
Crosstalk between Autophagy and Inflammatory Processes in Cancer


Abstract
:1. Introduction
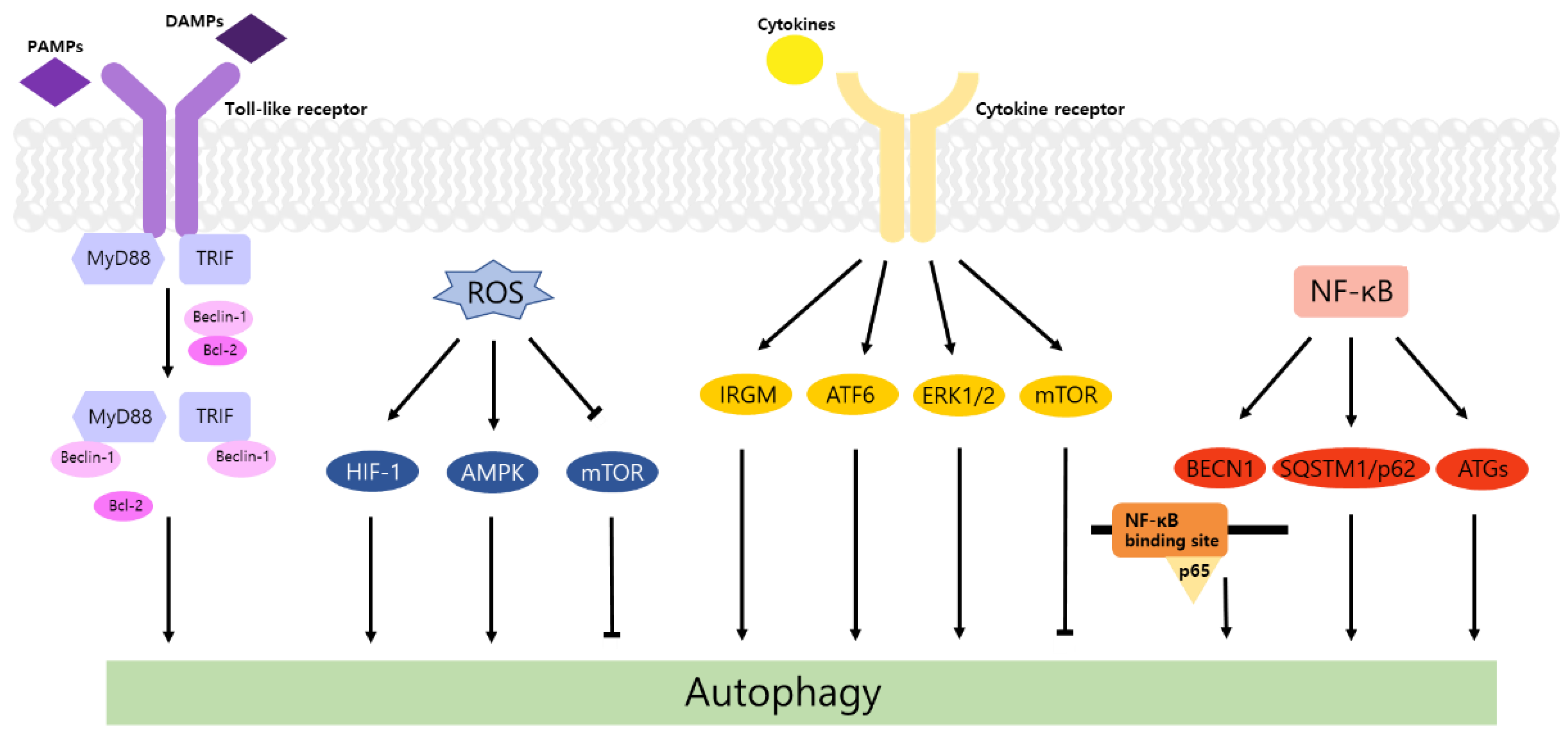
2. Autophagy and TLR Signaling in Cancer
3. Regulation of Autophagy by ROS in Inflammation and Cancer
4. Autophagy and Inflammatory Cytokines in Cancer
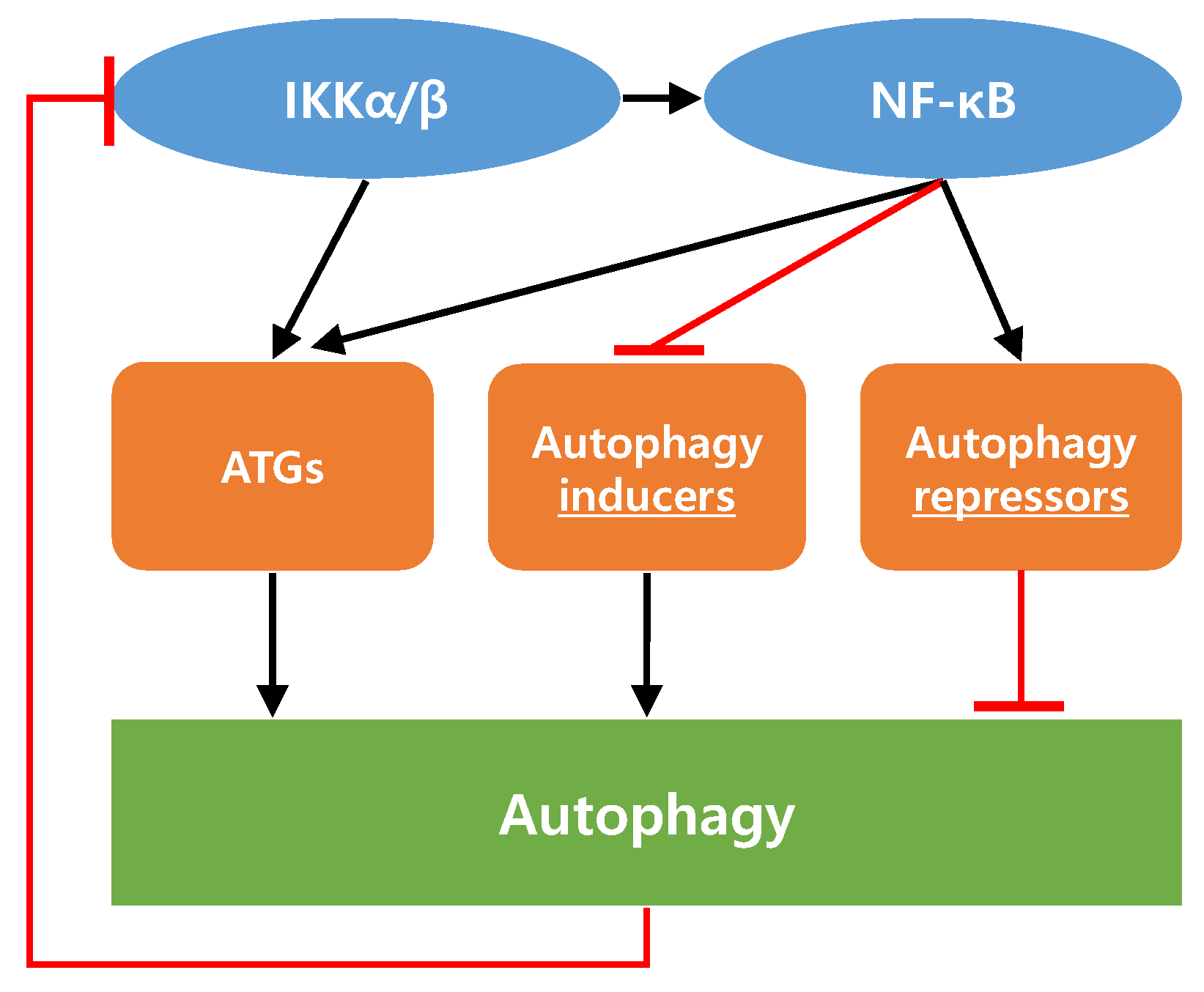
5. Interaction between IKK/NF-κB Axis and Autophagy in Cancer
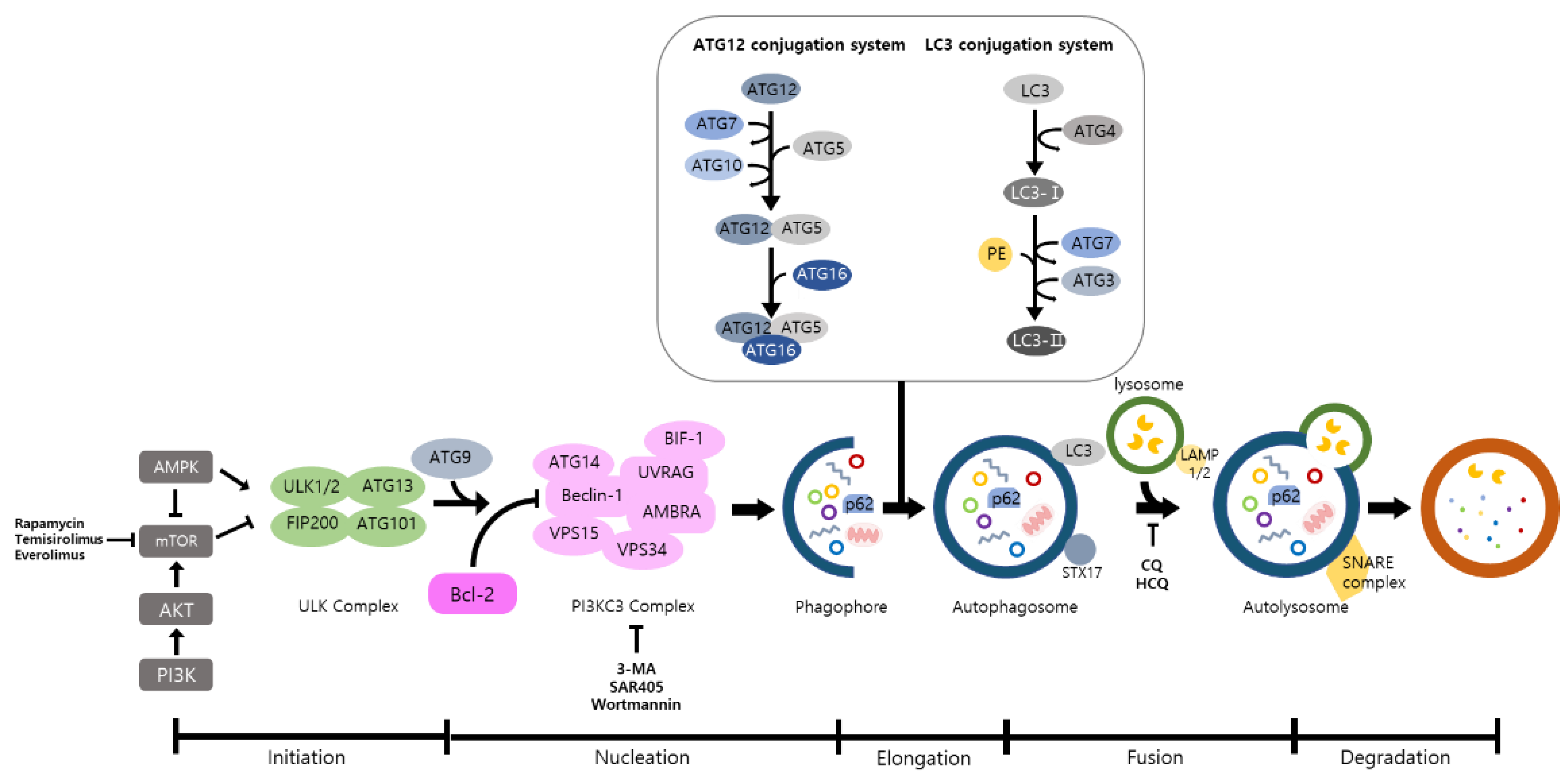
6. Autophagy Inhibitors and Activators for Cancer Treatment
6.1. Autophagy Inhibitors for Cancer Treatment
6.2. Autophagy Activators for Cancer Treatment
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| TLR | Toll-Like Receptor |
|---|---|
| DAMP | damage-associated molecular pattern |
| PAMP | pathogen-associated molecular pattern |
| MyD88 | myeloid differentiation primary response gene 88 |
| NF-κB | nuclear factor-κB |
| MAPK | mitogen-activated protein kinase |
| IRF | interferon-regulatory factor |
| TNF-α | tumor necrosis factor- α |
| IL-1β | interleukin-1β |
| TRIF | Toll/IL-1 receptor domain-containing adaptor-inducing interferon-β |
| ASK1 | apoptosis signal-regulating kinase 1 |
| ROS | reactive oxygen species |
| LC3 | light chain 3 |
| PI3KC3 | class III phosphatidylinositol 3-kinase |
| BECN1 | beclin 1 |
| TRAF6 | TNF receptor-associated factor 6 |
| CQ | chloroquine |
| MMP2 | matrix metalloproteinase 2 |
| CCL2 | CC chemokine ligand 2 |
| EMT | epithelial mesenchymal transition |
| HCQ | hydroxychloroquine |
| AMPK | AMP-activated protein kinase |
| HIF | hypoxia-inducible factor |
| IRGM | immunity-related GTPase family M protein |
| ATF6 | activating transcription factor 6 |
| ERK | extracellular signal-regulated kinase |
| TVX | trovafloxacin |
| STAT3 | signal transducer and activator of transcription 3 |
| CAF | cancer-associated fibroblast |
| TME | tumor microenvironment |
| ECM | extracellular matrix |
| IKK | IκB kinase |
| PI3K | phosphoinositide 3-kinase |
| ATG | autophagy related gene |
| 3-MA | 3-Methyladenine |
References
- Philip, M.; Rowley, D.A.; Schreiber, H. Inflammation as a tumor promoter in cancer induction. Semin. Cancer Biol. 2004, 14, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Gurel, B.; Lucia, M.S.; Thompson, I.M.; Goodman, P.J.; Tangen, C.M.; Kristal, A.R.; Parnes, H.L.; Hoque, A.; Lippman, S.M.; Sutcliffe, S.; et al. Chronic inflammation in benign prostate tissue is associated with high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer Epidemiol. Biomark. Prev. 2014, 23, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, C.J.; Glen, P.; McMillan, D.C. Chronic inflammation and pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 65–73. [Google Scholar] [CrossRef]
