
Lytic Release of Cellular ATP: Physiological Relevance and Therapeutic Applications

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Quantitative Evidence for Hypo-Induced Lytic ATP Release in Serum-Deprived A549 Cells
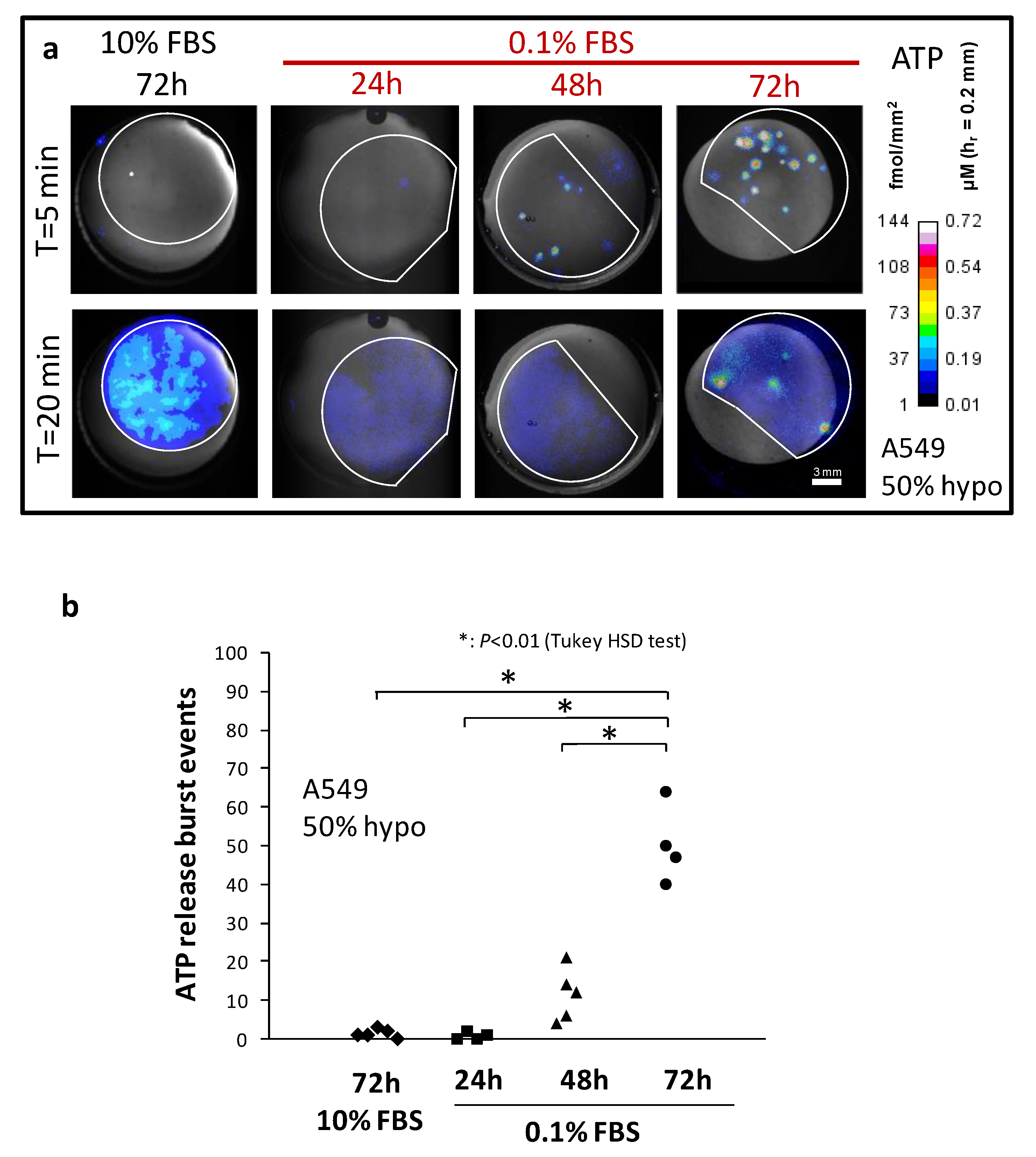
2.1. Serum Deprivation and Osmotic Fragility
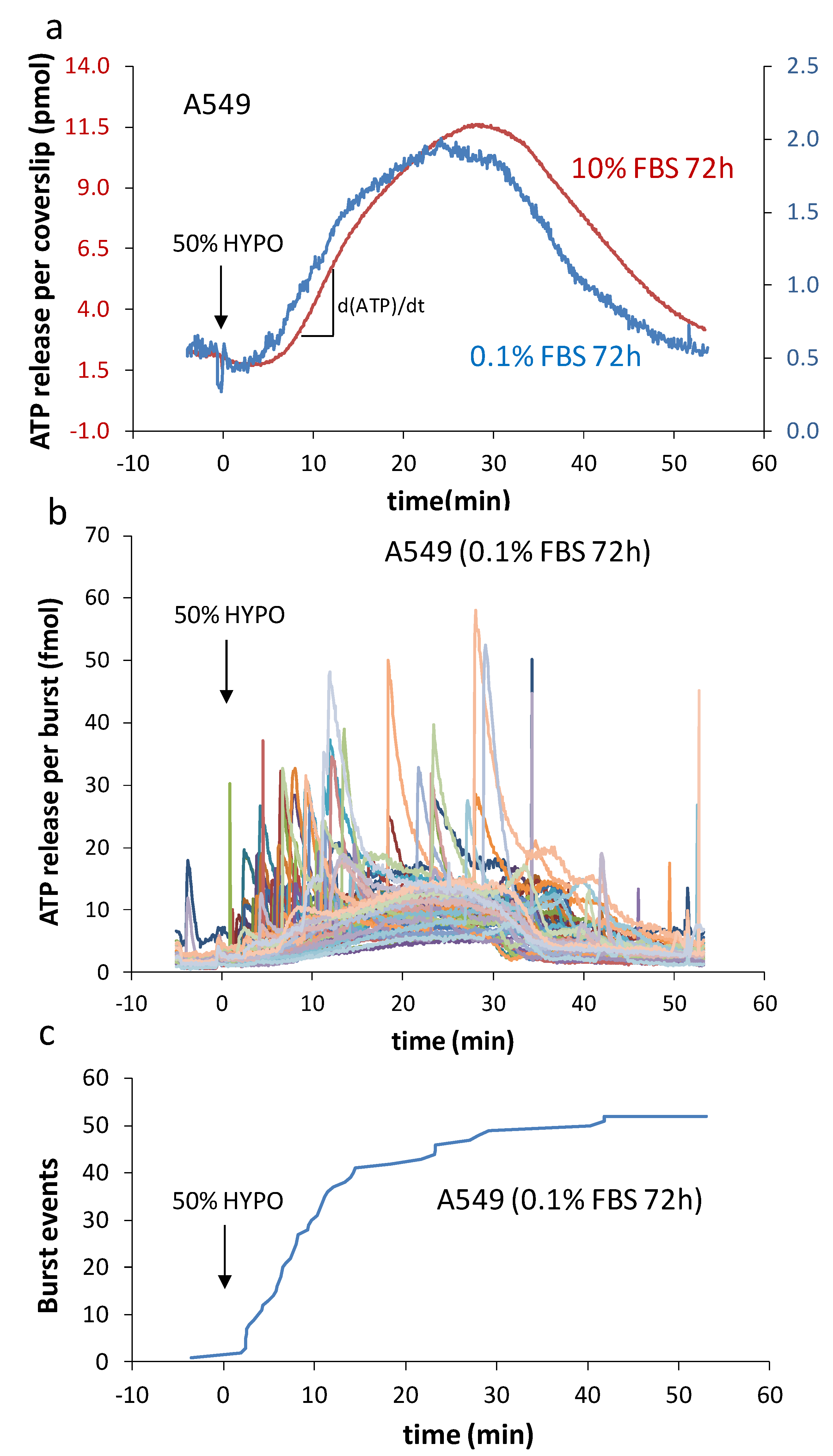
