
The Performance of HepG2 and HepaRG Systems through the Glass of Acetaminophen-Induced Toxicity

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Three-Dimensional (3D) Cell Cultures
2.3. APAP Treatment of HepG2 and HepaRG Cells
2.4. Determination of LC50 Values via MTT Assay
2.5. Evaluation of Cell Viability via Aspartate Aminotransferase (AST) Enzyme Activity
2.6. Reverse Transcription and Real-Time PCR Analysis
- For CYP2E1 cDNA:
- ○
- fw: 5′-AAGCAACCCGAGACACCATT-3′
- ○
- rv: 5′-ACACACTCGTTTTCCTGTGG-3′;
- For RIPK1 cDNA:
- ○
- fw: 5′-CGGCCTTGCCTCCTTTAAGA-3′
- ○
- rv: 5′-CCGACTTCTCTGTGGGCTTT-3′;
- For RIPK3 cDNA:
- ○
- fw: 5′-GCCCCAGAAGTCACTCCATC-3′
- ○
- rv: 5′-AGCCCCACTTCCTATGTTGC-3′
- ○
- and fw2: 5′-CATGGAGAACGGCTCCTTGT-3′
- ○
- rv2: 5′-GGTTCTGGTCGTGCAGGTAA-3′.
2.7. Measurement of Viable Cell Number Using Flow Cytometry
2.8. Isolation and Quantitation of Protein Samples
2.9. Western Blot
2.10. Determination of Caspase-3/7 Activation
2.11. GSH Measurement
2.12. Visualization of Cell Viability, Caspase-3/7 Activity, Reduced Glutathione, and Hepatocytes (of HepaRG) by Fluorescent Microscopy
2.13. Statistical Analyses
3. Results and Discussion
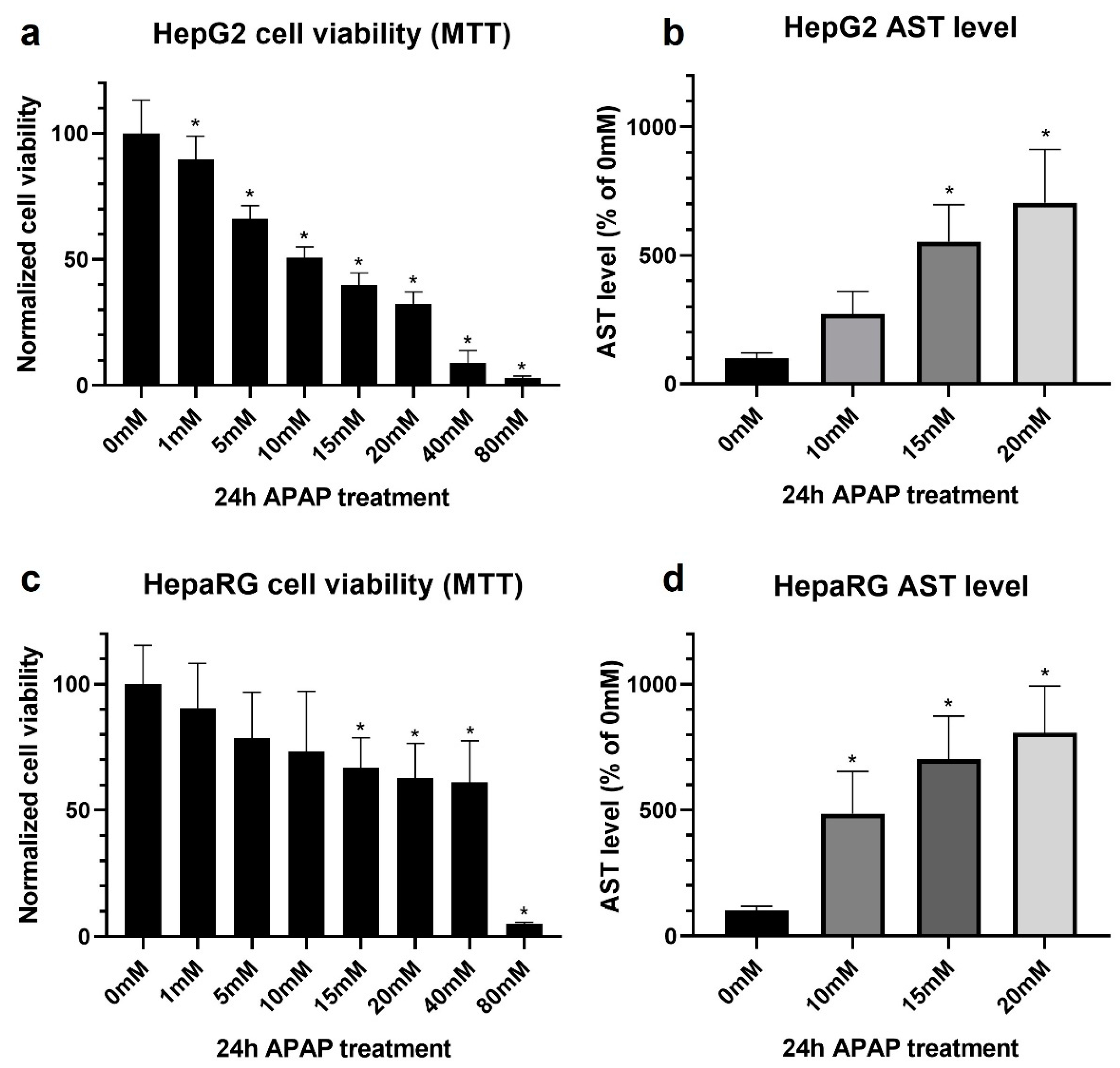
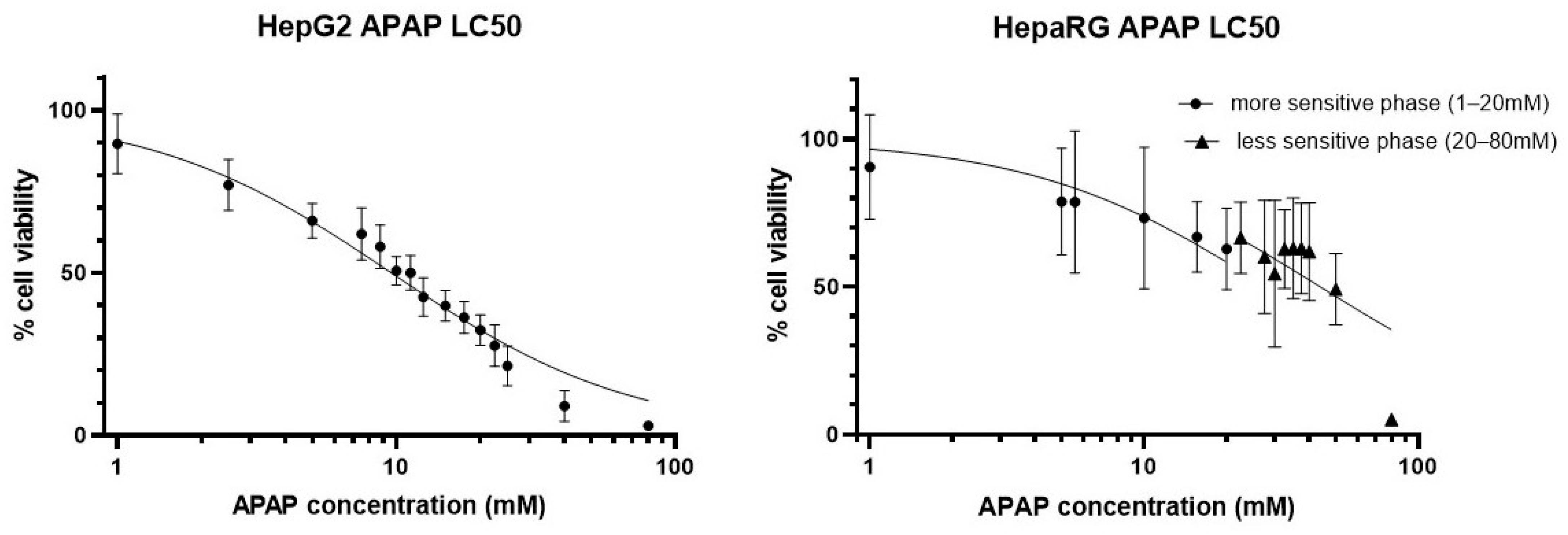
3.1. Acetaminophen Sensitivity of HepG2 and Differentiated HepaRG
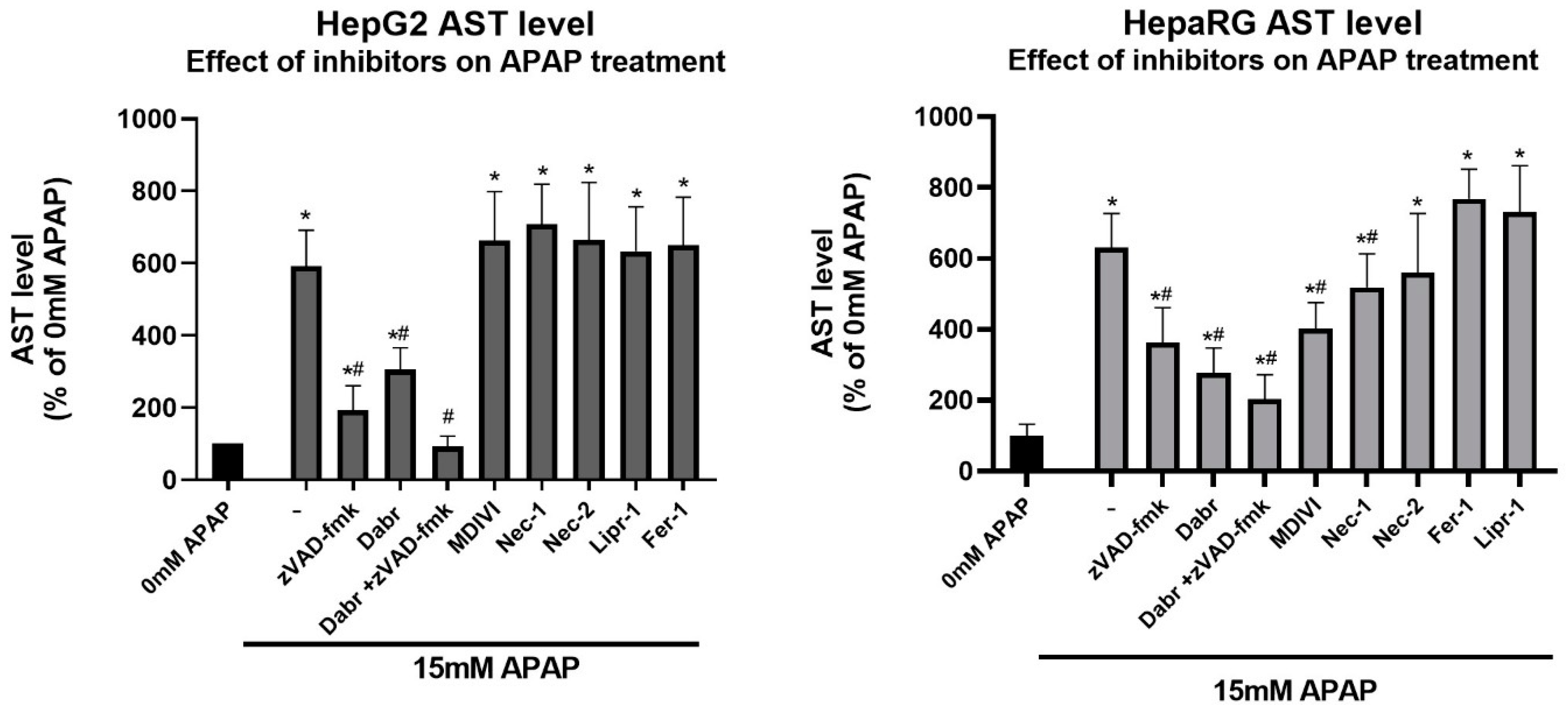
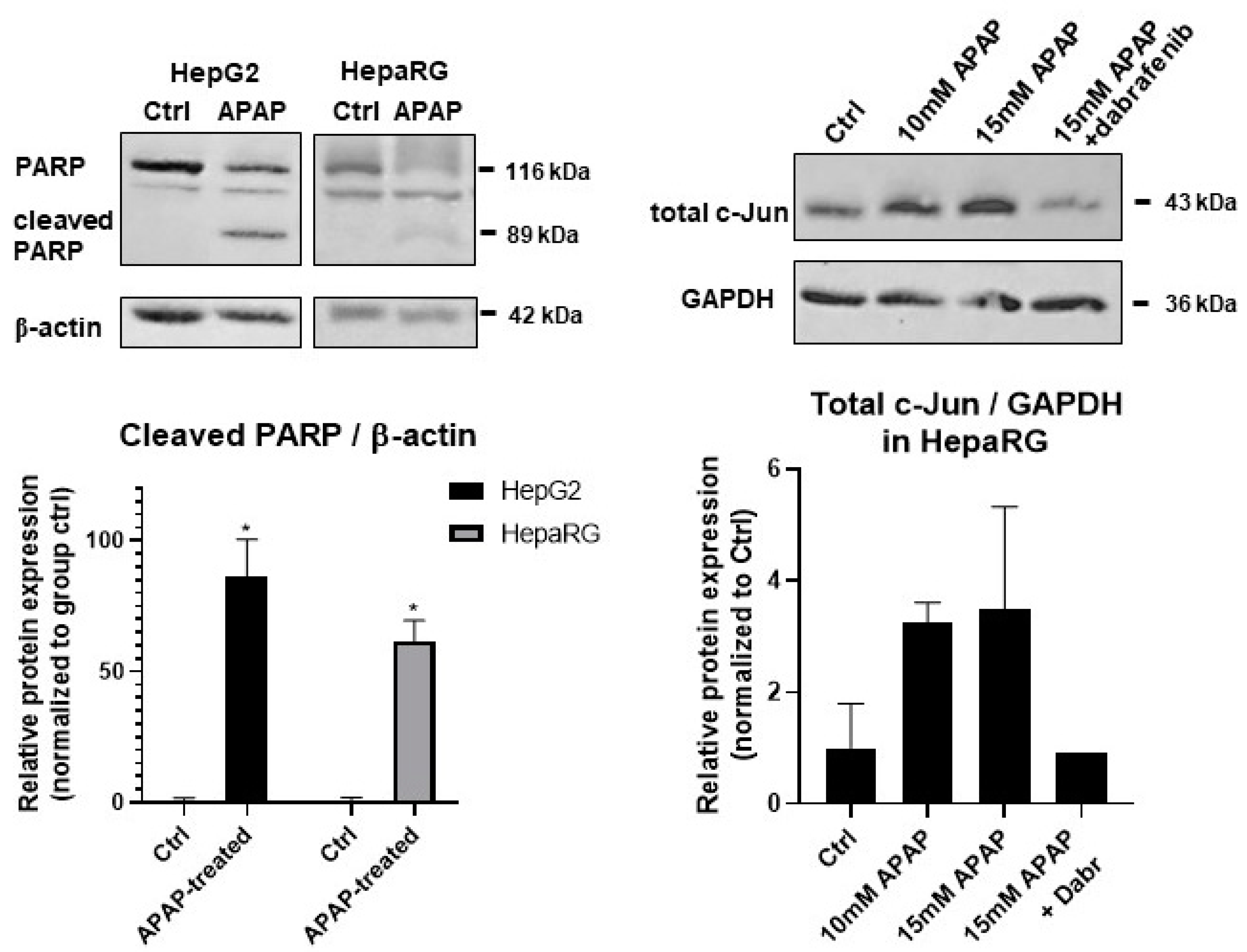
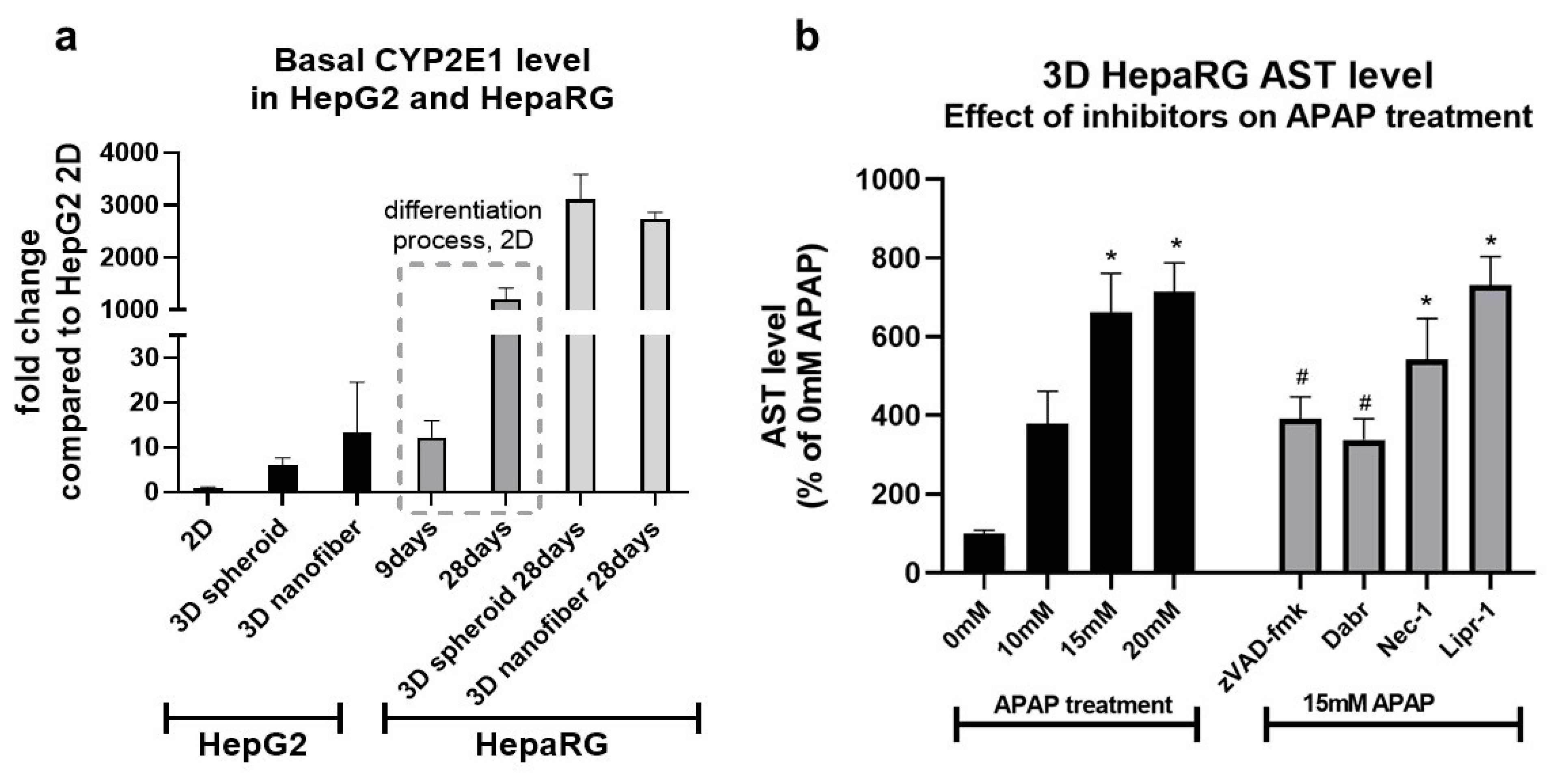
3.2. Investigation of Cell Death Pathways by Acetaminophen Cytotoxicity in HepG2 and Differentiated HepaRG
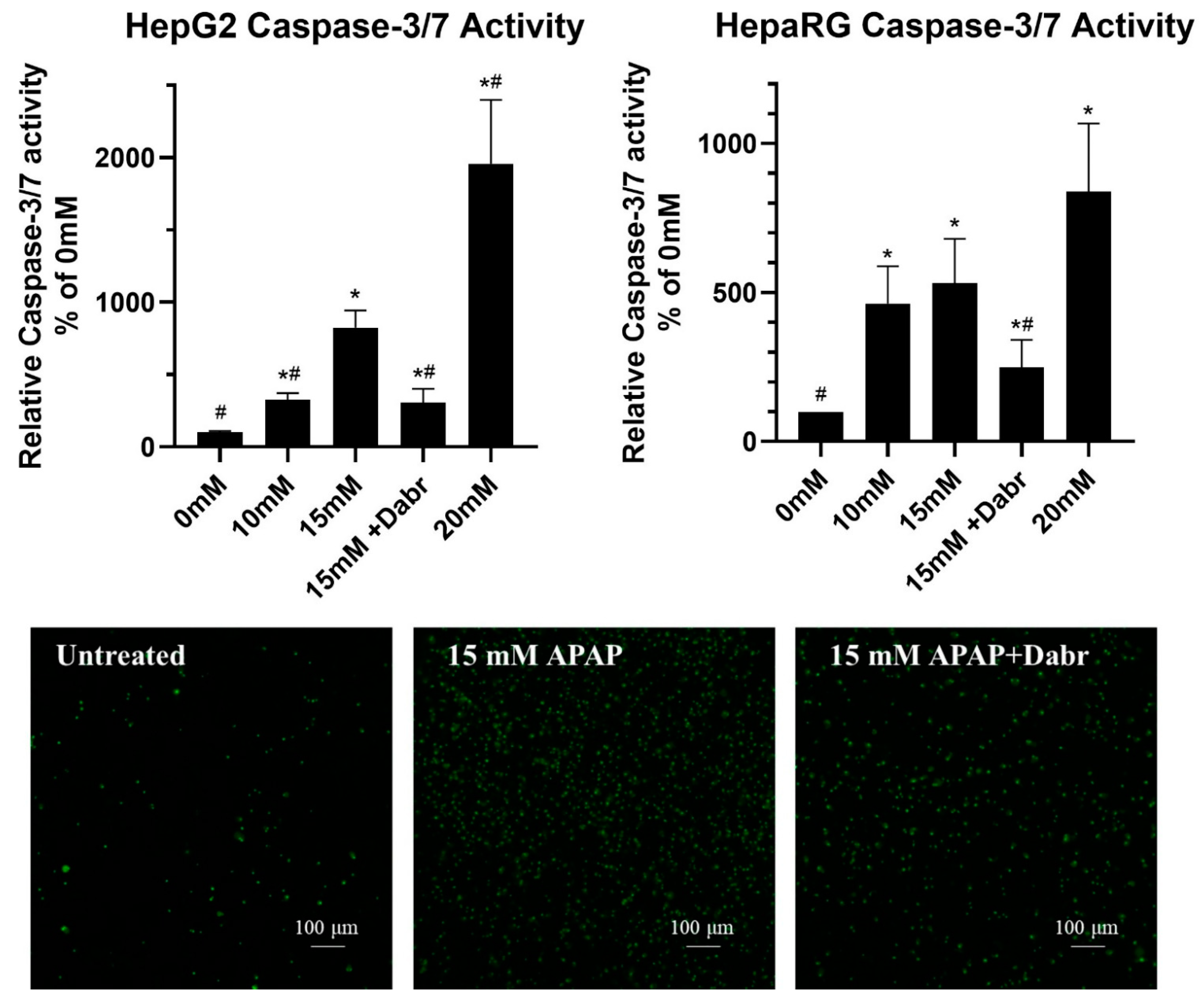
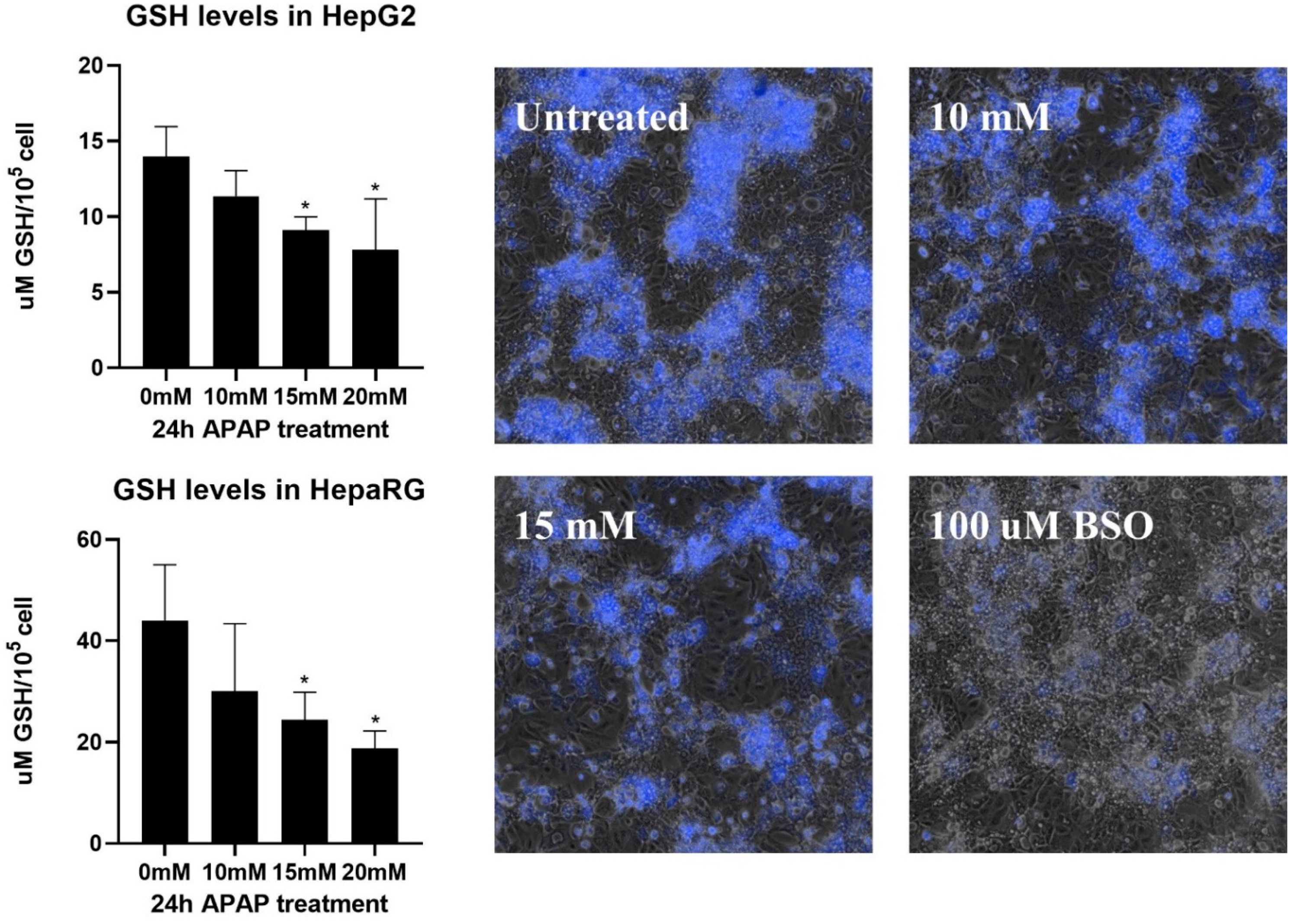
3.3. Caspase Activity and GSH Level in APAP-Treated HepG2 and Differentiated HepaRG Cells
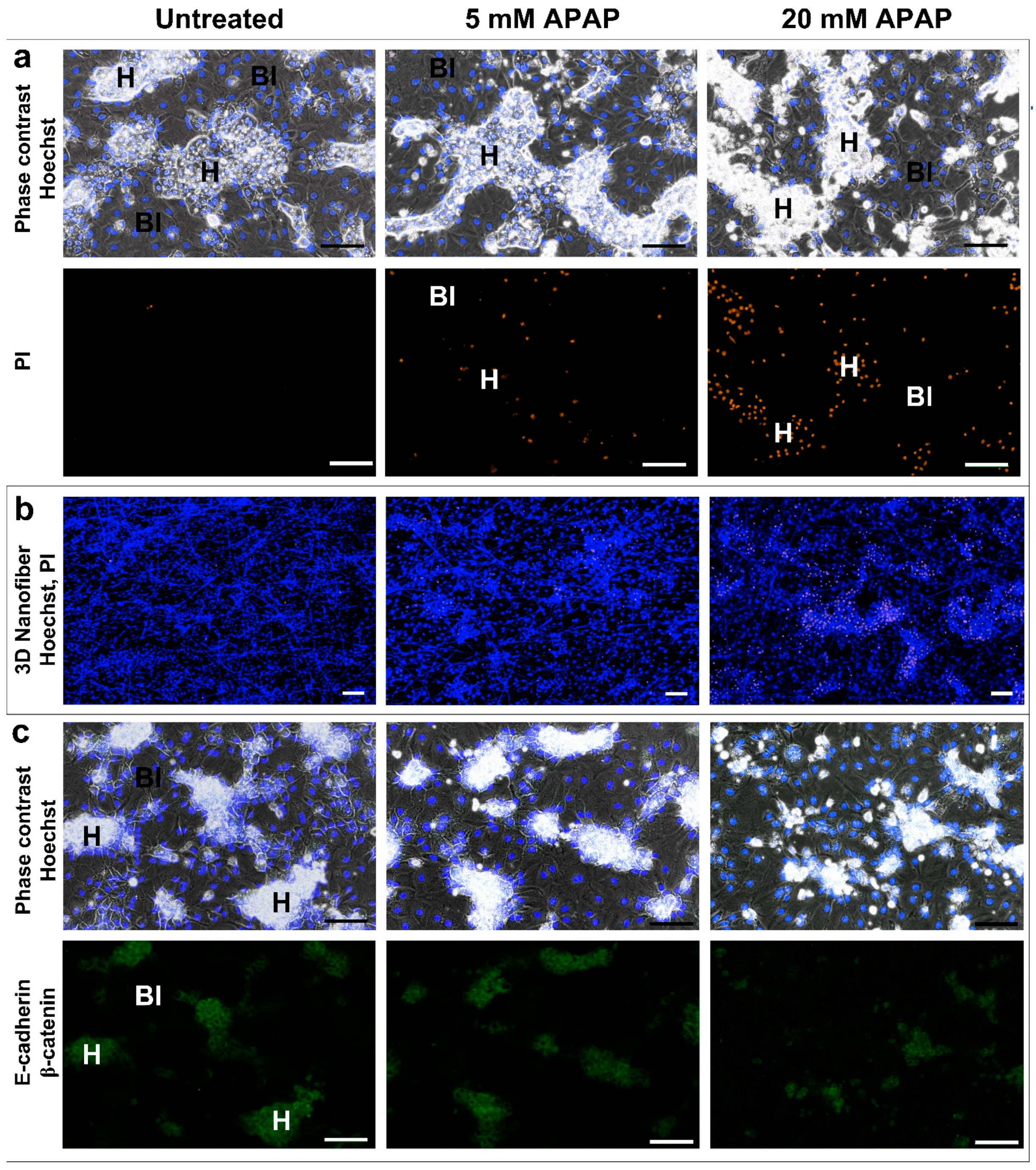
3.4. The Effect of 3D Culture Techniques (Spheroid and Nanofiber) on Acetaminophen Cytotoxicity in HepG2 and Differentiated HepaRG Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitory Compound | Abbreviation | Mechanism of Inhibition | Final Concentration in HepG2 cells | Final Concentration in HepaRG Cells |
|---|---|---|---|---|
| zVAD-fmk (solved in DMSO, Selleckchem) | Z-V-f | inhibition of caspases, apoptosis | 50 μM | 40 μM |
| Dabrafenib-mesylate (solved in DMSO, MCE®) | Dabr | inhibition of B-RafV600E, RIPK3, ZAK kinase | 10 μM | 10 μM |
| Necrostatin-1 (solved in DMSO, Santa Cruz Biotechnology) | Nec-1 | inhibition of RIPK1, necroptosis | 20 μM | 50 μM |
| Necrostatin-2 (solved in DMSO, MCE®) | Nec-2 | inhibition of necroptosis | 25 μM | 50 μM |