- Michaud, D.S. Chronic inflammation and bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2007, 25, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010, 6, 891–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A molecular switch node in the crosstalk between autophagy and apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083. [Google Scholar] [CrossRef]
- Saleem, S. Apoptosis, Autophagy, Necrosis and Their Multi Galore Crosstalk in Neurodegeneration. Neuroscience 2021, 469, 162–174. [Google Scholar] [CrossRef]
- Banskota, S.; Wang, H.; Kwon, Y.H.; Gautam, J.; Gurung, P.; Haq, S.; Hassan, F.M.N.; Bowdish, D.M.; Kim, J.A.; Carling, D.; et al. Salicylates ameliorate intestinal inflammation by activating macrophage AMPK. Inflamm. Bowel Dis. 2021, 27, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Monkkonen, T.; Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 2018, 14, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.S.; Chae, Y.; Choi, J.W.; Chang, J. Potential Therapeutic Approaches through Modulating the Autophagy Process for Skin Barrier Dysfunction. Int. J. Mol. Sci. 2021, 22, 7869. [Google Scholar] [CrossRef] [PubMed]
- Hayat, M.A. Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Academic Press: Cambridge, UK, 2017; Volume 12, ISBN 0128121475. [Google Scholar]
- Javaid, N.; Choi, S. Toll-like receptors from the perspective of cancer treatment. Cancers 2020, 12, 297. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yan, J.; Lin, H.; Hua, F.; Wang, X.; Liu, H.; Lv, X.; Yu, J.; Mi, S.; Wang, J.; et al. Toll-like receptor 4 activity protects against hepatocellular tumorigenesis and progression by regulating expression of DNA repair protein Ku70 in mice. Hepatology 2013, 57, 1869–1881. [Google Scholar] [CrossRef]
- Kim, M.J.; Min, Y.; Im, J.S.; Son, J.; Lee, J.S.; Lee, K.Y. p62 is Negatively Implicated in the TRAF6-BECN1 Signaling Axis for Autophagy Activation and Cancer Progression by Toll-Like Receptor 4 (TLR4). Cells 2020, 9, 1142. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Yadav, V.K.; Chu, Y.C.; Ong, J.R.; Huang, T.-Y.; Lee, K.-F.; Lee, K.-H.; Yeh, C.-T.; Lee, W.-H. Hydroxychloroquine (HCQ) Modulates Autophagy and Oxidative DNA Damage Stress in Hepatocellular Carcinoma to Overcome Sorafenib Resistance via TLR9/SOD1/hsa-miR-30a-5p/Beclin-1 Axis. Cancers 2021, 13, 3227. [Google Scholar] [CrossRef]
- Krumova, K.; Cosa, G. Chapter 1 Overview of Reactive Oxygen. In Singlet Oxygen: Application in Biosciences and Nanosciences Species; Royal Society of Chemistry: Cambridge, UK, 2016. [Google Scholar]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Ketelut-Carneiro, N.; Silva, G.K.; Rocha, F.A.; Milanezi, C.M.; Cavalcanti-Neto, F.F.; Zamboni, D.S.; Silva, J.S. IL-18 Triggered by the Nlrp3 Inflammasome Induces Host Innate Resistance in a Pulmonary Model of Fungal Infection. J. Immunol. 2015, 194, 4507–4517. [Google Scholar] [CrossRef]
