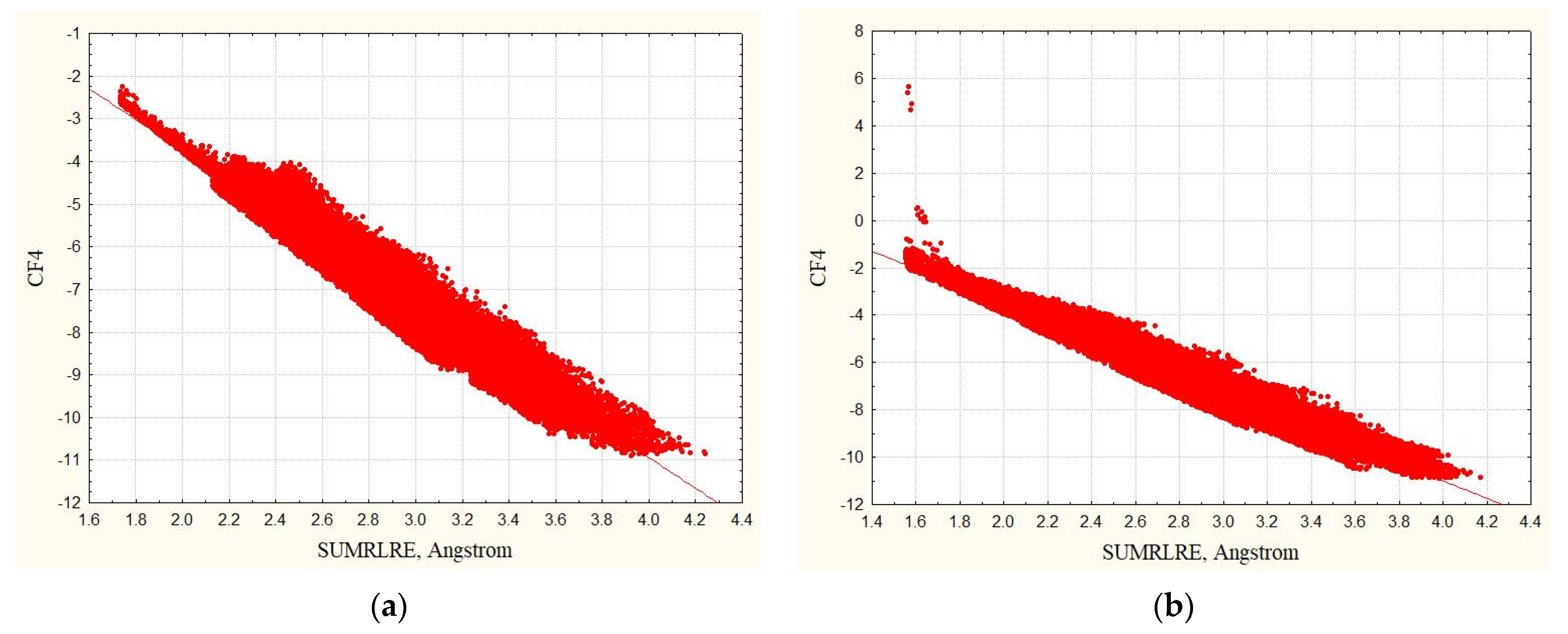
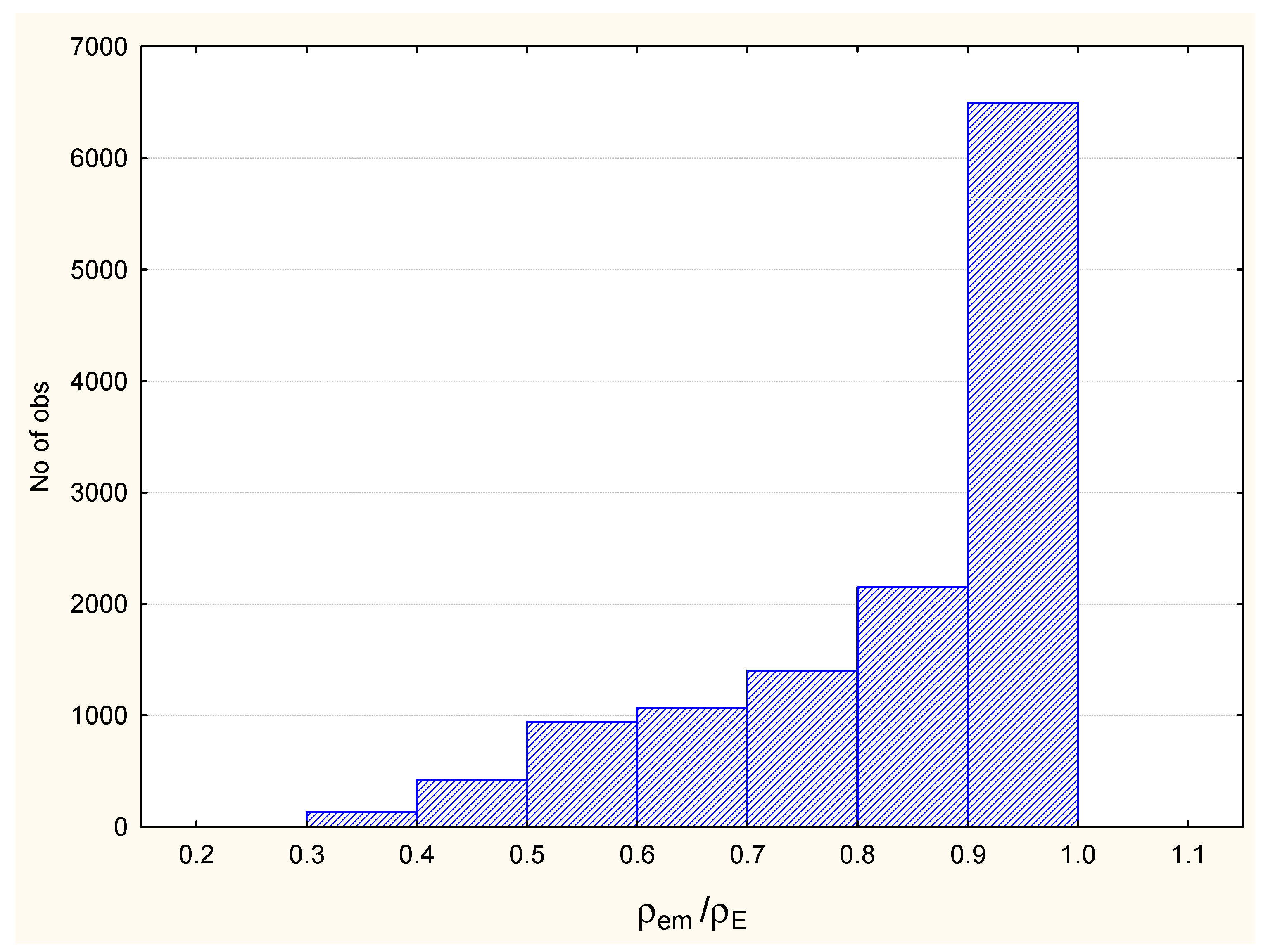
3.1. Complexes without Hydrogens
3.2. Complexes with Hydrogens
3.3. The Relationship of Experimental pIC50 Values with Statistical Characteristics of Equation (12)
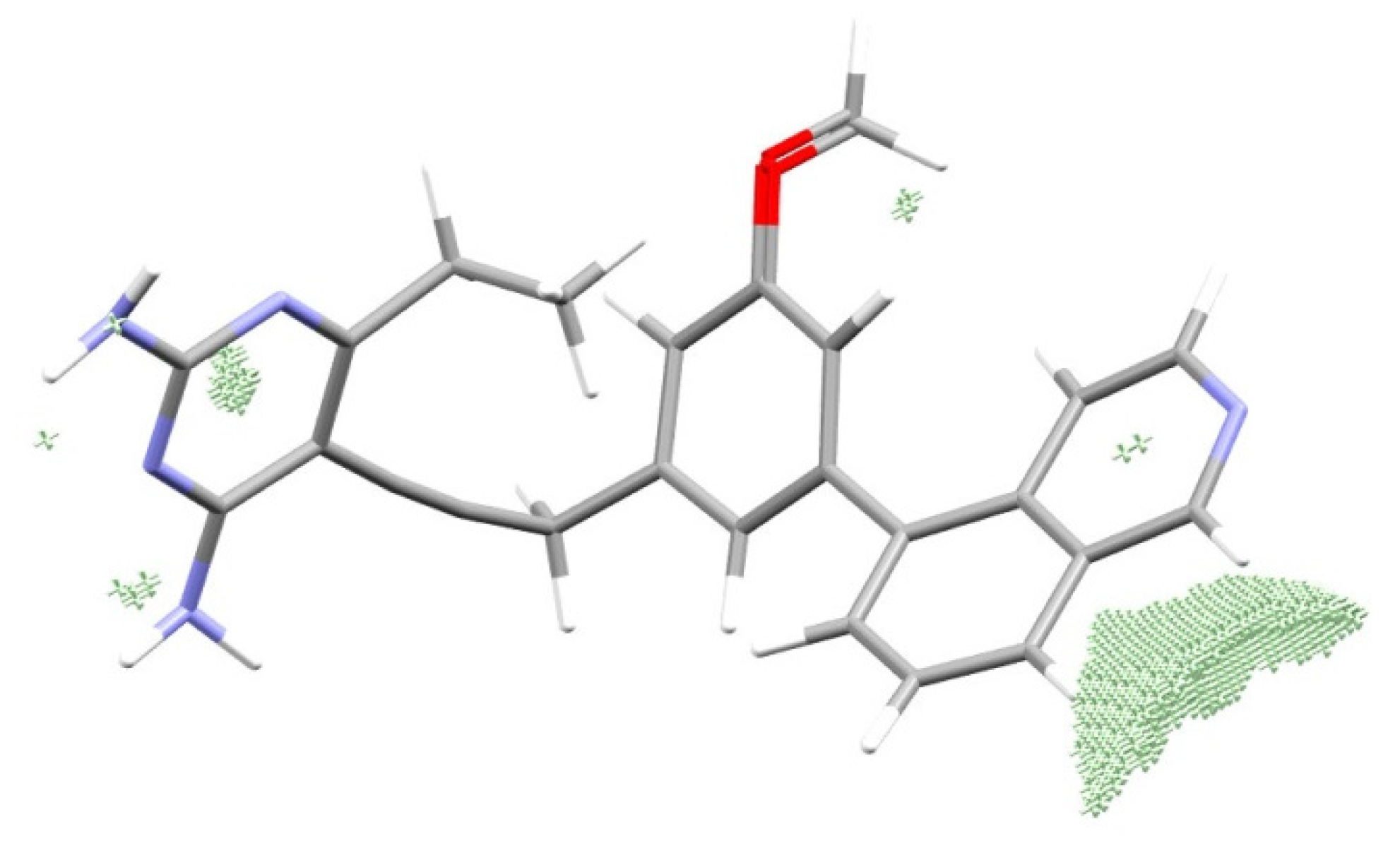
3.4. Reconstruction of the Pattern of the Electronic Structure of the Drug Suitable for the Given Enzyme
3.5. A Way towards Analytical Docking Procedure
- (1)
- Computation of the distances (Equations (38)–(40)) of the most effective contacts and the most preferred for a given enzyme.
- (2)
- The construction of contact zones for the ligand and the enzyme; these zones determine the direction of intermolecular interactions between the ligand and the enzyme.In order to find these zones, you need to find coordinates of points in the basin of eth atom (), which will satisfy the conditions:and will be the minimum points of the electron density function of contributions of neighbors of the eth atom.
