
Structure, Activity and Function of the SETDB1 Protein Methyltransferase


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
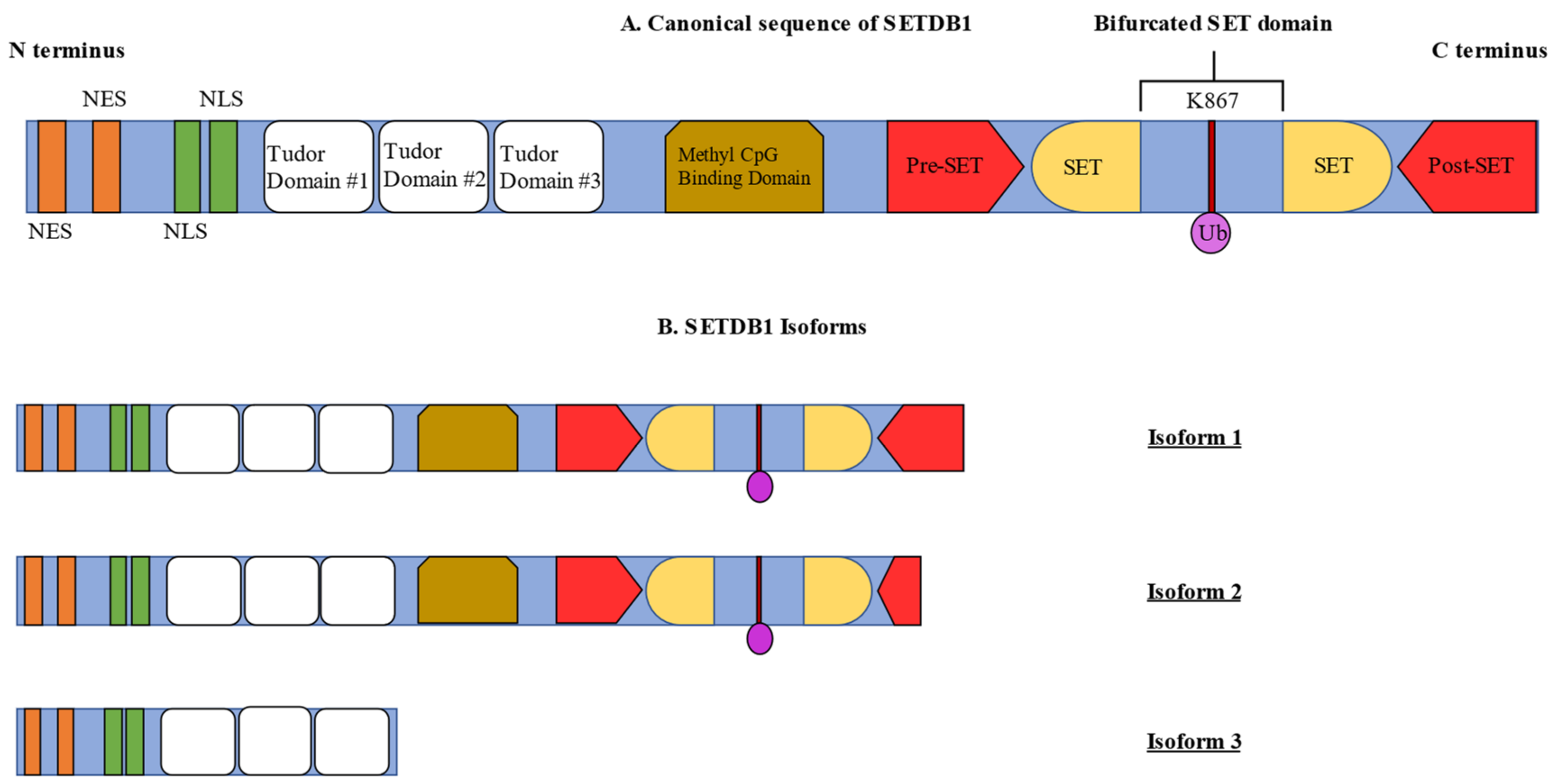
2. Structural Features of SETDB1
3. Biochemical Features of SETDB1
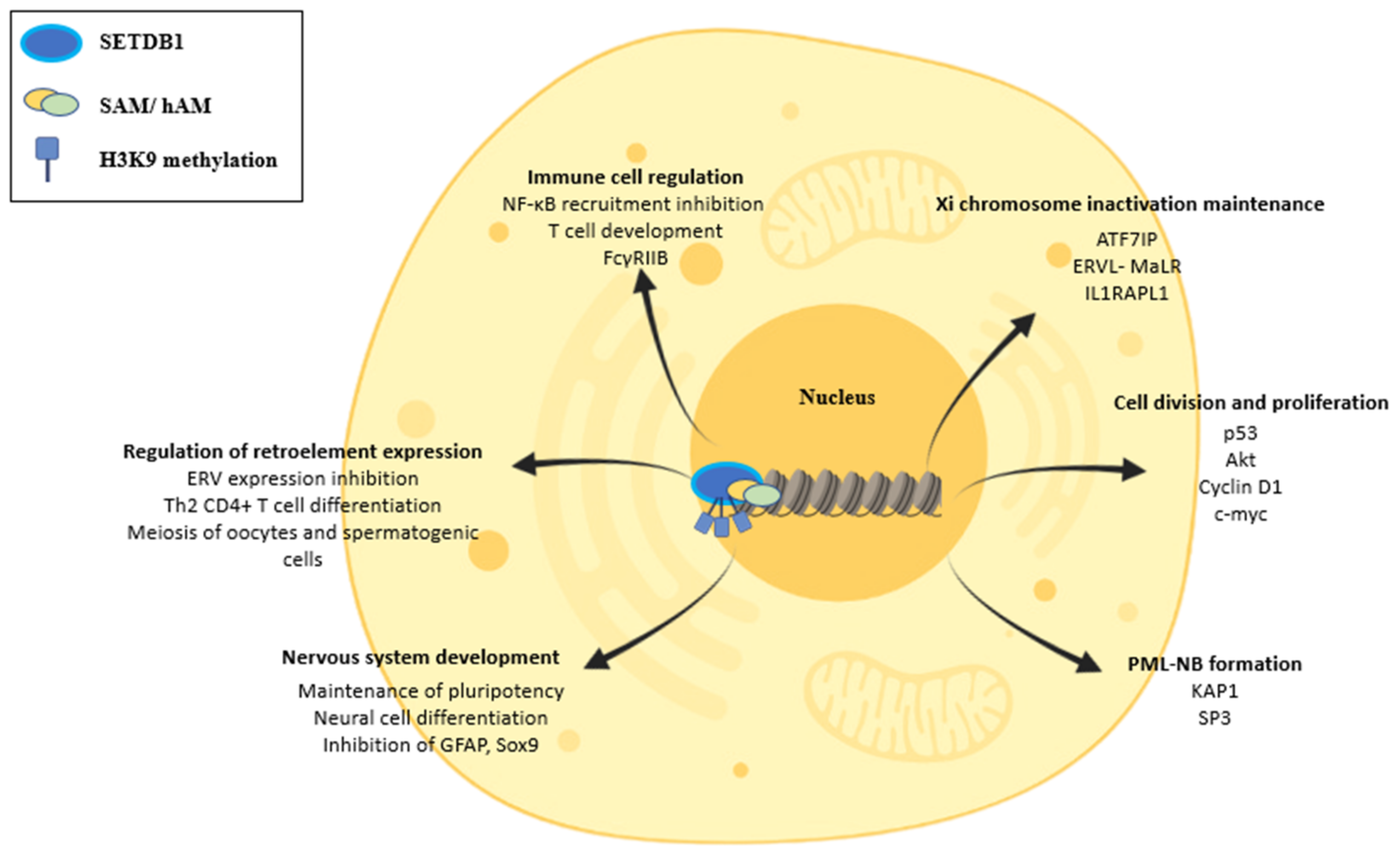
4. Physiologic Functions of SETDB1 and Cellular Features
4.1. SETDB1 Directly and Indirectly Affects Major Cellular Functions
4.2. SETDB1′s Role in Cell Division
4.3. SETDB1 in ERV Regulation and Its Implications
4.4. SETDB1 in the Regulation of the Inflammatory Response
4.5. SETDB1 Regulates the Formation of PML-NBs
4.6. SETDB1 Coordinates the Development of the Nervous System
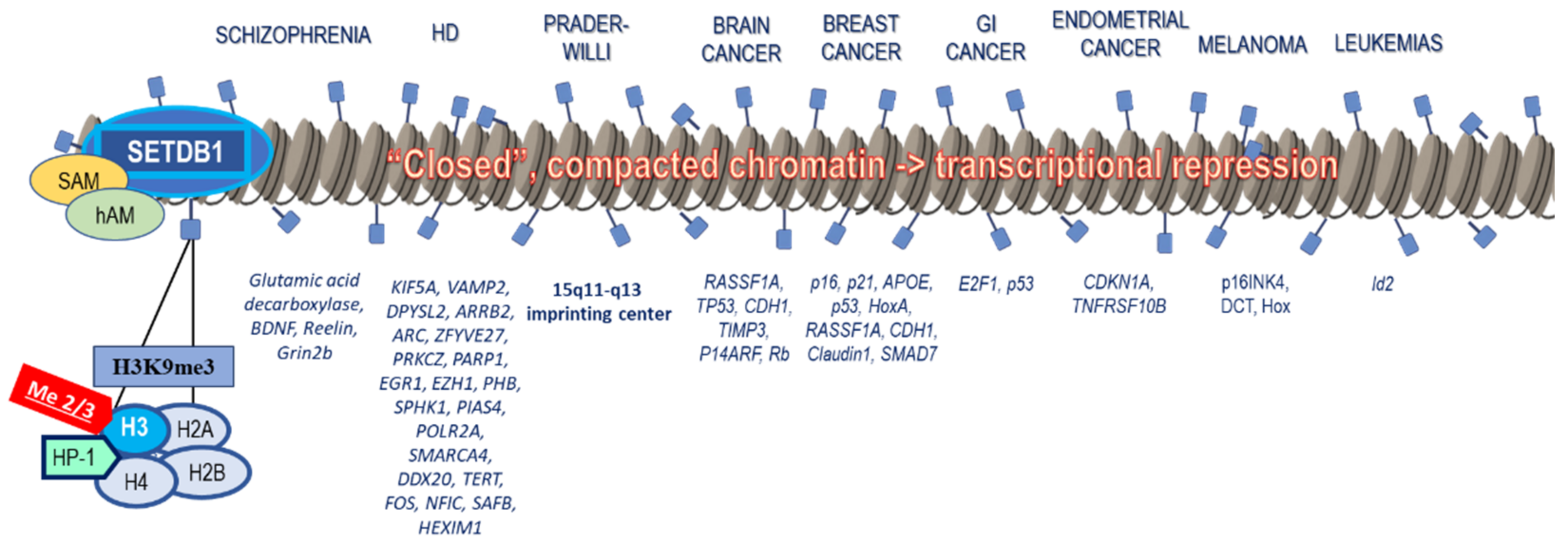
5. Connection of SETDB1 with Tumorigenesis
5.1. Brain and Head–Neck Cancer
5.2. Lung Cancer and Malignant Pleural Mesothelioma
5.3. Breast Cancer
5.4. Gastrointestinal Cancers
5.5. Reproductive System Cancers
5.6. Melanoma
5.7. Hematologic Cancers
5.8. Osteosarcoma
6. SETDB1 Connection to Other Diseases
6.1. SETDB1 Association with Neuropsychiatric Diseases
6.2. SETDB1 Association with Genetic Diseases
6.3. SETDB1 Association with Cardiovascular Diseases
6.4. SETDB1 Association with Gastrointestinal Diseases
7. Outlook and Directions for Future Research