- Liu, W.; Glunde, K.; Bhujwalla, Z.M.; Raman, V.; Sharma, A.; Phang, J.M. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012, 72, 3677–3686. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-L.; Liu, H.-W.; Shrestha, S.; Thiyagarajan, V.; Huang, H.-C.; Hseu, Y.-C. Antrodia salmonea induces apoptosis and enhances cytoprotective autophagy in colon cancer cells. Aging (Albany NY) 2021, 13, 15964–15989. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.D.; Kim, K.Y.; Park, K.I.; Kim, S.H.; Park, S.G.; Yu, S.N.; Kim, Y.W.; Kim, D.S.; Chung, K.T.; Ahn, S.C. Dual role of reactive oxygen species in autophagy and apoptosis induced by compound PN in prostate cancer cells. Mol. Cell. Toxicol. 2021, 17, 41–50. [Google Scholar] [CrossRef]
- Pelosse, M.; Cottet-Rousselle, C.; Bidan, C.M.; Dupont, A.; Gupta, K.; Berger, I.; Schlattner, U. Synthetic energy sensor AMPfret deciphers adenylate-dependent AMPK activation mechanism. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Mickymaray, S.; Alfaiz, F.A.; Paramasivam, A.; Veeraraghavan, V.P.; Periadurai, N.D.; Surapaneni, K.M.; Niu, G. Rhaponticin suppresses osteosarcoma through the inhibition of PI3K-Akt-mTOR pathway. Saudi J. Biol. Sci. 2021, 28, 3641–3649. [Google Scholar] [CrossRef] [PubMed]
- Abreu, S.; Kriegenburg, F.; Gómez-Sánchez, R.; Mari, M.; Sánchez-Wandelmer, J.; Skytte Rasmussen, M.; Soares Guimarães, R.; Zens, B.; Schuschnig, M.; Hardenberg, R.; et al. Conserved Atg8 recognition sites mediate Atg4 association with autophagosomal membranes and Atg8 deconjugation. EMBO Rep. 2017, 18, 765–780. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liu, J.H.; Jin, L.; Lin, S.M.; Yang, Y.; Sui, Y.X.; Shi, H. Over-expression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Lett. 2010, 294, 204–210. [Google Scholar] [CrossRef]
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Schink, M.K.O.; Theodossiou, T.A.; Johansen, T.; Juhász, G.; Bilder, D.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Novak, M.J.; Orraca, L.; Martinez-Gonzalez, J.; Kirakodu, S.; Chen, K.C.; Stromberg, A.; Gonzalez, O.A. Hypoxia-inducible transcription factors, HIF1A and HIF2A, increase in aging mucosal tissues. Immunology 2018, 154, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazon, A.; Goldrath, A.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, L.; Huang, C.J.; Choo, K.B.; Cheong, S.K.; Kamarul, T. Oxidative stress down-regulates mir-20b-5p, mir-106a-5p and E2F1 expression to suppress the g1/s transition of the cell cycle in multipotent stromal cells. Int. J. Med. Sci. 2020, 17, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.W.; Liu, V.W.S.; Tsao, G.S.W.; Yao, K.M.; Furukawa, T.; Chan, K.K.L.; Ngan, H.Y.S. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 2008, 29, 1742–1750. [Google Scholar] [CrossRef] [Green Version]