| MDIVI-1 (solved in DMSO MCE®) | MDIVI | inhibition of mitochondrial division, necroptosis | 25 μM | 50 μM |
| Liproxstatin-1 (solved in DMSO, Sigma®) | Lip-1 | inhibition of lipid peroxidation, ferroptosis | 1 μM | 1 μM |
| Ferrostatin-1 (solved in DMSO, Selleckchem) | Fer-1 | inhibition of lipid peroxidation, ferroptosis | 1 μM | 10 μM |
Appendix B

References
- Weaver, R.J.; Blomme, E.A.; Chadwick, A.E.; Copple, I.M.; Gerets, H.H.J.; Goldring, C.E.; Guillouzo, A.; Hewitt, P.G.; Ingelman-Sundberg, M.; Jensen, K.G.; et al. Managing the challenge of drug-induced liver injury: A roadmap for the development and deployment of preclinical predictive models. Nat. Rev. Drug Discov. 2020, 19, 131–148. [Google Scholar] [CrossRef]
- Maes, M.; Vinken, M.; Jaeschke, H. Experimental models of hepatotoxicity related to acute liver failure. Toxicol. Appl. Pharmacol. 2016, 290, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, A.; Jaeschke, H. Acetaminophen toxicity: Novel insights into mechanisms and future perspectives. Gene Expr. 2017, 18, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J. Acute liver failure including acetaminophen overdose. Med. Clin. N. Am. 2008, 92, 761–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlin, D.C.; Miwa, G.T.; Lu, A.Y.; Nelson, S.D. N-Acetyl-p-Benzoquinone imine: A cytochrome P-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. USA 1984, 81, 1327–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Gillette, J.R.; Brodie, B.B. Acetaminophen induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther. 1973, 187, 211–217. [Google Scholar]
- Rosen, G.M.; Rauckman, E.J.; Ellington, S.P.; Dahlin, D.C.; Christie, J.L.; Nelson, S.D. Reduction and Glutathione Conjugation Reactions of N-Acetyl-p-Benzoquinone Imine and Two Dimethylated Analogues. Mol. Pharmacol. 1984, 25, 151–157. [Google Scholar]
- Jaeschke, H.; Ramachandran, A.; Chao, X.; Ding, W.X. Emerging and Established Modes of Cell Death during Acetaminophen-Induced Liver Injury. Arch. Toxicol. 2019, 93, 3491–3502. [Google Scholar] [CrossRef] [PubMed]
- Lőrincz, T.; Jemnitz, K.; Kardon, T.; Mandl, J.; Szarka, A. Ferroptosis is involved in acetaminophen induced cell death. Pathol. Oncol. Res. 2015, 21, 1115–1121. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of N-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Karasawa, T.; Takahashi, M. Role of Ferroptosis in Acetaminophen-Induced Hepatotoxicity. Arch. Toxicol. 2020, 94, 1769–1770. [Google Scholar] [CrossRef]