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Albini, S.; Zakharova, V.; Ait-Si-Ali, S. Chapter 3—Histone Modifications. In Translational Epigenetics; Palacios, D., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 11, pp. 47–72. ISBN 25425358. [Google Scholar]
- Markouli, M.; Strepkos, D.; Chlamydas, S.; Piperi, C. Histone lysine methyltransferase SETDB1 as a novel target for central nervous system diseases. Prog. Neurobiol. 2020, 200, 101968. [Google Scholar] [CrossRef]
- Jenuwein, T.; Laible, G.; Dorn, R.; Reuter, G. SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell. Mol. Life Sci. 1998, 54, 80–93. [Google Scholar] [CrossRef]
- Stassen, M.J.; Bailey, D.; Nelson, S.; Chinwalla, V.; Harte, P.J. The Drosophila trithorax proteins contain a novel variant of the nuclear receptor type DNA binding domain and an ancient conserved motif found in other chromosomal proteins. Mech. Dev. 1995, 52, 209–223. [Google Scholar] [CrossRef]
- SETDB1 Orthologs—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/9869/ortholog/?scope=117570 (accessed on 21 July 2021).
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- SET DOMAIN PROTEIN, BIFURCATED, 1; SETDB1—OMIM. Available online: https://www.omim.org/entry/604396#references (accessed on 21 July 2021).
- SET Domain Bifurcated Histone Lysine Methyltransferase 1 [Homo Sapiens (Human)]—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/9869 (accessed on 21 July 2021).