- Chi, T.F.; Khoder-Agha, F.; Mennerich, D.; Kellokumpu, S.; Miinalainen, I.I.; Kietzmann, T.; Dimova, E.Y. Loss of USF2 promotes proliferation, migration and mitophagy in a redox-dependent manner. Redox Biol. 2020, 37, 1–9. [Google Scholar] [CrossRef]
- Banskota, S.; Regmi, S.C.; Kim, J.A. NOX1 to NOX2 switch deactivates AMPK and induces invasive phenotype in colon cancer cells through overexpression of MMP-7. Mol. Cancer 2015, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Pramanik, K.C.; Boreddy, S.R.; Srivastava, S.K. Role of mitochondrial Electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PLoS ONE 2011, 6, 20151. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.C.; Lee, C.C.; Lin, H.H.; Chen, M.C.; Lin, C.C.; Chang, J.Y. Autophagy-regulated ROS from xanthine oxidase acts as an early effector for triggering late mitochondria-dependent apoptosis in cathepsin S-targeted tumor cells. PLoS ONE 2015, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mandal, A.K.; Son, Y.O.K.; Pratheeshkumar, P.; Wise, J.T.F.; Wang, L.; Zhang, Z.; Shi, X.; Chen, Z. Roles of ROS, Nrf2, and autophagy in cadmium-carcinogenesis and its prevention by sulforaphane. Toxicol. Appl. Pharmacol. 2018, 353, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ramani, T.; Auletta, C.S.; Weinstock, D.; Mounho-Zamora, B.; Ryan, P.C.; Salcedo, T.W.; Bannish, G. Cytokines: The good, the bad, and the deadly. Int. J. Toxicol. 2015, 34, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Chien, S.Y.; Chen, C.L.; Hsieh, C.Y.; Tseng, P.C.; Wang, Y.C. IFN-γ induces mimic extracellular trap cell death in lung epithelial cells through autophagy-regulated DNA damage. J. Interf. Cytokine Res. 2016, 36, 100–112. [Google Scholar] [CrossRef]
- Ahn, J.H.; Jegal, H.; Choi, M.S.; Kim, S.; Park, S.M.; Ahn, J.; Han, H.Y.; Cho, H.S.; Yoon, S.; Oh, J.H. TNFα enhances trovafloxacin-induced in vitro hepatotoxicity by inhibiting protective autophagy. Toxicol. Lett. 2021, 342, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1β secretion by targeting Pro-IL-1β for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraresi, A.; Phadngam, S.; Morani, F.; Galetto, A.; Alabiso, O.; Chiorino, G.; Isidoro, C. Resveratrol inhibits IL-6-induced ovarian cancer cell migration through epigenetic up-regulation of autophagy. Mol. Carcinog. 2017, 56, 1164–1181. [Google Scholar] [CrossRef]