- (3)
- In order to find the contact zones in the ligand, the same can be performed for the periphery ligand atoms.
- (4)
- The correct binding position of the ligand can then be determined from the condition of maximum overlap between the contact zones of the enzyme and the ligand.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ehrlich, P. Croonian Lecture—On Immunity with Special Reference to Cell Life. Proc. R. Soc. Lond. 1900, 66, 424–448. [Google Scholar]
- Bohr, N. The Recent Developments in Atomic Theory and the Quantum Postulate. In Atti del Congresso Internazionale dei Fisici; N. Zanicelli: Bologna, Italy, 1928. [Google Scholar]
- Heisenberg, W. Über den anschaulichen Inhalt der quantentheoretischen Kinematik und Mechanik. Z. Für Phys. 1927, 43, 172–198. [Google Scholar] [CrossRef]
- Kennard, E.H. Zur Quantenmechanik Einfacher Bewegungstypen. Zeitschrift für Phys. 1927, 44, 326–352. [Google Scholar] [CrossRef]
- Guth, E. Gruppentheorie Und Quantenmechanik-H. Weyl S. Hirzel, Leipzig 1928 VIII+288 Seiten, RM 20. Mon. Für Math. Und Phys. 1929, 36, A48–A52. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H.C. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Rimac, H.; Grishina, M.A.; Potemkin, V.A. Electron density analysis of CDK complexes using the AlteQ method. Future Med. Chem. 2020, 12, 1387–1397. [Google Scholar] [CrossRef]
- Kandagalla, S.; Rimac, H.; Potemkin, V.A.; Grishina, M.A. Complementarity principle in terms of electron density for the study of epidermal growth factor receptor complexes. Future Med. Chem. 2021, 13, 863–875. [Google Scholar] [CrossRef]
- Rimac, H.; Grishina, M.; Potemkin, V. Use of the Complementarity Principle in Docking Procedures: A New Approach for Evaluating the Correctness of Binding Poses. J. Chem. Inf. Model. 2021, 61, 1801–1813. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Christie, C.; Duarte, J.M.; Feng, Z.; Westbrook, J.; Young, J.; Zardecki, C. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018, 27, 316–330. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Potemkin, V.A.; Grishina, M.A. A New Paradigm for Pattern Recognition of Drugs. J. Comput. Aided. Mol. Des. 2008, 22, 489–505. [Google Scholar] [CrossRef]
- Potemkin, V.; Grishina, M. Electron-based descriptors in the study of physicochemical properties of compounds. Comput. Theor. Chem. 2018, 1123, 1–10. [Google Scholar] [CrossRef]
- Potemkin, V.; Potemkin, A.; Grishina, M. Internet Resources for Drug Discovery and Design. Curr. Top. Med. Chem. 2018, 18, 1955–1975. [Google Scholar] [CrossRef]
- Potemkin, V.; Palko, N.; Grishina, M. Quantum theory of atoms in molecules for photovoltaics. Sol. Energy 2019, 190, 475–487. [Google Scholar] [CrossRef]
- Grishina, M.A.; Potemkin, V.A. Topological Analysis of Electron Density in Large Biomolecular Systems. Curr. Drug Discov. Technol. 2019, 16, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Cario, H.; Smith, D.E.C.; Blom, H.; Blau, N.; Bode, H.; Holzmann, K.; Pannicke, U.; Hopfner, K.P.; Rump, E.-M.; Ayric, Z.; et al. Dihydrofolate Reductase Deficiency Due to a Homozygous DHFR Mutation Causes Megaloblastic Anemia and Cerebral Folate Deficiency Leading to Severe Neurologic Disease. Am. J. Hum. Genet. 2011, 88, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, A.F.; Mathis, A.D.; Ingle, C.; Park, J.O.; Chen, L.; Rabinowitz, J.D.; Junier, I.; Rivoire, O.; Reynolds, K.A. A Two-Enzyme Adaptive Unit within Bacterial Folate Metabolism. Cell Rep. 2019, 27, 3359–3370.e7. [Google Scholar] [CrossRef]