- Torrano, J.; Al Emran, A.; Hammerlindl, H.; Schaider, H. Emerging roles of H3K9me3, SETDB1 and SETDB2 in therapy-induced cellular reprogramming. Clin. Epigenet. 2019, 11, 43. [Google Scholar] [CrossRef] [Green Version]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., III. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Jurkowska, R.Z.; Qin, S.; Kungulovski, G.; Tempel, W.; Liu, Y.; Bashtrykov, P.; Stiefelmaier, J.; Jurkowski, T.P.; Kudithipudi, S.; Weirich, S.; et al. H3K14ac is linked to methylation of H3K9 by the triple Tudor domain of SETDB1. Nat. Commun. 2017, 8, 2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terns, M.P.; Terns, R.M. Macromolecular complexes: SMN—The master assembler. Curr. Biol. 2001, 11, R862–R864. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhang, F.; Ding, J.; Liang, Y.; Zhan, Z.; Zhan, Y.; Chen, L.-H.; Ding, Y. Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53. J. Cancer 2017, 8, 3318–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Rauch, T.; Chen, Z.-X.; Szabó, P.E.; Riggs, A.D.; Pfeifer, G.P. The Histone Methyltransferase SETDB1 and the DNA Methyltransferase DNMT3A Interact Directly and Localize to Promoters Silenced in Cancer Cells. J. Biol. Chem. 2006, 281, 19489–19500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Park, J.S.; Kang, Y.-K. Regulated nuclear entry of over-expressed Setdb1. Genes Cells 2013, 18, 694–703. [Google Scholar] [CrossRef]
- Wu, H.; Min, J.; Lunin, V.V.; Antoshenko, T.; Dombrovski, L.; Zeng, H.; Allali-Hassani, A.; Campagna-Slater, V.; Vedadi, M.; Arrowsmith, C.H.; et al. Structural biology of human H3K9 methyltransferases. PLoS ONE 2010, 5, e8570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimoto, K.; Kawamata, N.; Uchihara, Y.; Okubo, M.; Fujimoto, R.; Gotoh, E.; Kakinouchi, K.; Mizohata, E.; Hino, N.; Okada, Y.; et al. Ubiquitination of Lysine 867 of the Human SETDB1 Protein Upregulates Its Histone H3 Lysine 9 (H3K9) Methyltransferase Activity. PLoS ONE 2016, 11, e0165766. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xia, L.; Wu, D.Y.; Wang, H.; Chansky, H.A.; Schubach, W.H.; Hickstein, D.D.; Zhang, Y. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene 2002, 21, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, M.L.; Chansky, H.A.; Zielinska-Kwiatkowska, A.; Matsui, Y.; Yang, L. Genomic structure and expression of the mouse ESET gene encoding an ERG-associated histone methyltransferase with a SET domain. Biochim. Biophys. Acta Gene Struct. Expr. 2003, 1629, 8–14. [Google Scholar] [CrossRef]
- Lazaro-Camp, V.J.; Salari, K.; Meng, X.; Yang, S. SETDB1 in cancer: Overexpression and its therapeutic implications. Am. J. Cancer Res. 2021, 11, 1803–1827. [Google Scholar]
- Wang, H.; An, W.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Chatton, B.; Tempst, P.; Roeder, R.G.; Zhang, Y. mAM Facilitates Conversion by ESET of Dimethyl to Trimethyl Lysine 9 of Histone H3 to Cause Transcriptional Repression. Mol. Cell 2003, 12, 475–487. [Google Scholar] [CrossRef]
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable Elements and Their KRAB-ZFP Controllers Regulate Gene Expression in Adult Tissues. Dev. Cell 2016, 36, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Tie, C.H.; Fernandes, L.; Conde, L.; Robbez-Masson, L.; Sumner, R.P.; Peacock, T.; Rodriguez-Plata, M.T.; Mickute, G.; Gifford, R.; Towers, G.J.; et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 2018, 19, e45000. [Google Scholar] [CrossRef]
- Wolf, G.; Yang, P.; Füchtbauer, A.C.; Füchtbauer, E.-M.; Silva, A.M.; Park, C.; Wu, W.; Nielsen, A.L.; Pedersen, F.S.; Macfarlan, T.S. The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev. 2015, 29, 538–554. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Sun, D.; Jakovcevski, M.; Jiang, Y. Epigenetic mechanism of SETDB1 in brain: Implications for neuropsychiatric disorders. Transl. Psychiatry 2020, 10, 115. [Google Scholar] [CrossRef]
- Minkovsky, A.; Sahakyan, A.; Rankin-Gee, E.; Bonora, G.; Patel, S.; Plath, K. The Mbd1-Atf7ip-Setdb1 pathway contributes to the maintenance of X chromosome inactivation. Epigenet. Chromatin 2014, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozawa, R.-S.; Nagao, K.; Masuda, H.-T.; Iwasaki, O.; Hirota, T.; Nozaki, N.; Kimura, H.; Obuse, C. Human POGZ modulates dissociation of HP1alpha from mitotic chromosome arms through Aurora B activation. Nat. Cell Biol. 2010, 12, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Gotoh, E.; Kawamata, N.; Ishimoto, K.; Uchihara, Y.; Iwanari, H.; Sugiyama, A.; Kawamura, T.; Mochizuki, Y.; Tanaka, T.; et al. Analysis of the subcellular localization of the human histone methyltransferase SETDB1. Biochem. Biophys. Res. Commun. 2015, 465, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Watanabe, S.; Sakamoto, Y.; Aoto, T.; Pujita, N.; Nakao, M. Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J. Biol. Chem. 2005, 280, 13928–13935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timms, R.T.; Tchasovnikarova, I.A.; Antrobus, R.; Dougan, G.; Lehner, P.J. ATF7IP-Mediated Stabilization of the Histone Methyltransferase SETDB1 Is Essential for Heterochromatin Formation by the HUSH Complex. Cell Rep. 2016, 17, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Sarraf, S.A.; Stancheva, I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell 2004, 15, 595–605. [Google Scholar] [CrossRef]
- Tsusaka, T.; Shimura, C.; Shinkai, Y. ATF7IP regulates SETDB1 nuclear localization and increases its ubiquitination. EMBO Rep. 2019, 20, e48297. [Google Scholar] [CrossRef]
- Yang, L.; Mei, Q.; Zielinska-Kwiatkowska, A.; Matsui, Y.; Blackburn, M.L.; Benedetti, D.; Krumm, A.A.; Taborsky, G.J., Jr.; Chansky, H.A. An ERG (ets-related gene)-associated histone methyltransferase interacts with histone deacetylases 1/2 and transcription co-repressors mSin3A/B. Biochem. J. 2003, 369, 651–657. [Google Scholar] [CrossRef]