- Thongchot, S.; Jamjuntra, P.; Therasakvichya, S.; Warnnissorn, M.; Ferraresi, A.; Thuwajit, P.; Isidoro, C.; Thuwajit, C. Interleukin-8 released by cancer-associated fibroblasts attenuates the autophagy and promotes the migration of ovarian cancer cells. Int. J. Oncol. 2021, 58, 1–14. [Google Scholar] [CrossRef]
- Sivaprasad, U.; Basu, A. Inhibition of ERK attenuates autophagy and potentiates tumour necrosis factor-α-induced cell death in MCF-7 cells. J. Cell. Mol. Med. 2008, 12, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Angrini, M.; Varthaman, A.; Cremer, I. Toll-like receptors (TLRs) in the tumor microenvironment (TME): A dragon-like weapon in a non-fantasy game of thrones. In Tumor Microenvironment; Springer: Cham, Switzerland, 2020; pp. 145–173. [Google Scholar]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506. [Google Scholar] [CrossRef] [Green Version]
- Cotzomi-Ortega, I.; Rosas-Cruz, A.; Ramírez-Ramírez, D.; Reyes-Leyva, J.; Rodriguez-Sosa, M.; Aguilar-Alonso, P.; Maycotte, P. Autophagy inhibition induces the secretion of macrophage migration inhibitory factor (MIF) with autocrine and paracrine effects on the promotion of malignancy in breast cancer. Biology 2020, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, S.; Gupta, D. Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed. Pharmacother. 2018, 108, 1866–1878. [Google Scholar] [CrossRef]
- Pun, N.T.; Subedi, A.; Kim, M.J.; Park, P.H. Globular adiponectin causes tolerance to LPS-induced TNF-α expression via autophagy induction in RAW 264.7 macrophages: Involvement of SIRT1/FoxO3A axis. PLoS ONE 2015, 10, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-B, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.F.; Perkins, N.D. Nuclear factor-κB, p53, and mitochondria: Regulation of cellular metabolism and the Warburg effect. Trends Biochem. Sci. 2012, 37, 317–324. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Hyttinen, J.M.T.; Kauppinen, A.; Kaarniranta, K. Context-dependent regulation of autophagy by IKK-NF- B signaling: Impact on the aging process. Int. J. Cell Biol. 2012, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, A.S. Regulation of cell death and autophagy by IKK and NF-κB: Critical mechanisms in immune function and cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef]
- Copetti, T.; Bertoli, C.; Dalla, E.; Demarchi, F.; Schneider, C. p65/RelA Modulates BECN1 Transcription and Autophagy. Mol. Cell. Biol. 2009, 29, 2594–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Qiang, L.; Sample, A.; Shah, P.; He, Y.Y. NF-κB signaling activation induced by chloroquine requires autophagosome, p62 protein, and c-Jun N-terminal Kinase (JNK) signaling and promotes tumor cell resistance. J. Biol. Chem. 2017, 292, 3379–3388. [Google Scholar] [CrossRef] [Green Version]