- Hamid Boulares, A.; Zoltoski, A.J.; Stoica, B.A.; Cuvillier, O.; Smulson, M.E. Acetaminophen Induces a Caspase-Dependent and Bcl-XL Sensitive Apoptosis in Human Hepatoma Cells and Lymphocytes. Pharmacol. Toxicol. 2002, 90, 38–50. [Google Scholar] [CrossRef]
- Kass, G.E.N.; Macanas-Pirard, P.; Lee, P.C.; Hinton, R.H. The role of apoptosis in acetaminophen-induced injury. In Annals of the New York Academy of Sciences; New York Academy of Sciences: New York, NY, USA, 2003; Volume 1010, pp. 557–559. [Google Scholar]
- Manov, I.; Hirsh, M.; Iancu, T.C. N-Acetylcysteine Does Not Protect HepG2 Cells against Acetaminophen-Induced Apoptosis. Basic Clin. Pharmacol. Toxicol. 2004, 94, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Wang, H.; Jaeschke, H.; Ding, W.X. Role and Mechanisms of Autophagy in Acetaminophen-Induced Liver Injury. Liver Int. 2018, 38, 1363–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-Induced Hepatotoxicity in Mice Is Dependent on Tlr9 and the Nalp3 Inflammasome. J. Clin. Investig. 2009, 119, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Farhood, A.; Jaeschke, H. Role of Caspase-1 and Interleukin-1β in Acetaminophen-Induced Hepatic Inflammation and Liver Injury. Toxicol. Appl. Pharmacol. 2010, 247, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; McGill, M.R.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Jaeschke, H. Mechanisms of Acetaminophen-Induced Cell Death in Primary Human Hepatocytes. Toxicol. Appl. Pharmacol. 2014, 279, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Aritomi, K.; Ishitsuka, Y.; Tomishima, Y.; Shimizu, D.; Abe, N.; Shuto, T.; Irikura, M.; Kai, H.; Irie, T. Evaluation of Three-Dimensional Cultured HepG2 Cells in a Nano Culture Plate System: An in Vitro Human Model of Acetaminophen Hepatotoxicity. J. Pharmacol. Sci. 2014, 124, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Ramaiahgari, S.C.; den Braver, M.W.; Herpers, B.; Terpstra, V.; Commandeur, J.N.M.; van de Water, B.; Price, L.S. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch. Toxicol. 2014, 88, 1083–1095. [Google Scholar] [CrossRef]
- Ellero, A.A.; van den Bout, I.; Vlok, M.; Cromarty, A.D.; Hurrell, T. Continual proteomic divergence of HepG2 cells as a consequence of long-term spheroid culture. Sci. Rep. 2021, 11, 10917. [Google Scholar] [CrossRef]