- Arooj, M.; Sakkiah, S.; Cao, G.p.; Lee, K.W. An Innovative Strategy for Dual Inhibitor Design and Its Application in Dual Inhibition of Human Thymidylate Synthase and Dihydrofolate Reductase Enzymes. PLoS ONE 2013, 8, e60470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etienne, M.-C.; Ilc, K.; Formento, J.-L.; Laurent-Puig, P.; Formento, P.; Cheradame, S.; Fischel, J.-L.; Milano, G. Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphisms: Relationships with 5-fluorouracil sensitivity. Br. J. Cancer 2004, 90, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Longley, D.B.; Latif, T.; Boyer, J.; Allen, W.L.; Maxwell, P.J.; Johnston, P.G. The interaction of thymidylate synthase expression with p53-regulated signaling pathways in tumor cells. Semin. Oncol. 2003, 30 (Suppl. 6), as0093775403001192. [Google Scholar] [CrossRef]
- Hawser, S.; Lociuro, S.; Islam, K. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharmacol. 2006, 71, 941–948. [Google Scholar] [CrossRef]
- Cody, V.; Galitsky, N.; Luft, J.R.; Pangborn, W.; Blakley, R.L.; Gangjee, A. Comparison of ternary crystal complexes of F31 variants of human dihydrofolate reductase with NADPH and a classical antitumor furopyrimidine. Anti-Cancer Drug Des. 1998, 13, 307–315. Available online: http://www.ncbi.nlm.nih.gov/pubmed/9627670 (accessed on 17 September 2021).
- Cody, V.; Pace, J.; Namjoshi, O.A.; Gangjee, A. Structure–activity correlations for three pyrido[2,3-d]pyrimidine antifolates binding to human and Pneumocystis carinii dihydrofolate reductase. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 799–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangjee, A.; Li, W.; Kisliuk, R.L.; Cody, V.; Pace, J.; Piraino, J.; Makin, J. Design, Synthesis, and X-ray Crystal Structure of Classical and Nonclassical 2-Amino-4-oxo-5-substituted-6-ethylthieno[2,3-d]pyrimidines as Dual Thymidylate Synthase and Dihydrofolate Reductase Inhibitors and as Potential Antitumor Agents. J. Med. Chem. 2009, 52, 4892–4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangjee, A.; Vidwans, A.P.; Vasudevan, A.; Queener, S.F.; Kisliuk, R.L.; Cody, V.; Li, R.; Galitsky, N.; Luft, J.R.; Pangborn, W. Structure-Based Design and Synthesis of Lipophilic 2,4-Diamino-6-Substituted Quinazolines and Their Evaluation as Inhibitors of Dihydrofolate Reductases and Potential Antitumor Agents. J. Med. Chem. 1998, 41, 3426–3434. [Google Scholar] [CrossRef]
- Klon, A.E.; Héroux, A.; Ross, L.J.; Pathak, V.; Johnson, C.A.; Piper, J.R.; Borhani, D.W. Atomic Structures of Human Dihydrofolate Reductase Complexed with NADPH and Two Lipophilic Antifolates at 1.09Å and 1.05Å Resolution. J. Mol. Biol. 2002, 320, 677–693. [Google Scholar] [CrossRef]