- Broemer, M.; Krappmann, D.; Scheidereit, C. Requirement of Hsp90 activity for IκB kinase (IKK) biosynthesis and for constitutive and inducible IKK and NF-κB activation. Oncogene 2004, 23, 5378–5386. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Paimela, T.; Suuronen, T.; Kaarniranta, K. Innate immunity meets with cellular stress at the IKK complex: Regulation of the IKK complex by HSP70 and HSP90. Immunol. Lett. 2008, 117, 9–15. [Google Scholar] [CrossRef]
- Qing, G.; Yan, P.; Xiao, G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IκB kinase (IKK). Cell Res. 2006, 16, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Comb, W.C.; Cogswell, P.; Sitcheran, R.; Baldwin, A.S. IKK-dependent, NF-κB-independent control of autophagic gene expression. Oncogene 2011, 30, 1727–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010, 29, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Tamatani, M.; Che, Y.H.; Matsuzaki, H.; Ogawa, S.; Okado, H.; Miyake, S.I.; Mizuno, T.; Tohyama, M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFκB activation in primary hippocampal neurons. J. Biol. Chem. 1999, 274, 8531–8538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Edelstein, L.C.; Gélinas, C. The Rel/NF-κB Family Directly Activates Expression of the Apoptosis Inhibitor Bcl-xL. Mol. Cell. Biol. 2000, 20, 2687–2695. [Google Scholar] [CrossRef]
- Gang, H.; Dhingra, R.; Wang, Y.; Mughal, W.; Gordon, J.W.; Kirshenbaum, L.A. Epigenetic regulation of E2F-1-dependent Bnip3 transcription and cell death by nuclear factor-κB and histone deacetylase-1. Pediatr. Cardiol. 2011, 32, 263–266. [Google Scholar] [CrossRef]
- Liu, J.; Lin, A. Wiring the cell signaling circuitry by the NF-κB and JNK1 crosstalk and its applications in human diseases. Oncogene 2007, 26, 3267–3278. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminskyy, V.O.; Piskunova, T.; Zborovskaya, I.B.; Tchevkina, E.M.; Zhivotovsky, B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy 2012, 8, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Woo, S.U.; Kang, J.H.; Kim, K.; Kwon, M.H.; Park, S.; Shin, H.J.; Gwak, H.S.; Chwae, Y.J. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy 2010, 6, 1125–1138. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Peng, Y.; Wang, J.; Gu, A.X.; Li, Q.; Mao, D.W.; Guo, L.Y. Effect of autophagy on the resveratrol-induced apoptosis of ovarian cancer SKOV3 cells. J. Cell. Biochem. 2019, 120, 7788–7793. [Google Scholar] [CrossRef]
- Zhao, F.; Feng, G.; Zhu, J.; Su, Z.; Guo, R.; Liu, J.; Zhang, H.; Zhai, Y. 3-Methyladenine-enhanced susceptibility to sorafenib in hepatocellular carcinoma cells by inhibiting autophagy. Anticancer Drugs 2021, 32, 386–393. [Google Scholar] [CrossRef]