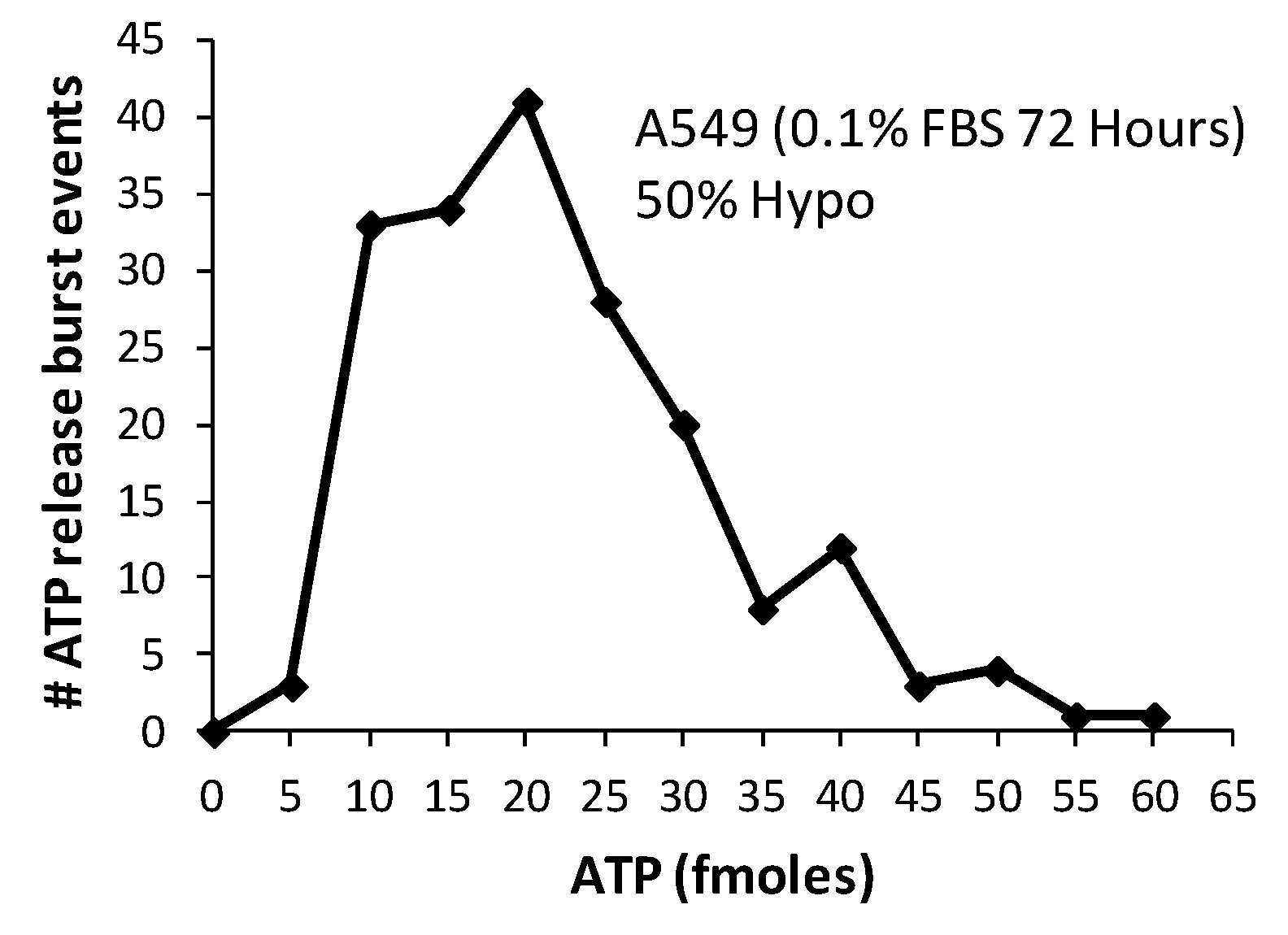
2.2. Characteristics of Burst Events
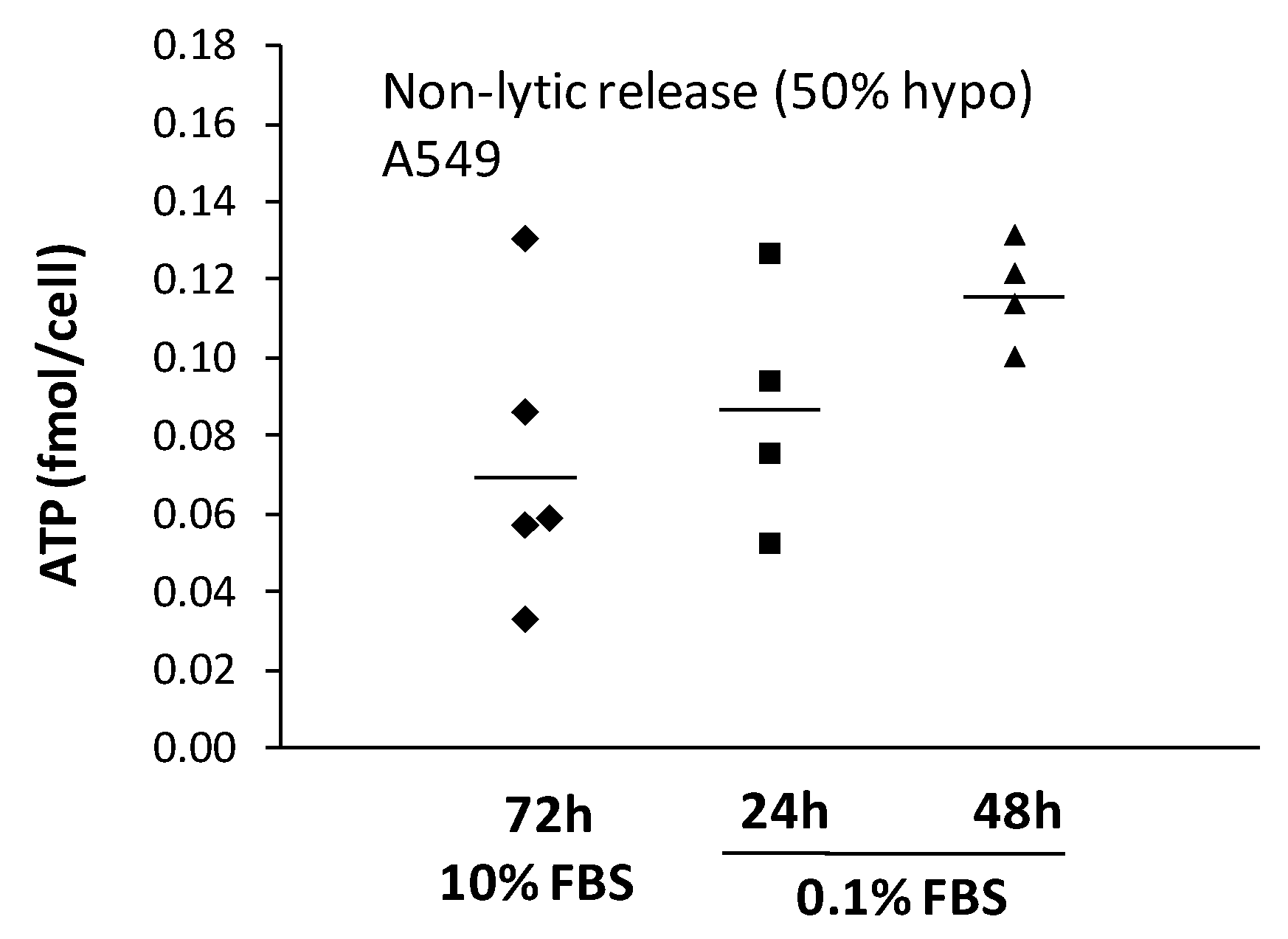
2.3. Non-Lytic ATP Release
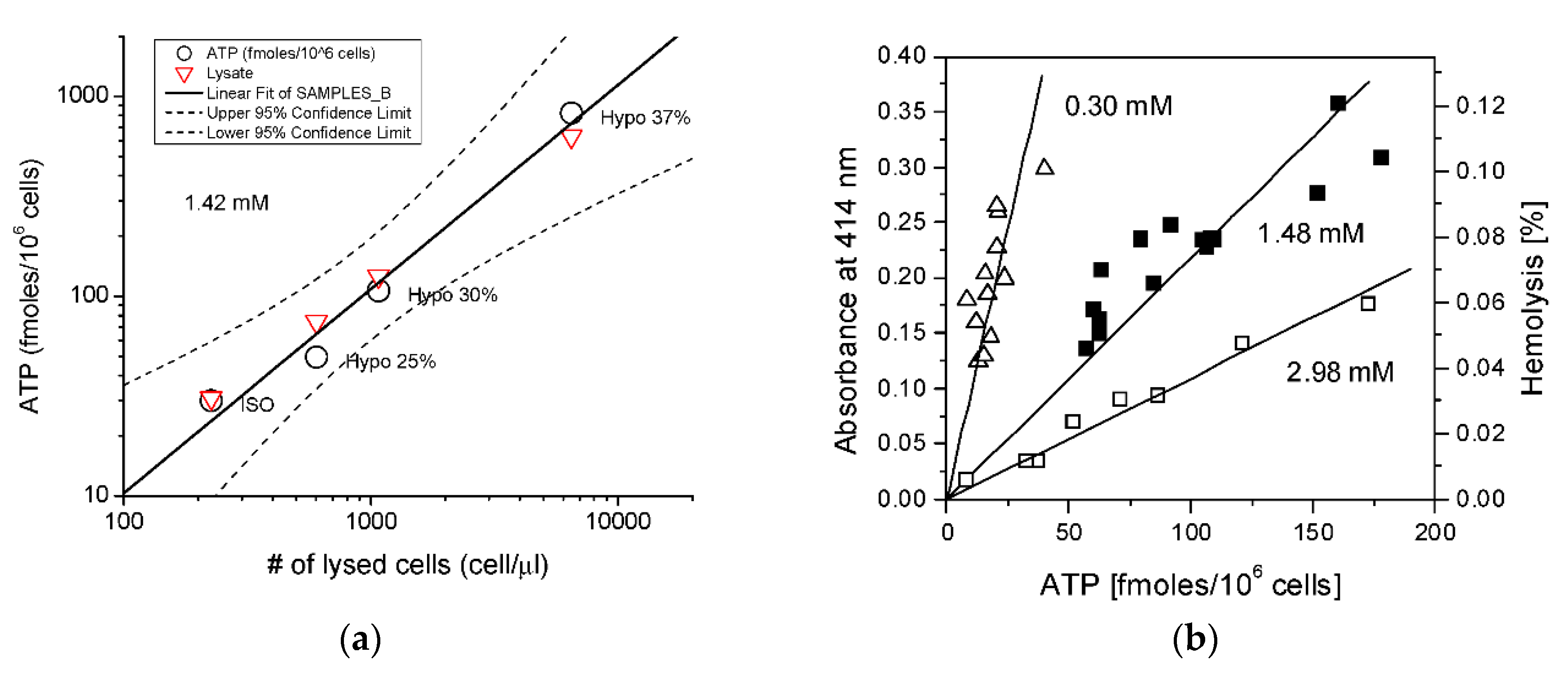