- Fei, Q.; Yang, X.; Jiang, H.; Wang, Q.; Yu, Y.; Yu, Y.; Yi, W.; Zhou, S.; Chen, T.; Lu, C.; et al. SETDB1 modulates PRC2 activity at developmental genes independently of H3K9 trimethylation in mouse ES cells. Genome Res. 2015, 25, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Dai, X.; Laurent, B.; Zheng, N.; Gan, W.; Zhang, J.; Guo, A.; Yuan, M.; Liu, P.; Asara, J.M.; et al. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat. Cell Biol. 2019, 21, 226–237. [Google Scholar] [CrossRef]
- Xiao, J.-F.; Sun, Q.-Y.; Ding, L.-W.; Chien, W.; Liu, X.-Y.; Mayakonda, A.; Jiang, Y.-Y.; Loh, X.-Y.; Ran, X.-B.; Doan, N.B.; et al. The c-MYC–BMI1 axis is essential for SETDB1-mediated breast tumourigenesis. J. Pathol. 2018, 246, 89–102. [Google Scholar] [CrossRef]
- Maksakova, I.A.; Romanish, M.T.; Gagnier, L.; Dunn, C.A.; van de Lagemaat, L.N.; Mager, D.L. Retroviral elements and their hosts: Insertional mutagenesis in the mouse germ line. PLoS Genet. 2006, 2, e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.-L.; Nishi, M.; Ohtsuka, T.; Matsui, T.; Takemoto, K.; Kamio-Miura, A.; Aburatani, H.; Shinkai, Y.; Kageyama, R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 2012, 139, 3806–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takikita, S.; Muro, R.; Takai, T.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Oda, H.; Kitajima, M.; Oshima, K.; Hattori, M.; et al. A Histone Methyltransferase ESET Is Critical for T Cell Development. J. Immunol. 2016, 197, 2269–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohmann, F.; Loureiro, J.; Su, H.; Fang, Q.; Lei, H.; Lewis, T.; Yang, Y.; Labow, M.; Li, E.; Chen, T.; et al. KMT1E Mediated H3K9 Methylation Is Required for the Maintenance of Embryonic Stem Cells by Repressing Trophectoderm Differentiation. Stem Cells 2010, 28, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Adoue, V.; Binet, B.; Malbec, A.; Fourquet, J.; Romagnoli, P.; van Meerwijk, J.P.M.; Amigorena, S.; Joffre, O.P. The Histone Methyltransferase SETDB1 Controls T Helper Cell Lineage Integrity by Repressing Endogenous Retroviruses. Immunity 2019, 50, 629–644.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepsa, A.; Levidou, G.; Gargalionis, A.; Adamopoulos, C.; Spyropoulou, A.; Dalagiorgou, G.; Thymara, I.; Boviatsis, E.; Themistocleous, M.S.; Petraki, K.; et al. Emerging role of linker histone variant H1x as a biomarker with prognostic value in astrocytic gliomas. A multivariate analysis including trimethylation of H3K9 and H4K20. PLoS ONE 2015, 10, e0115101. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Matsumura, Y.; Kano, Y.; Yoshida, A.; Kawamura, T.; Hirakawa, H.; Inagaki, T.; Tanaka, T.; Kimura, H.; Yanagi, S.; et al. Ubiquitination-dependent and -independent repression of target genes by SETDB1 reveal a context-dependent role for its methyltransferase activity during adipogenesis. Genes Cells 2021, 26, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone methyltransferase SETDB1: A common denominator of tumorigenesis with therapeutic potential. Cancer Res. 2020, 81, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Takemoto, K.; Shinkai, Y. A somatic role for the histone methyltransferase Setdb1 in endogenous retrovirus silencing. Nat. Commun. 2018, 9, 1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, K.; Okuda, A.; Yusa, K.; Shinkai, Y. A CRISPR knockout screen identifies SETDB1-target retroelement silencing factors in embryonic stem cells. Genome Res. 2018, 28, 846–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Brind’Amour, J.; Karimi, M.M.; Shirane, K.; Bogutz, A.; Lefebvre, L.; Sasaki, H.; Shinkai, Y.; Lorincz, M.C. Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev. 2014, 28, 2041–2055. [Google Scholar] [CrossRef] [Green Version]
- Eymery, A.; Liu, Z.; Ozonov, E.A.; Stadler, M.B.; Peters, A.H.F.M. The methyltransferase Setdb1 is essential for meiosis and mitosis in mouse oocytes and early embryos. Development 2016, 143, 2767–2779. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Zhao, H.; Dan, J.; Kim, S.; Hardikar, S.; Hollowell, D.; Lin, K.; Lu, Y.; Takata, Y.; Shen, J.; et al. Maternal Setdb1 Is Required for Meiotic Progression and Preimplantation Development in Mouse. PLoS Genet. 2016, 12, e1005970. [Google Scholar] [CrossRef] [Green Version]
- Cheng, E.-C.; Hsieh, C.-L.; Liu, N.; Wang, J.; Zhong, M.; Chen, T.; Li, E.; Lin, H. The Essential Function of SETDB1 in Homologous Chromosome Pairing and Synapsis during Meiosis. Cell Rep. 2021, 34. [Google Scholar] [CrossRef]
- Sun, Z.; Chadwick, B.P. Loss of SETDB1 decompacts the inactive X chromosome in part through reactivation of an enhancer in the IL1RAPL1 gene. Epigenet. Chromatin 2018, 11, 45. [Google Scholar] [CrossRef]
- Smolko, A.E.; Shapiro-Kulnane, L.; Salz, H.K. The H3K9 methyltransferase SETDB1 maintains female identity in Drosophila germ cells. Nat. Commun. 2018, 9, 4155. [Google Scholar] [CrossRef] [Green Version]
- Hachiya, R.; Shiihashi, T.; Shirakawa, I.; Iwasaki, Y.; Matsumura, Y.; Oishi, Y.; Nakayama, Y.; Miyamoto, Y.; Manabe, I.; Ochi, K.; et al. The H3K9 methyltransferase Setdb1 regulates TLR4-mediated inflammatory responses in macrophages. Sci. Rep. 2016, 6, 28845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellaire, G.; Bazett-Jones, D.P. PML nuclear bodies: Dynamic sensors of DNA damage and cellular stress. Bioessays 2004, 26, 963–977. [Google Scholar] [CrossRef]
- Cho, S.; Park, J.S.; Kang, Y.-K. Dual functions of histone-lysine N-methyltransferase Setdb1 protein at promyelocytic leukemia-nuclear body (PML-NB): Maintaining PML-NB structure and regulating the expression of its associated genes. J. Biol. Chem. 2011, 286, 41115–41124. [Google Scholar] [CrossRef] [Green Version]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Felicella, M.M.; Coyne, T.; Phillips, J.J.; Gorovets, D.; Huse, J.T.; Kofler, J.; Lu, C.; Tihan, T.; Sullivan, L.M.; et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J. Neuropathol. Exp. Neurol. 2013, 72, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Spyropoulou, A.; Gargalionis, A.; Dalagiorgou, G.; Adamopoulos, C.; Papavassiliou, K.A.; Lea, R.W.; Piperi, C.; Papavassiliou, A.G. Role of Histone Lysine Methyltransferases SUV39H1 and SETDB1 in Gliomagenesis: Modulation of Cell Proliferation, Migration, and Colony Formation. Neuromol. Med. 2014, 16, 70–82. [Google Scholar] [CrossRef]