- Chen, W.C.; Hsu, K.Y.; Hung, C.M.; Lin, Y.C.; Yang, N.S.; Ho, C.T.; Kuo, S.C.; Way, T.D. The anti-tumor efficiency of pterostilbene is promoted with a combined treatment of Fas signaling or autophagy inhibitors in triple negative breast cancer cells. Food Funct. 2014, 5, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Song, H.X.; Lu, Y.; Li, X.; Chen, T.; Zhang, Y.; Xue, J.X.; Liu, H.; Kan, B.; Yang, G.; et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis. Esophagus 2011, 24, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.X.; Yang, G.; Hao, F.; Urba, W.J.; Hu, H.M. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008, 68, 6889–6895. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, B. SAR405, a PIK3C3/VPS34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 2015, 11, 725–726. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Huang, Z.; Wu, H.; Zhou, W.; Jin, P.; Wei, P.; Zhang, Y.; Zheng, F.; Zhang, J.; Xu, J.; et al. Inhibition of autophagy enhances the anticancer activity of silver nanoparticles. Autophagy 2014, 10, 2006–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.R.; Yoo, Y.J.; Ban, Y.H.; Yoon, Y.J. Biosynthesis of rapamycin and its regulation: Past achievements and recent progress. J. Antibiot. 2010, 63, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Zhao, J.; Xue, J.; Zhao, X.; Liu, P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am. J. Cancer Res. 2016, 6, 2162–2177. [Google Scholar]
- Miow, Q.H.; Tan, T.Z.; Ye, J.; Lau, J.A.; Yokomizo, T.; Thiery, J.P.; Mori, S. Epithelial-mesenchymal status renders differential responses to cisplatin in ovarian cancer. Oncogene 2014, 34, 1899–1907. [Google Scholar] [CrossRef]
- Kan, J.; Yen, M.; Wang, J.; Wu, D. Nesfatin-1 / Nucleobindin-2 enhances cell migration, invasion, and epithelial-mesenchymal transition via LKB1/AMPK/TORC1/ZEB1 pathways in colon cancer. Oncotarget 2016, 7, 31336–31349. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Li, Y.; Xu, J.; Ren, Q.; Yao, J.; Tian, X. MicroRNA-409-3p regulates cell invasion and metastasis by targeting ZEB1 in breast cancer. IUBMB Life 2016, 68, 394–402. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carayol, N.; Vakana, E.; Sassano, A.; Kaur, S.; Goussetis, D.J.; Glaser, H.; Druker, B.J.; Donato, N.J.; Altman, J.K.; Barr, S.; et al. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 12469–12474. [Google Scholar] [CrossRef] [Green Version]
- Inamura, S.; Hideaki, I.T.O.; Taga, M.; Tsuchiyama, K.; Hoshino, H.; Kobayashi, M.; Yokoyama, O. Low-dose docetaxel enhanced the anticancer effect of temsirolimus by overcoming autophagy in prostate cancer cells. Anticancer Res. 2019, 39, 5417–5425. [Google Scholar] [CrossRef]