3. Hemolytic ATP Release in RBCs
3.1. Contribution of Hemolysis to ATP Release
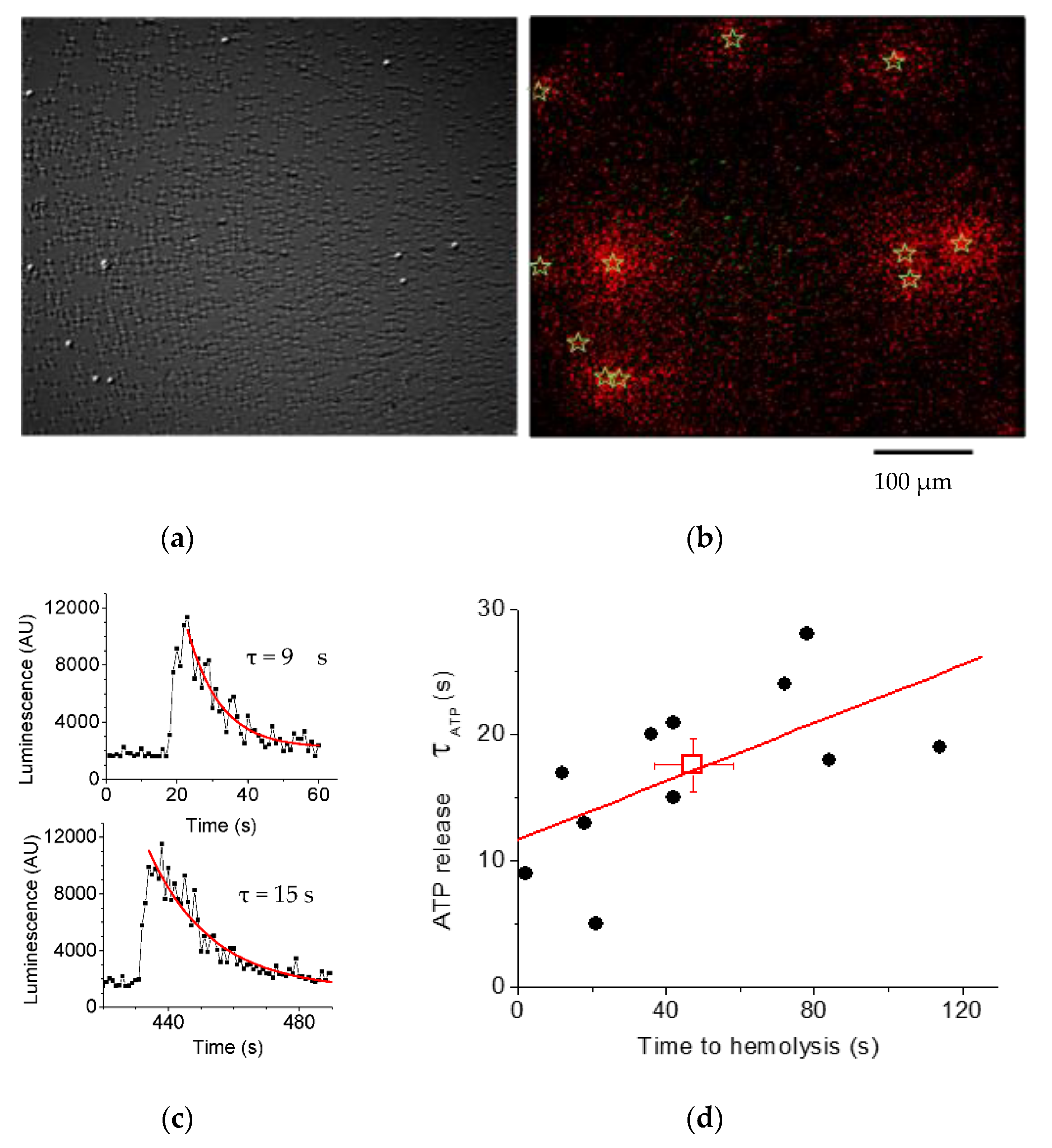
3.2. Luminescence Imaging Demonstrates ATP