- Mandon, M.; Huet, S.; Dubreil, E.; Fessard, V.; Le Hégarat, L. Three-Dimensional HepaRG Spheroids as a Liver Model to Study Human Genotoxicity in Vitro with the Single Cell Gel Electrophoresis Assay. Sci. Rep. 2019, 9, 10548. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Kawai, K.; Kanaki, T.; Yamazaki, H.; Chesné, C.; Guguen-Guillouzo, C.; Suemizu, H. Functional Polymer-Dependent 3D Culture Accelerates the Differentiation of HepaRG Cells into Mature Hepatocytes. Hepatol. Res. 2016, 46, 1045–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunness, P.; Mueller, D.; Shevchenko, V.; Heinzle, E.; Ingelman-Sundberg, M.; Noor, F. 3D Organotypic Cultures of Human HepaRG Cells: A Tool for In Vitro Toxicity Studies. Toxicol. Sci. 2013, 133, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Berger, B.; Donzelli, M.; Maseneni, S.; Boess, F.; Roth, A.; Krähenbühl, S.; Haschke, M. Comparison Of Liver Cell Models Using The Basel Phenotyping Cocktail. Front. Pharmacol. 2016, 7, 443. [Google Scholar] [CrossRef] [Green Version]
- Oda, S.; Uchida, Y.; Aleo, M.D.; Koza-Taylor, P.H.; Matsui, Y.; Hizue, M.; Marroquin, L.D.; Whritenour, J.; Uchida, E.; Yokoi, T. An in Vitro Coculture System of Human Peripheral Blood Mononuclear Cells with Hepatocellular Carcinoma-Derived Cells for Predicting Drug-Induced Liver Injury. Arch. Toxicol. 2021, 95, 149–168. [Google Scholar] [CrossRef]
- Chen, S.; Wu, Q.; Li, X.; Li, D.; Mei, N.; Ning, B.; Puig, M.; Ren, Z.; Tolleson, W.H.; Guo, L. Characterization of Cytochrome P450s (CYP)-Overexpressing HepG2 Cells for Assessing Drug and Chemical-Induced Liver Toxicity. J. Environ. Sci. Health Part C Toxicol. Carcinog. 2021, 39, 68–86. [Google Scholar] [CrossRef]
- Bai, J.; Cederbaum, A.I. Adenovirus Mediated Overexpression of CYP2E1 Increases Sensitivity of HepG2 Cells to Acetaminophen Induced Cytotoxicity. Mol. Cell. Biochem. 2004, 262, 165–176. [Google Scholar] [CrossRef]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a Human Hepatoma Cell Line by Hepatitis B Virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [Green Version]
- Cerec, V.; Glaise, D.; Garnier, D.; Morosan, S.; Turlin, B.; Drenou, B.; Gripon, P.; Kremsdorf, D.; Guguen-Guillouzo, C.; Corlu, A. Transdifferentiation of Hepatocyte-like Cells from the Human Hepatoma HepaRG Cell Line through Bipotent Progenitor. Hepatology 2007, 45, 957–967. [Google Scholar] [CrossRef]
- Parent, R.; Marion, M.-J.; Furio, L.; Trépo, C.; Petit, M.-A. Origin and Characterization of a Human Bipotent Liver Progenitor Cell Line. Gastroenterology 2004, 126, 1147–1156. [Google Scholar] [CrossRef]
- McGill, M.R.; Yan, H.-M.M.; Ramachandran, A.; Murray, G.J.; Rollins, D.E.; Jaeschke, H. HepaRG Cells: A Human Model to Study Mechanisms of Acetaminophen Hepatotoxicity. Hepatology 2011, 53, 974–982. [Google Scholar] [CrossRef] [Green Version]