- Shiratori, H.; Kawai, K.; Hata, K.; Tanaka, T.; Nishikawa, T.; Otani, K.; Sasaki, K.; Kaneko, M.; Murono, K.; Emoto, S.; et al. The combination of temsirolimus and chloroquine increases radiosensitivity in colorectal cancer cells. Oncol. Rep. 2019, 42, 377–385. [Google Scholar] [CrossRef]
- Trivedi, N.D.; Armstrong, S.; Wang, H.; Hartley, M.; Deeken, J.; Ruth He, A.; Subramaniam, D.; Melville, H.; Albanese, C.; Marshall, J.L.; et al. A phase I trial of the mTOR inhibitor temsirolimus in combination with capecitabine in patients with advanced malignancies. Cancer Med. 2021, 10, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- El Guerrab, A.; Bamdad, M.; Bignon, Y.J.; Penault-Llorca, F.; Aubel, C. Co-targeting EGFR and mTOR with gefitinib and everolimus in triple-negative breast cancer cells. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Feldman, D.R.; Ged, Y.; Lee, C.H.; Knezevic, A.; Molina, A.M.; Chen, Y.B.; Chaim, J.; Coskey, D.T.; Murray, S.; Tickoo, S.K.; et al. Everolimus plus bevacizumab is an effective first-line treatment for patients with advanced papillary variant renal cell carcinoma: Final results from a phase II trial. Cancer 2020, 126, 5247–5255. [Google Scholar] [CrossRef]
- Zhu, M.; Molina, J.R.; Dy, G.K.; Croghan, G.A.; Qi, Y.; Glockner, J.; Hanson, L.J.; Roos, M.M.; Tan, A.D.; Adjei, A.A. A phase I study of the VEGFR kinase inhibitor vatalanib in combination with the mTOR inhibitor, everolimus, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1755–1762. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cancer Types | Signaling Pathways | Autophagy | Therapeutic Effects | Refs. |
|---|---|---|---|---|
| Hepatocellular Carcinoma | TLR4 | ↑ | Positive | [16] |
| TLR9 | ↑ | Negative | [18] | |
| TNF-α | ↓ | Positive | [42] | |
| Colorectal cancer | ROS | ↑ | Positive | [23] |
| Prostate Cancer | ROS | ↑ | Positive | [24] |
| Lung Cancer | IFN-γ | ↑ | Positive | [41] |
| Breast Cancer | TNF-α | ↑ | Negative | [46] |
| IL-6 | ↓ | Positive | [50] | |
| Ovarian Cancer | IL-6 | ↓ | Negative | [44] |
| IL-8 | ↓ | Negative | [45] | |
| Head and Neck Squamous Cell Carcinoma | IL-6, IL-8 | ↑ | Negative | [48] |
| Pancreatic Cancer | IL-6 | ↑ | Positive | [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.-J.; Kim, H.-J.; Choi, M.S.; Chang, J.-E. Crosstalk between Autophagy and Inflammatory Processes in Cancer. Life 2021, 11, 903. https://doi.org/10.3390/life11090903
Lee E-J, Kim H-J, Choi MS, Chang J-E. Crosstalk between Autophagy and Inflammatory Processes in Cancer. Life. 2021; 11(9):903. https://doi.org/10.3390/life11090903
Chicago/Turabian StyleLee, Eun-Ji, Hyun-Jeong Kim, Min Sik Choi, and Ji-Eun Chang. 2021. "Crosstalk between Autophagy and Inflammatory Processes in Cancer" Life 11, no. 9: 903. https://doi.org/10.3390/life11090903