Release Exclusively from Lysing Cells
4. Ultrasound and Microbubble-Induced ATP Release for Enhancing Cancer Immunotherapy
4.1. Microbubbles for Imaging and Therapy
4.2. Sonoporation
4.3. Beyond Sonoporation
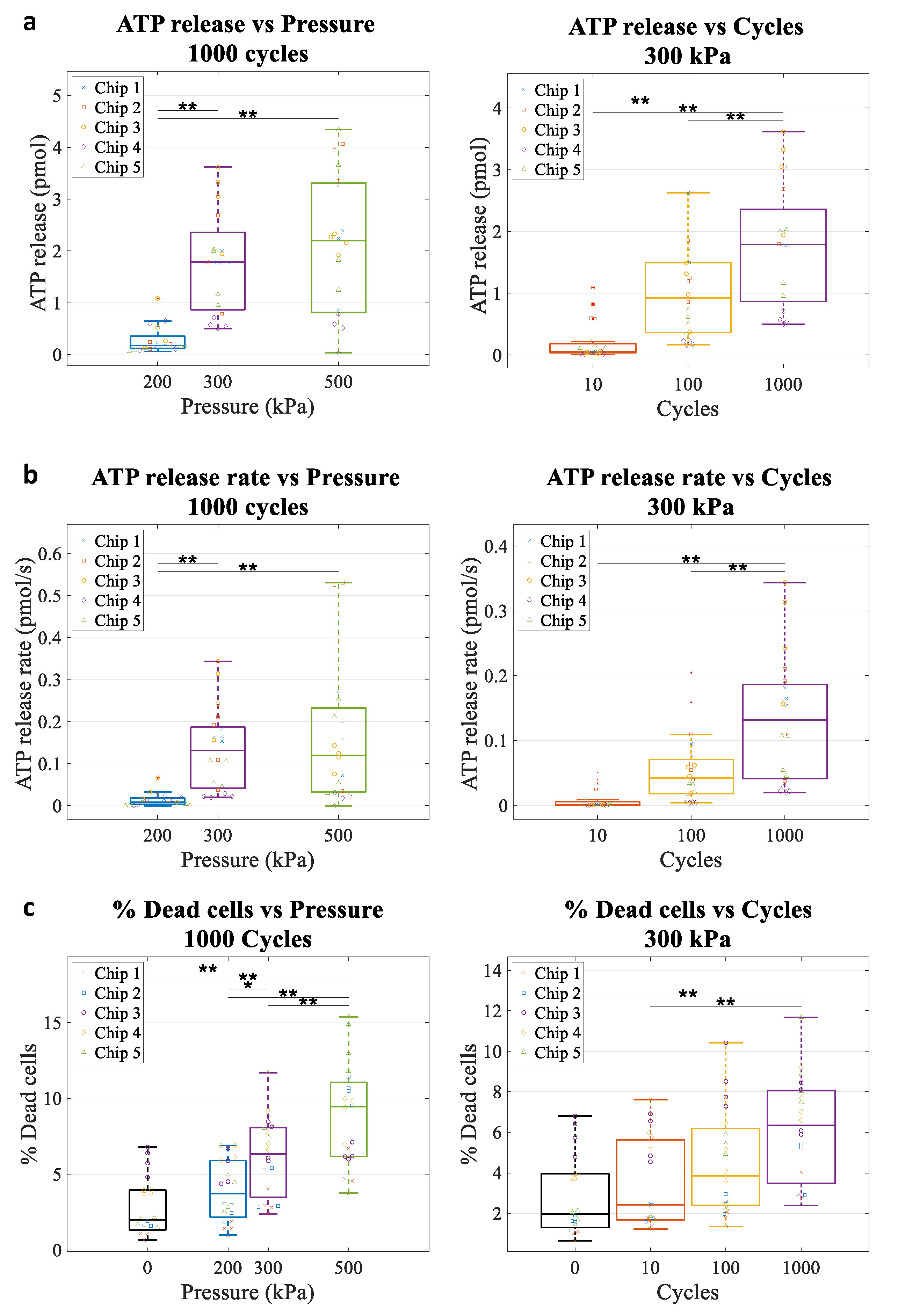
4.4. Microbubble-Driven ATP Release
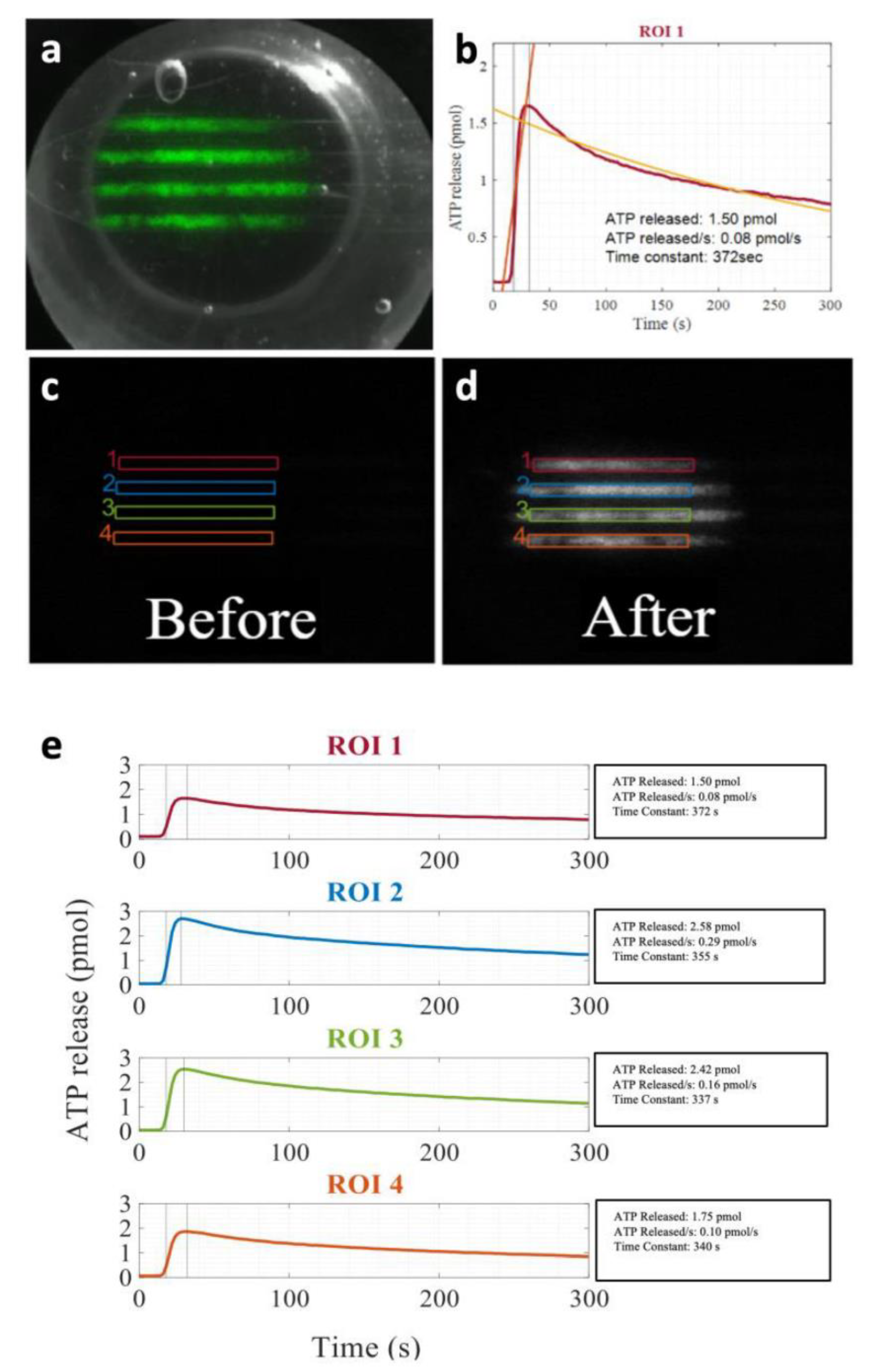
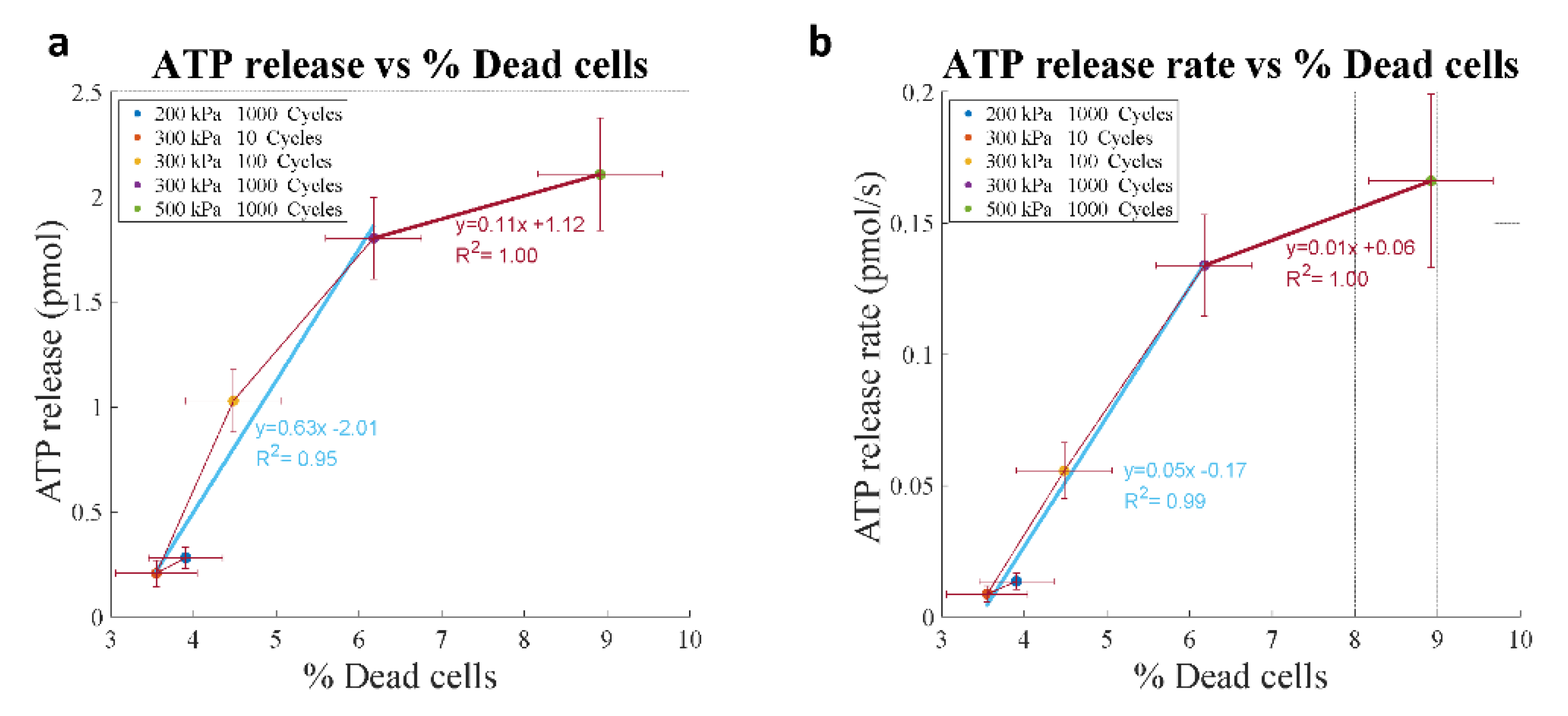