- Lamb, K.M.; G-Dayanandan, N.; Wright, D.L.; Anderson, A.C. Elucidating Features That Drive the Design of Selective Antifolates Using Crystal Structures of Human Dihydrofolate Reductase. Biochemistry 2013, 52, 7318–7326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhou, X.; Kisliuk, R.L.; Piraino, J.; Cody, V.; Gangjee, A. Design, synthesis, biological evaluation and X-ray crystal structure of novel classical 6,5,6-tricyclic benzo[4,5]thieno[2,3-d]pyrimidines as dual thymidylate synthase and dihydrofolate reductase inhibitors. Bioorganic Med. Chem. 2011, 19, 3585–3594. [Google Scholar] [CrossRef]
- Naumovich, V.; Grishina, M.; Novak, J.; Pathak, P.; Potemkin, V.; Shahbaaz, M.; Abdellattif, M.H. Electronic properties investigation of human dihydrofolate reductase complexes with ligands. J. Biomol. Struct. Dyn. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A quantum theory. In The International Series of Monographs on Chemistry; Clarendon Press: Oxford, UK, 1990; Volume 22. [Google Scholar] [CrossRef]
- Bader, R.F.W. Principle of stationary action and the definition of a proper open system. Phys. Rev. B 1994, 49, 13348–13356. [Google Scholar] [CrossRef]
- Grishina, M.; Bolshakov, O.; Potemkin, A.; Potemkin, V. Theoretical investigation of electron structure and surface morphology of titanium dioxide anatase nano-particles. Comp. Theor. Chem. 2016, 1091, 122–136. [Google Scholar] [CrossRef]
- Gillespie, R.J. The valence-shell electron-pair repulsion (VSEPR) theory of directed valency. J. Chem. Educ. 1963, 40, 295. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Nyholm, R.S. Inorganic stereochemistry. Q. Rev. Chem. Soc. 1957, 11, 339–380. [Google Scholar] [CrossRef]
- Chemosophia.Com. Available online: www.chemosophia.com (accessed on 17 September 2021).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | ||
|---|---|---|
| 1boz | 0.2131 | 0.1737 |
| 1hfp | 0.3188 | 0.2494 |
| 1kms | 0.3118 | 0.2465 |
| 3ghc | 0.4099 | 0.3255 |
| 3gi2 | 0.4606 | 0.3623 |
| 3ntz | 0.3673 | 0.2947 |
| 3nu0 | 0.3943 | 0.3162 |
| 4kfj | 0.4430 | 0.3256 |
| 4qhv | 0.3174 | 0.2387 |
| 1hfp (Tautomer 1) | 1hfp (Tautomer 2) | 1hfp (Tautomer 3) | |
|---|---|---|---|
| MM3-MERA force field energy, kcal/mol | −2406.70678 | −2398.00307 | −2341.35311 |
| Rel, Å | 1.9126 | 1.5514932 | 1.6928 |
| , e/Å3 | 0.000004206 | 0.000004206 | 0.00004766 |
| , e/Å3 | 0.000002932 | 0.000156418 | 0.001223 |
| 2.75 1 | 2.88 1 | |
| 2.88 1 | 2.96 1 | |
| 2.83 1 | 2.90 1 |
| 1.63 1 | 1.75 1 | 1.97 1 | |
| 1.75 1 | 1.85 | 2.02 | |
| 1.97 1 | 2.02 | 2.13 |
| 1.94 1–3.34 | 1.83 1–3.66 | 1.90 1–3.49 | |
| 3.52–3.91 | 2.76 1–3.75 | ||
| 2.76 1–3.75 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potemkin, V.; Grishina, M. The Complementarity Principle—One More Step towards Analytical Docking on the Example of Dihydrofolate Reductase Complexes. Life 2021, 11, 983. https://doi.org/10.3390/life11090983
Potemkin V, Grishina M. The Complementarity Principle—One More Step towards Analytical Docking on the Example of Dihydrofolate Reductase Complexes. Life. 2021; 11(9):983. https://doi.org/10.3390/life11090983
Chicago/Turabian StylePotemkin, Vladimir, and Maria Grishina. 2021. "The Complementarity Principle—One More Step towards Analytical Docking on the Example of Dihydrofolate Reductase Complexes" Life 11, no. 9: 983. https://doi.org/10.3390/life11090983