- Ramachandran, A.; Gong, E.M.; Pelton, K.; Ranpura, S.A.; Mulone, M.; Seth, A.; Gomez, P., III; Adam, R.M. FosB regulates stretch-induced expression of extracellular matrix proteins in smooth muscle. Am. J. Pathol. 2011, 179, 2977–2989. [Google Scholar] [CrossRef]
- Özdaş, S. Knockdown of SET Domain, Bifurcated 1 suppresses head and neck cancer cell viability and wound-healing ability in vitro. Turk. J. Biol. 2019, 43, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Huang, W.; Liu, M.; Zhu, J.; Jiang, D.; Xiong, Y.; Zhen, Y.; Yang, D.; Chen, Z.; Peng, L.; et al. Enhanced expression of SETDB1 possesses prognostic value and promotes cell proliferation, migration and invasion in nasopharyngeal carcinoma. Oncol. Rep. 2018, 40, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, C.; Yu, X.; Jin, C.; Fu, D.; Ni, Q. Activation of multiple tumor suppressor genes by MBD1 siRNA in pancreatic cancer cell line BxPC-3. Zhonghua Yi Xue Za Zhi 2008, 88, 1948–1951. [Google Scholar]
- Jiang, Y.; Jakovcevski, M.; Bharadwaj, R.; Connor, C.; Schroeder, F.A.; Lin, C.L.; Straubhaar, J.; Martin, G.; Akbarian, S. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J. Neurosci. 2010, 30, 7152–7167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batham, J.; Lim, P.S.; Rao, S. SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis. Cancers 2019, 11, 1143. [Google Scholar] [CrossRef] [Green Version]
- Noh, H.-J.; Kim, K.-A.; Kim, K.-C. p53 Down-regulates SETDB1 gene expression during paclitaxel induced-cell death. Biochem. Biophys. Res. Commun. 2014, 446, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paredes, M.; Martinez de Paz, A.; Simó-Riudalbas, L.; Sayols, S.; Moutinho, C.; Moran, S.; Villanueva, A.; Vázquez-Cedeira, M.; Lazo, P.A.; Carneiro, F.; et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 2014, 33, 2807–2813. [Google Scholar] [CrossRef]
- Wang, G.; Long, J.; Gao, Y.; Zhang, W.; Han, F.; Xu, C.; Sun, L.; Yang, S.-C.; Lan, J.; Hou, Z.; et al. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat. Cell Biol. 2019, 21, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Fei, Q.; Shang, K.; Zhang, J.; Chuai, S.; Kong, D.; Zhou, T.; Fu, S.; Liang, Y.; Li, C.; Chen, Z.; et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat. Commun. 2015, 6, 8651. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Matsuura, S.; Kurabe, N.; Kahyo, T.; Mori, H.; Kawase, A.; Karayama, M.; Inui, N.; Funai, K.; Shinmura, K.; et al. Clinicopathological and Survival Analysis of Japanese Patients with Resected Non-Small-Cell Lung Cancer Harboring NKX2-1, SETDB1, MET, HER2, SOX2, FGFR1, or PIK3CA Gene Amplification. J. Thorac. Oncol. 2015, 10, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- Lafuente-Sanchis, A.; Zúñiga, Á.; Galbis, J.M.; Cremades, A.; Estors, M.; Martínez-Hernández, N.J.; Carretero, J. Prognostic value of ERCC1, RRM1, BRCA1 and SETDB1 in early stage of non-small cell lung cancer. Clin. Transl. Oncol. 2016, 18, 798–804. [Google Scholar] [CrossRef]
- Xu, L.; Liao, W.-L.; Lu, Q.-J.; Zhang, P.; Zhu, J.; Jiang, G.-N. Hypoxic tumor-derived exosomal circular RNA SETDB1 promotes invasive growth and EMT via the miR-7/Sp1 axis in lung adenocarcinoma. Mol. Ther. Nucleic Acids 2021, 23, 1078–1092. [Google Scholar] [CrossRef]
- Na, H.-H.; Noh, H.-J.; Cheong, H.-M.; Kang, Y.; Kim, K.-C. SETDB1 mediated FosB expression increases the cell proliferation rate during anticancer drug therapy. BMB Rep. 2016, 49, 238–243. [Google Scholar] [CrossRef]
- Wu, P.-C.; Lu, J.-W.; Yang, J.-Y.; Lin, I.-H.; Ou, D.-L.; Lin, Y.-H.; Chou, K.-H.; Huang, W.-F.; Wang, W.-P.; Huang, Y.-L.; et al. H3K9 Histone Methyltransferase, KMT1E/SETDB1, Cooperates with the SMAD2/3 Pathway to Suppress Lung Cancer Metastasis. Cancer Res. 2014, 74, 7333. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-K.; Kim, K.-C. DZNep, inhibitor of S-adenosylhomocysteine hydrolase, down-regulates expression of SETDB1 H3K9me3 HMTase in human lung cancer cells. Biochem. Biophys. Res. Commun. 2013, 438, 647–652. [Google Scholar] [CrossRef]
- Park, J.-A.; Na, H.-H.; Jin, H.-O.; Kim, K.-C. Increased Expression of FosB through Reactive Oxygen Species Accumulation Functions as Pro-Apoptotic Protein in Piperlongumine Treated MCF7 Breast Cancer Cells. Mol. Cells 2019, 42, 884–892. [Google Scholar] [CrossRef]
- Kang, H.C.; Kim, H.K.; Lee, S.; Mendez, P.; Kim, J.W.; Woodard, G.; Yoon, J.-H.; Jen, K.-Y.; Fang, L.T.; Jones, K.; et al. Whole exome and targeted deep sequencing identify genome-wide allelic loss and frequent SETDB1 mutations in malignant pleural mesotheliomas. Oncotarget 2016, 7, 8321–8331. [Google Scholar] [CrossRef] [Green Version]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Kimball, S.; Liu, H.; Holowatyj, A.; Yang, Z.-Q. Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget 2015, 6, 2466–2482. [Google Scholar] [CrossRef] [Green Version]