- Anthérieu, S.; Chesné, C.; Li, R.; Guguen-Guillouzo, C.; Guillouzo, A. Optimization of the HepaRG Cell Model for Drug Metabolism and Toxicity Studies. Toxicol. In Vitro 2012, 26, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Geng, X.; Wang, J.; Miao, Y.; Lu, Y.; Li, B. The HepaRG Cell Line, a Superior in Vitro Model to L-02, HepG2 and HiHeps Cell Lines for Assessing Drug-Induced Liver Injury. Cell Biol. Toxicol. 2016, 32, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Tascher, G.; Burban, A.; Camus, S.; Plumel, M.; Chanon, S.; Le Guevel, R.; Shevchenko, V.; van Dorsselaer, A.; Lefai, E.; Guguen-Guillouzo, C.; et al. In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes. Cells 2019, 8, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, B.C.; Devonshire, A.S.; Schutte, M.E.; Foy, C.A.; Minguez, J.; Przyborski, S.; Maltman, D.; Bokhari, M.; Marshall, D. Validation of Reference Gene Stability for APAP Hepatotoxicity Studies in Different in Vitro Systems and Identification of Novel Potential Toxicity Biomarkers. Toxicol. In Vitro 2010, 24, 1962–1970. [Google Scholar] [CrossRef] [PubMed]
- Lőrincz, T.; Szarka, A. The Determination of Hepatic Glutathione at Tissue and Subcellular Level. J. Pharmacol. Toxicol. Methods 2017, 88, 32–39. [Google Scholar] [CrossRef]
- Hajdinák, P.; Czobor, Á.; Lőrincz, T.; Szarka, A. The Problem of Glutathione Determination: A Comparative Study on the Measurement of Glutathione from Plant Cells. Period. Polytech. Chem. Eng. 2019, 63, 1–10. [Google Scholar] [CrossRef]
- May, J.E.; Xu, J.; Morse, H.R.; Avent, N.D.; Donaldson, C. Toxicity Testing: The Search for an in Vitro Alternative to Animal Testing. Br. J. Biomed. Sci. 2009, 66, 160–165. [Google Scholar] [CrossRef]
- Han, W.; Wu, Q.; Zhang, X.; Duan, Z. Innovation for Hepatotoxicity in Vitro Research Models: A Review. J. Appl. Toxicol. 2019, 39, 146–162. [Google Scholar] [CrossRef] [Green Version]
- Hawksworth, G. Advantages and Disadvantages of Using Human Cells for Pharmacological and Toxicological Studies. Hum. Exp. Toxicol. 1994, 13, 568–573. [Google Scholar] [CrossRef]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knöspel, F. Cell Sources for in Vitro Human Liver Cell Culture Models. Exp. Biol. Med. 2016, 241, 1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.; Yi, X.; Liao, W.; Chen, Q.; Yang, W.; Li, Y.; Li, S.; Gao, Y.; Peng, Q.; Zhou, S. Advancements in Stem Cell-Derived Hepatocyte-like Cell Models for Hepatotoxicity Testing. Stem Cell Res. Ther. 2021, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Pathways of Acetaminophen Metabolism at the Therapeutic versus Toxic Doses. Pharmacogenet. Genomics 2015, 25, 416. [Google Scholar] [CrossRef] [Green Version]