- Gauchier, M.; Kan, S.; Barral, A.; Sauzet, S.; Agirre, E.; Bonnell, E.; Saksouk, N.; Barth, T.K.; Ide, S.; Urbach, S.; et al. SETDB1-dependent heterochromatin stimulates alternative lengthening of telomeres. Sci. Adv. 2019, 5, eaav3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Ying, S.U.; Chenjian, H.O.U.; Chen, L.; Zhou, D.; Kehan, R.E.N.; Zhou, Z.; Zhang, R.; Xiuping, L.I.U. SETDB1 induces epithelial-mesenchymal transition in breast carcinoma by directly binding with Snail promoter. Oncol. Rep. 2019, 41, 1284–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, T.Y.; Kim, K.; Kim, S.-K.; Oh, J.-H.; Min, J.-K.; Jung, C.-R.; Son, M.-Y.; Kim, D.-S.; Cho, H.-S. SETDB1 regulates SMAD7 expression for breast cancer metastasis. BMB Rep. 2019, 52, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, Inhibited Indirectly by LincRNA HOTAIR, Directly Inhibits SETDB1 and Reverses the EMT of Breast Cancer Stem Cells by Downregulating the STAT3 Pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Mozdarani, H.; Ezzatizadeh, V.; Rahbar Parvaneh, R. The emerging role of the long non-coding RNA HOTAIR in breast cancer development and treatment. J. Transl. Med. 2020, 18, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regina, C.; Compagnone, M.; Peschiaroli, A.; Lena, A.; Annicchiarico-Petruzzelli, M.; Piro, M.C.; Melino, G.; Candi, E. Setdb1, a novel interactor of ΔNp63, is involved in breast tumorigenesis. Oncotarget 2016, 7, 28836–28848. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Tan, Y.; Liu, H.; Ooi, S.; Li, L.; Wright, K.; Bennett, S.; Addison, C.L.; Wang, L. Cardamonin reduces chemotherapy-enriched breast cancer stem-like cells in vitro and in vivo. Oncotarget 2016, 7, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Creighton, C.J.; Chang, J.C.; Rosen, J.M. Epithelial-Mesenchymal Transition (EMT) in Tumor-Initiating Cells and Its Clinical Implications in Breast Cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Rauscher, F.J.; Cress, W.D.; Chen, J. Regulation of E2F1 function by the nuclear corepressor KAP1. J. Biol. Chem. 2007, 282, 29902–29909. [Google Scholar] [CrossRef] [Green Version]
- Yokoe, T.; Toiyama, Y.; Okugawa, Y.; Tanaka, K.; Ohi, M.; Inoue, Y.; Mohri, Y.; Miki, C.; Kusunoki, M. KAP1 Is Associated With Peritoneal Carcinomatosis in Gastric Cancer. Ann. Surg. Oncol. 2010, 17, 821–828. [Google Scholar] [CrossRef]
- Ho, Y.-J.; Lin, Y.-M.; Huang, Y.-C.; Chang, J.; Yeh, K.-T.; Lin, L.-I.; Gong, Z.; Tzeng, T.-Y.; Lu, J.-W. Significance of histone methyltransferase SETDB1 expression in colon adenocarcinoma. APMIS 2017, 125, 985–995. [Google Scholar] [CrossRef]
- Yu, L.; Ye, F.; Li, Y.-Y.; Zhan, Y.-Z.; Liu, Y.; Yan, H.-M.; Fang, Y.; Xie, Y.-W.; Zhang, F.-J.; Chen, L.-H.; et al. Histone methyltransferase SETDB1 promotes colorectal cancer proliferation through the STAT1-CCND1/CDK6 axis. Carcinogenesis 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, C.; Hwang, C.Y.; Kim, D.; Han, Y.; Hong, S.N.; Kim, S.-H.; Cho, K.-H. Network Inference Analysis Identifies SETDB1 as a Key Regulator for Reverting Colorectal Cancer Cells into Differentiated Normal-Like Cells. Mol. Cancer Res. 2020, 18, 118. [Google Scholar] [CrossRef]
- Longerich, T. Dysregulation of the epigenetic regulator SETDB1 in liver carcinogenesis—More than one way to skin a cat. Chin. Clin. Oncol. 2016, 5, 72. [Google Scholar] [CrossRef]
- Wong, C.-M.; Wei, L.; Law, C.-T.; Ho, D.W.-H.; Tsang, F.H.-C.; Au, S.L.-K.; Sze, K.M.-F.; Lee, J.M.-F.; Wong, C.C.-L.; Ng, I.O.-L. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology 2016, 63, 474–487. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, S.; Fukuda, A.; Matsumoto, Y.; Hanyu, Y.; Sono, M.; Fukunaga, Y.; Masuda, T.; Araki, O.; Nagao, M.; Yoshikawa, T.; et al. et al SETDB1 Inhibits p53-Mediated Apoptosis and is Required for Formation of Pancreatic Ductal Adenocarcinomas in Mice. Gastroenterology 2020, 159, 682–696.e13. [Google Scholar] [CrossRef] [PubMed]
- Parpart, S.; Roessler, S.; Dong, F.; Rao, V.; Takai, A.; Ji, J.; Qin, L.-X.; Ye, Q.-H.; Jia, H.-L.; Tang, Z.-Y.; et al. Modulation of miR-29 expression by α-fetoprotein is linked to the hepatocellular carcinoma epigenome. Hepatology 2014, 60, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.; Dang, H.; You, H.; Steinway, S.; Takahashi, Y.; Wang, H.-G.; Liao, J.; Stiles, B.; Albert, R.; Rountree, C.B. miR-200b restoration and DNA methyltransferase inhibitor block lung metastasis of mesenchymal-phenotype hepatocellular carcinoma. Oncogenesis 2012, 1, e15. [Google Scholar] [CrossRef] [PubMed]
- Federico, A. Molecular and functional characterization of the role of the histone methyltransferase SETDB1 in malignant melanoma. Fac. Nat. Sci. Fac. Math. 2019. [Google Scholar] [CrossRef]
- Wu, M.; Fan, B.; Guo, Q.; Li, Y.; Chen, R.; Lv, N.; Diao, Y.; Luo, Y. Knockdown of SETDB1 inhibits breast cancer progression by miR-381-3p-related regulation. Biol. Res. 2018, 51, 39. [Google Scholar] [CrossRef]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.-M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881.e13. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, J.; Zhang, X.; Liu, G. Serum circSETDB1 is a promising biomarker for predicting response to platinum-taxane-combined chemotherapy and relapse in high-grade serous ovarian cancer. Onco Targets Ther. 2019, 12, 7451–7457. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, Y.; Tamiya, T.; Takada, I.; Fukaya, T.; Sugiyama, Y.; Inoue, N.; Kimura, A.; Morita, R.; Kashiwagi, I.; Takimoto, T.; et al. Histone 3 lysine 9 (H3K9) methyltransferase recruitment to the interleukin-2 (IL-2) promoter is a mechanism of suppression of IL-2 transcription by the transforming growth factor-β-smad pathway. J. Biol. Chem. 2011, 286, 35456–35465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Y.; Kawaler, E.A.; Cui Zhou, D.; Gritsenko, M.A.; Huang, C.; Blumenberg, L.; Karpova, A.; Petyuk, V.A.; Savage, S.R.; Satpathy, S.; et al. Proteogenomic Characterization of Endometrial Carcinoma. Cell 2020, 180, 729–748.e26. [Google Scholar] [CrossRef]
- Sun, Y.; Wei, M.; Ren, S.-C.; Chen, R.; Xu, W.-D.; Wang, F.-B.; Lu, J.; Shen, J.; Yu, Y.-W.; Hou, J.-G.; et al. Histone methyltransferase SETDB1 is required for prostate cancer cell proliferation, migration and invasion. Asian J. Androl. 2014, 16, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Robbez-Masson, L.; Tie, C.H.C.; Rowe, H.M. Cancer cells, on your histone marks, get SETDB1, silence retrotransposons, and go! J. Cell Biol. 2017, 216, 3429–3431. [Google Scholar] [CrossRef] [PubMed]