- McGill, M.R. The Past and Present of Serum Aminotransferases and the Future of Liver Injury Biomarkers. EXCLI J. 2016, 15, 817–828. [Google Scholar] [CrossRef]
- Jang, M.; Neuzil, P.; Volk, T.; Manz, A.; Kleber, A. On-Chip Three-Dimensional Cell Culture in Phaseguides Improves Hepatocyte Functions in Vitro. Biomicrofluidics 2015, 9, 034113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, L.T.; Minsky, N.W.; Espinosa, L.E.M.; Aranda, R.S.; Meseguer, J.P.; Pérez, P.C. In Vitro Assessment of Hepatoprotective Agents against Damage Induced by Acetaminophen and CCl4. BMC Complement. Altern. Med. 2017, 17, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.F.; He, W.; Zhang, C.; Liu, X.J.; Lu, Y.; Wang, H.; Zhang, Z.H.; Chen, X.; Xu, D.X. Role of Receptor Interacting Protein (RIP)1 on Apoptosis-Inducing Factor-Mediated Necroptosis during Acetaminophen-Evoked Acute Liver Failure in Mice. Toxicol. Lett. 2014, 225, 445–453. [Google Scholar] [CrossRef]
- Ramachandran, A.; McGill, M.R.; Xie, Y.; Ni, H.-M.; Ding, W.-X.; Jaeschke, H. Receptor Interacting Protein Kinase 3 Is a Critical Early Mediator of Acetaminophen-Induced Hepatocyte Necrosis in Mice. Hepatology 2013, 58, 2099–2108. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Gadang, V.; Jaeschke, A. Critical Role for Mixed-Lineage Kinase 3 in Acetaminophen-Induced Hepatotoxicity. Mol. Pharmacol. 2012, 82, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Li, J.X.; Feng, J.M.; Wang, Y.; Li, X.H.; Chen, X.X.; Su, Y.; Shen, Y.Y.; Chen, Y.; Xiong, B.; Yang, C.H.; et al. The B-RafV600E Inhibitor Dabrafenib Selectively Inhibits RIP3 and Alleviates Acetaminophen-Induced Liver Injury. Cell Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef] [Green Version]
- Dara, L.; Johnson, H.; Suda, J.; Win, S.; Gaarde, W.; Han, D.; Kaplowitz, N. Receptor Interacting Protein Kinase 1 Mediates Murine Acetaminophen Toxicity Independent of the Necrosome and Not through Necroptosis. Hepatology 2015, 62, 1847–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.-Y.; Chen, M.-C.; Tseng, Y.-S.; Chen, M.-C.; Zhou, Z.; Yang, J.-J.; Lin, Y.-M.; Viswanadha, V.P.; Wang, G.; Huang, C.-Y. Fisetin Activates Hippo Pathway and JNK/ERK/AP-1 Signaling to Inhibit Proliferation and Induce Apoptosis of Human Osteosarcoma Cells via ZAK Overexpression. Environ. Toxicol. 2019, 34, 902–911. [Google Scholar] [CrossRef]
- Vin, H.; Ojeda, S.S.; Ching, G.; Leung, M.L.; Chitsazzadeh, V.; Dwyer, D.W.; Adelmann, C.H.; Restrepo, M.; Richards, K.N.; Stewart, L.R.; et al. BRAF Inhibitors Suppress Apoptosis through Off-Target Inhibition of JNK Signaling. eLife 2013, 2013, e00969. [Google Scholar] [CrossRef]
- Zhang, J.; Min, R.W.M.; Le, K.; Zhou, S.; Aghajan, M.; Than, T.A.; Win, S.; Kaplowitz, N. The Role of MAP2 Kinases and P38 Kinase in Acute Murine Liver Injury Models. Cell Death Dis. 2017, 8, e2903–e2909. [Google Scholar] [CrossRef] [Green Version]
- Sarikhani, M.; Mishra, S.; Arumugam, P.; Chaithanya, D.; Donald, K. SIRT2 Regulates Oxidative Stress-Induced Cell Death through Deacetylation of c-Jun NH 2 -Terminal Kinase. Cell Death Differ. 2018, 25, 1638–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, K.; Hatano, E.; Iwaisako, K.; Takeiri, M.; Noma, N.; Ohmae, S.; Toriguchi, K.; Tanabe, K.; Tanaka, H.; Seo, S.; et al. Necrostatin-1 Protects against Reactive Oxygen Species (ROS)-Induced Hepatotoxicity in Acetaminophen-Induced Acute Liver Failure. FEBS Open Bio 2014, 4, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, N.J.; Lechón, M.J.G.; Houston, J.B.; Hallifax, D.; Brown, H.S.; Maurel, P.; Kenna, J.G.; Gustavsson, L.; Lohmann, C.; Skonberg, C.; et al. Primary Hepatocytes: Current Understanding of the Regulation of Metabolic Enzymes and Transporter Proteins, and Pharmaceutical Practice for the Use of Hepatocytes in Metabolism, Enzyme Induction, Transporter, Clearance, and Hepatotoxicity Studies. Drug Metab. Rev. 2007, 39, 159–234. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.; Gerrard, D.T.; Jenkins, R.; Berry, A.; Durkin, K.; Sundstrom, L.; Goldring, C.E.; Park, B.K.; Kitteringham, N.R.; Hanley, K.P.; et al. Proteome-Wide Analyses of Human Hepatocytes during Differentiation and Dedifferentiation. Hepatology 2013, 58, 799–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleine, M.; Riemer, M.; Krech, T.; DeTemple, D.; Jäger, M.D.; Lehner, F.; Manns, M.P.; Klempnauer, J.; Borlak, J.; Bektas, H.; et al. Explanted Diseased Livers—A Possible Source of Metabolic Competent Primary Human Hepatocytes. PLoS ONE 2014, 9, e101386. [Google Scholar] [CrossRef] [PubMed]
- Kiamehr, M.; Heiskanen, L.; Laufer, T.; Düsterloh, A.; Kahraman, M.; Käkelä, R.; Laaksonen, R.; Aalto-Setälä, K. Dedifferentiation of Primary Hepatocytes Is Accompanied with Reorganization of Lipid Metabolism Indicated by Altered Molecular Lipid and MiRNA Profiles. Int. J. Mol. Sci. 2019, 20, 2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Kang, K.; Jeong, J.; Paik, S.S.; Kim, J.S.; Park, S.A.; Kim, W.D.; Park, J.; Choi, D. Three-Dimensional (3D) Printing of Mouse Primary Hepatocytes to Generate 3D Hepatic Structure. Ann. Surg. Treat. Res. 2017, 92, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, A.; Razpotnik, A.; Rouimi, P.; de Sousa, G.; Cravedi, J.P.; Rahmani, R. Cellular Impact of Combinations of Endosulfan, Atrazine, and Chlorpyrifos on Human Primary Hepatocytes and HepaRG Cells after Short and Chronic Exposures. Cell Biol. Toxicol. 2014, 30, 17–29. [Google Scholar] [CrossRef]
- Turpeinen, M.; Tolonen, A.; Chesne, C.; Guillouzo, A.; Uusitalo, J.; Pelkonen, O. Functional Expression, Inhibition and Induction of CYP Enzymes in HepaRG Cells. Toxicol. In Vitro 2009, 23, 748–753. [Google Scholar] [CrossRef]
- Hajdinák, P.; Czobor, Á.; Szarka, A. The Potential Role of Acrolein in Plant Ferroptosis-like Cell Death. PLoS ONE 2019, 14, e0227278. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summer, K.H.; Wiebel, F.J. Glutathione and Glutathione S-Transferase Activities of Mammalian Cells in Culture. Toxicol. Lett. 1981, 9, 409–413. [Google Scholar] [CrossRef]
- Manyike, P.T.; Kharasch, E.D.; Kalhorn, T.F.; Slattery, J.T. Contribution of CYP2E1 and CYP3A to Acetaminophen Reactive Metabolite Formation. Clin. Pharmacol. Ther. 2000, 67, 275–282. [Google Scholar] [CrossRef]
- Hart, S.N.; Li, Y.; Nakamoto, K.; Subileau, E.; Steen, D.; Zhong, X. A Comparison of Whole Genome Gene Expression Profiles of HepaRG Cells and HepG2 Cells to Primary Human Hepatocytes and Human Liver Tissues. Drug Metab. Dispos. 2010, 38, 988–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luckert, C.; Schulz, C.; Lehmann, N.; Thomas, M.; Hofmann, U.; Hammad, S.; Hengstler, J.G.; Braeuning, A.; Lampen, A.; Hessel, S. Comparative Analysis of 3D Culture Methods on Human HepG2 Cells. Arch. Toxicol. 2017, 91, 393–406. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lőrincz, T.; Deák, V.; Makk-Merczel, K.; Varga, D.; Hajdinák, P.; Szarka, A. The Performance of HepG2 and HepaRG Systems through the Glass of Acetaminophen-Induced Toxicity. Life 2021, 11, 856. https://doi.org/10.3390/life11080856
Lőrincz T, Deák V, Makk-Merczel K, Varga D, Hajdinák P, Szarka A. The Performance of HepG2 and HepaRG Systems through the Glass of Acetaminophen-Induced Toxicity. Life. 2021; 11(8):856. https://doi.org/10.3390/life11080856
Chicago/Turabian StyleLőrincz, Tamás, Veronika Deák, Kinga Makk-Merczel, Dóra Varga, Péter Hajdinák, and András Szarka. 2021. "The Performance of HepG2 and HepaRG Systems through the Glass of Acetaminophen-Induced Toxicity" Life 11, no. 8: 856. https://doi.org/10.3390/life11080856