- Mita, P.; Savas, J.N.; Briggs, E.M.; Ha, S.; Gnanakkan, V.; Yates, J.R., III; Robins, D.M.; David, G.; Boeke, J.D.; Garabedian, M.J.; et al. URI Regulates KAP1 Phosphorylation and Transcriptional Repression via PP2A Phosphatase in Prostate Cancer Cells. J. Biol. Chem. 2016, 291, 25516–25528. [Google Scholar] [CrossRef] [Green Version]
- Orouji, E.; Federico, A.; Larribère, L.; Novak, D.; Lipka, D.B.; Assenov, Y.; Sachindra, S.; Hüser, L.; Granados, K.; Gebhardt, C.; et al. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int. J. Cancer 2019, 145, 3462–3477. [Google Scholar] [CrossRef] [PubMed]
- Kostaki, M.; Manona, A.D.; Stavraka, I.; Korkolopoulou, P.; Levidou, G.; Trigka, E.-A.; Christofidou, E.; Champsas, G.; Stratigos, A.J.; Katsambas, A.; et al. High-frequency p16INK4A promoter methylation is associated with histone methyltransferase SETDB1 expression in sporadic cutaneous melanoma. Exp. Dermatol. 2014, 23, 332–338. [Google Scholar] [CrossRef]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef]
- Idilli, A.I.; Precazzini, F.; Mione, M.C.; Anelli, V. Zebrafish in Translational Cancer Research: Insight into Leukemia, Melanoma, Glioma and Endocrine Tumor Biology. Genes 2017, 8, 236. [Google Scholar] [CrossRef] [Green Version]
- Koide, S.; Takubo, K.; Oshima, M.; Miyagi, S.; Saraya, A.; Wang, C.; Matsui, H.; Kimura, H.; Shinkai, Y.; Suda, T.; et al. Histone methyltransferase Setdb1 regulates energy metabolism in hematopoietic stem and progenitor cells. Exp. Hematol. 2015, 43, S73. [Google Scholar] [CrossRef]
- Ropa, J.; Saha, N.; Hu, H.; Peterson, L.F.; Talpaz, M.; Muntean, A.G. SETDB1 mediated histone H3 lysine 9 methylation suppresses MLL-fusion target expression and leukemic transformation. Haematologica 2019, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropa, J.; Saha, N.; Chen, Z.; Serio, J.; Chen, W.; Mellacheruvu, D.; Zhao, L.; Basrur, V.; Nesvizhskii, A.I.; Muntean, A.G. PAF1 complex interactions with SETDB1 mediate promoter H3K9 methylation and transcriptional repression of Hoxa9 and Meis1 in acute myeloid leukemia. Oncotarget 2018, 9, 22123–22136. [Google Scholar] [CrossRef] [Green Version]
- Cuellar, T.L.; Herzner, A.-M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-K. SETDB1 in early embryos and embryonic stem cells. Curr. Issues Mol. Biol. 2015, 17, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzinova, M.B.; Benezra, R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003, 13, 410–418. [Google Scholar] [CrossRef]
- Zhou, J.-D.; Ma, J.-C.; Zhang, T.-J.; Li, X.-X.; Zhang, W.; Wu, D.-H.; Wen, X.-M.; Xu, Z.-J.; Lin, J.; Qian, J. High bone marrow ID2 expression predicts poor chemotherapy response and prognosis in acute myeloid leukemia. Oncotarget 2017, 8, 91979–91989. [Google Scholar] [CrossRef] [PubMed]
- Mayers, J.G.; Gokgoz, N.; Wunder, J.S.; Andrulis, I.L. Abstract 5507: Investigation of the effects of alterations in the glutamate receptor, GRIK2 on osteosarcoma tumorigenesis. Cancer Res. 2017, 77, 5507. [Google Scholar] [CrossRef]
- Mayers, J.G.; Gokgoz, N.; Wunder, J.S.; Andrulis, I.L. Abstract B39: Investigation of the effects of alterations in the glutamate receptor, GRIK2, on osteosarcoma tumorigenesis. Clin. Cancer Res. 2018, 24, B39. [Google Scholar] [CrossRef]
- Chase, K.A.; Gavin, D.P.; Guidotti, A.; Sharma, R.P. Histone methylation at H3K9: Evidence for a restrictive epigenome in schizophrenia. Schizophr. Res. 2013, 149, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Hossein Fatemi, S.; Stary, J.M.; Earle, J.A.; Araghi-Niknam, M.; Eagan, E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr. Res. 2005, 72, 109–122. [Google Scholar] [CrossRef]
- Jindal, R.D.; Pillai, A.K.; Mahadik, S.P.; Eklund, K.; Montrose, D.M.; Keshavan, M.S. Decreased BDNF in patients with antipsychotic naïve first episode schizophrenia. Schizophr. Res. 2010, 119, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Avramopoulos, D.; Lasseter, V.K.; Fallin, M.D.; Wolyniec, P.S.; McGrath, J.A.; Nestadt, G.; Valle, D.; Pulver, A.E. Stage II follow-up on a linkage scan for bipolar disorder in the Ashkenazim provides suggestive evidence for chromosome 12p and the GRIN2B gene. Genet. Med. 2007, 9, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, R.; Peter, C.J.; Jiang, Y.; Roussos, P.; Vogel-Ciernia, A.; Shen, E.Y.; Mitchell, A.C.; Mao, W.; Whittle, C.; Dincer, A.; et al. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron 2014, 84, 997–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jury, N.; Abarzua, S.; Diaz, I.; Guerra, M.V.; Ampuero, E.; Cubillos, P.; Martinez, P.; Herrera-Soto, A.; Arredondo, C.; Rojas, F.; et al. Widespread loss of the silencing epigenetic mark H3K9me3 in astrocytes and neurons along with hippocampal-dependent cognitive impairment in C9orf72 BAC transgenic mice. Clin. Epigenet. 2020, 12, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Sun, X.-Y.; Yang, X.; Liu, L.; Tong, K.; Gao, Y.; Hao, J.-R.; Cao, J.; Gao, C. Histone H3K9 Trimethylation Downregulates the Expression of Brain-Derived Neurotrophic Factor in the Dorsal Hippocampus and Impairs Memory Formation During Anaesthesia and Surgery. Front. Mol. Neurosci. 2019, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Goldstein, J.; Wang, P.; Gadi, I.K.; Labreche, H.; Rehder, C.; Wang, W.-P.; McConkie, A.; Xu, X.; Jiang, Y.-H. Chromosomal microarray analysis in clinical evaluation of neurodevelopmental disorders-reporting a novel deletion of SETDB1 and illustration of counseling challenge. Pediatr. Res. 2016, 80, 371–381. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Cukier, H.N.; Lee, J.M.; Ma, D.; Young, J.I.; Mayo, V.; Butler, B.L.; Ramsook, S.S.; Rantus, J.A.; Abrams, A.J.; Whitehead, P.L.; et al. The expanding role of MBD genes in autism: Identification of a MECP2 duplication and novel alterations in MBD5, MBD6, and SETDB1. Autism Res. 2012, 5, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Veazey, K.J.; Parnell, S.E.; Miranda, R.C.; Golding, M.C. Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenet. Chromatin 2015, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Basavarajappa, B.S.; Subbanna, S. Epigenetic Mechanisms in Developmental Alcohol-Induced Neurobehavioral Deficits. Brain Sci. 2016, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.D.; Idrus, N.M.; Monk, B.R.; Dominguez, H.D. Prenatal choline supplementation mitigates behavioral alterations associated with prenatal alcohol exposure in rats. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in β-endorphin-producing POMC neurons of the hypothalamus. Alcohol. Clin. Exp. Res. 2013, 37, 1133–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, K.A.; Sharma, R.P. Nicotine induces chromatin remodelling through decreases in the methyltransferases GLP, G9a, Setdb1 and levels of H3K9me2. Int. J. Neuropsychopharmacol. 2013, 16, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valcárcel-Ocete, L.; Alkorta-Aranburu, G.; Iriondo, M.; Fullaondo, A.; García-Barcina, M.; Fernández-García, J.M.; Lezcano-García, E.; Losada-Domingo, J.M.; Ruiz-Ojeda, J.; Álvarez de Arcaya, A.; et al. Exploring Genetic Factors Involved in Huntington Disease Age of Onset: E2F2 as a New Potential Modifier Gene. PLoS ONE 2015, 10, e0131573. [Google Scholar] [CrossRef]
- Irmak, D.; Fatima, A.; Gutiérrez-Garcia, R.; Rinschen, M.M.; Wagle, P.; Altmüller, J.; Arrigoni, L.; Hummel, B.; Klein, C.; Frese, C.K.; et al. Mechanism suppressing H3K9 trimethylation in pluripotent stem cells and its demise by polyQ-expanded huntingtin mutations. Hum. Mol. Genet. 2018, 27, 4117–4134. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hwang, Y.J.; Kim, Y.; Lee, M.Y.; Hyeon, S.J.; Lee, S.; Kim, D.H.; Jang, S.J.; Im, H.; Min, S.-J.; et al. Remodeling of heterochromatin structure slows neuropathological progression and prolongs survival in an animal model of Huntington’s disease. Acta Neuropathol. 2017, 134, 729–748. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lee, J.; Olofsson, B.A.; Mwidau, A.; Dedeoglu, A.; Escudero, M.; Flemington, E.; Azizkhan-Clifford, J.; Ferrante, R.J.; Ratan, R.R. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 4281–4286. [Google Scholar] [CrossRef] [Green Version]
- Steffan, J.S.; Bodai, L.; Pallos, J.; Poelman, M.; McCampbell, A.; Apostol, B.L.; Kazantsev, A.; Schmidt, E.; Zhu, Y.-Z.; Greenwald, M.; et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001, 413, 739–743. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.; Taye, A.A.; Whitty, L.; Penney, E.; Steffan, J.S.; Fischbeck, K.H. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 15179–15184. [Google Scholar] [CrossRef] [Green Version]
- Simó-Riudalbas, L.; Esteller, M. Targeting the histone orthography of cancer: Drugs for writers, erasers and readers. Br. J. Pharmacol. 2015, 172, 2716–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.J.; Hyeon, S.J.; Kim, Y.; Lim, S.; Lee, M.Y.; Kim, J.; Londhe, A.M.; Gotina, L.; Kim, Y.; Pae, A.N.; et al. Modulation of SETDB1 activity by APQ ameliorates heterochromatin condensation, motor function, and neuropathology in a Huntington’s disease mouse model. J. Enzym. Inhib. Med. Chem. 2021, 36, 856–868. [Google Scholar] [CrossRef]
- Jiang, Y.; Matevossian, A.; Guo, Y.; Akbarian, S. Setdb1-mediated histone H3K9 hypermethylation in neurons worsens the neurological phenotype of Mecp2-deficient mice. Neuropharmacology 2011, 60, 1088–1097. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Wang, S.E.; Jiang, Y.-H. Epigenetic therapy of Prader-Willi syndrome. Transl. Res. 2019, 208, 105–118. [Google Scholar] [CrossRef]
- Cruvinel, E.; Budinetz, T.; Germain, N.; Chamberlain, S.; Lalande, M.; Martins-Taylor, K. Reactivation of maternal SNORD116 cluster via SETDB1 knockdown in Prader-Willi syndrome iPSCs. Hum. Mol. Genet. 2014, 23, 4674–4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buiting, K.; Saitoh, S.; Gross, S.; Dittrich, B.; Schwartz, S.; Nicholls, R.D.; Horsthemke, B. Inherited microdeletions in the Angelman and Prader–Willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 1995, 9, 395–400. [Google Scholar] [CrossRef]
- Langouët, M.; Glatt-Deeley, H.R.; Chung, M.S.; Dupont-Thibert, C.M.; Mathieux, E.; Banda, E.C.; Stoddard, C.E.; Crandall, L.; Lalande, M. Zinc finger protein 274 regulates imprinted expression of transcripts in Prader-Willi syndrome neurons. Hum. Mol. Genet. 2017, 27, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Demarest, T.G.; Babbar, M.; Kim, E.W.; Okur, M.N.; De, S.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome group B deficiency reduces H3K9me3 chromatin remodeler SETDB1 and exacerbates cellular aging. Nucleic Acids Res. 2019, 47, 8548–8562. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mysliwiec, M.R.; Bresnick, E.H.; Lee, Y. Endothelial Jarid2/Jumonji is required for normal cardiac development and proper Notch1 expression. J. Biol. Chem. 2011, 286, 17193–17204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mysliwiec, M.R.; Carlson, C.D.; Tietjen, J.; Hung, H.; Ansari, A.Z.; Lee, Y. Jarid2 (Jumonji, AT rich interactive domain 2) regulates NOTCH1 expression via histone modification in the developing heart. J. Biol. Chem. 2012, 287, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Južnić, L.; Peuker, K.; Strigli, A.; Brosch, M.; Herrmann, A.; Häsler, R.; Koch, M.; Matthiesen, L.; Zeissig, Y.; Löscher, B.-S.; et al. SETDB1 is required for intestinal epithelial differentiation and the prevention of intestinal inflammation. Gut 2021, 70, 485–498. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markouli, M.; Strepkos, D.; Piperi, C. Structure, Activity and Function of the SETDB1 Protein Methyltransferase. Life 2021, 11, 817. https://doi.org/10.3390/life11080817
Markouli M, Strepkos D, Piperi C. Structure, Activity and Function of the SETDB1 Protein Methyltransferase. Life. 2021; 11(8):817. https://doi.org/10.3390/life11080817
Chicago/Turabian StyleMarkouli, Mariam, Dimitrios Strepkos, and Christina Piperi. 2021. "Structure, Activity and Function of the SETDB1 Protein Methyltransferase" Life 11, no. 8: 817. https://doi.org/10.3390/life11080817