4.5. Kinetics of ATP Release In Vitro
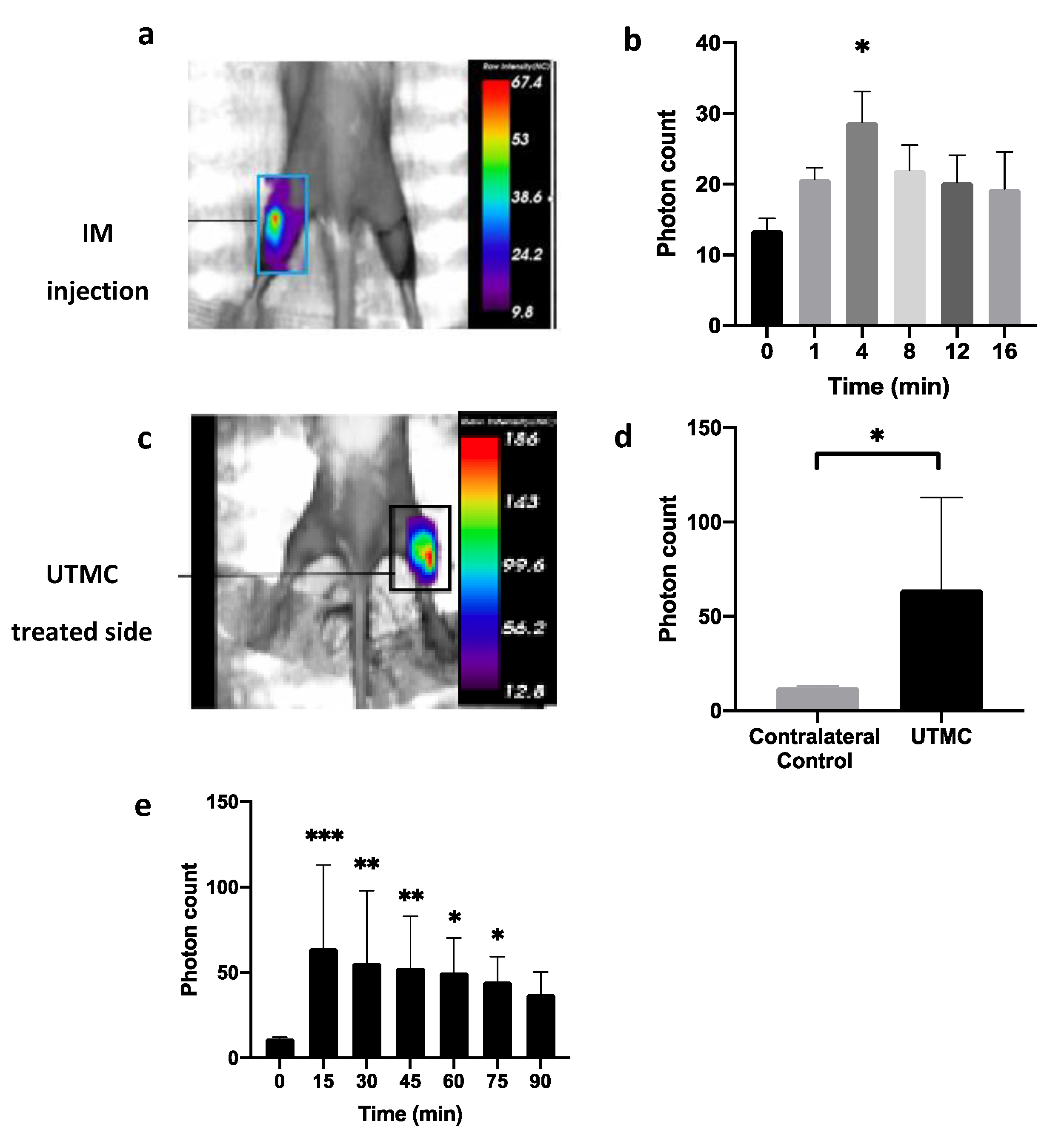
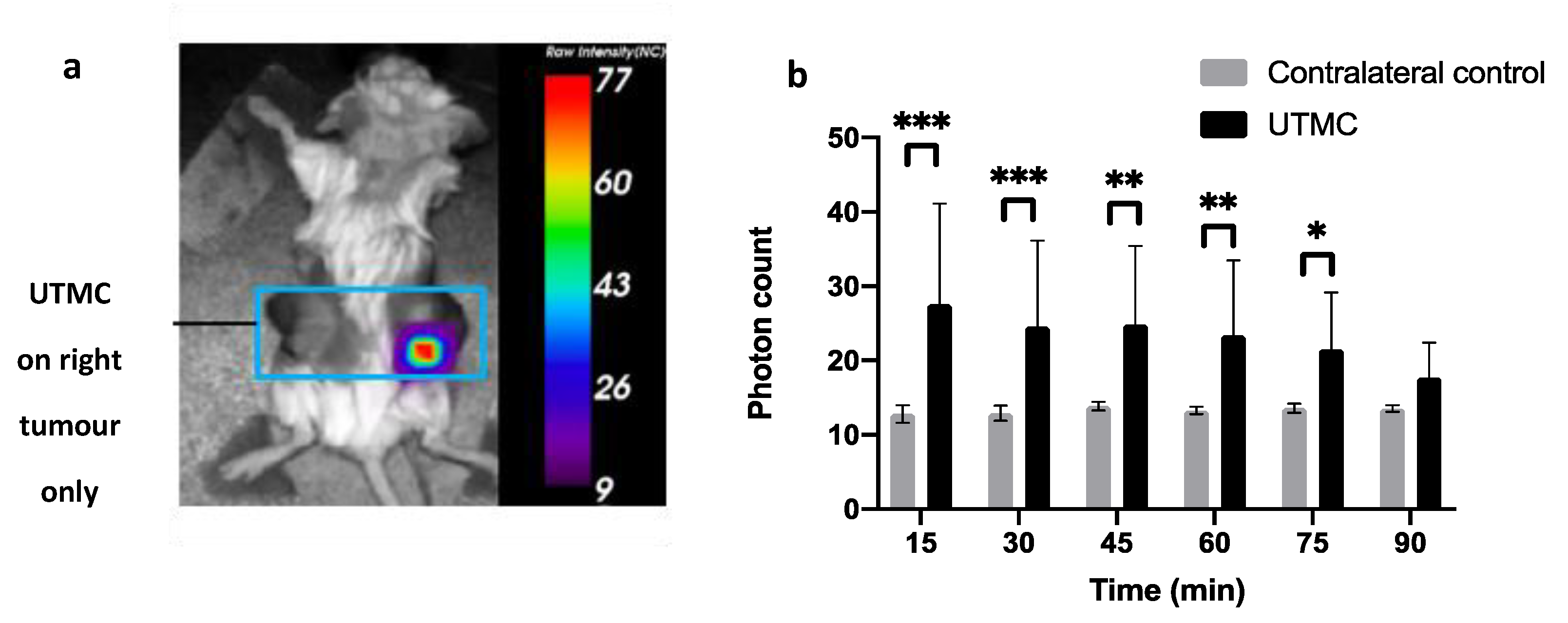
4.6. ATP Release In Vivo in Muscle and in Tumors
5. Concluding Remarks
6. Methods
6.1. Methods for Section 2
6.2. Methods for Section 4
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zimmermann, H. Extracellular ATP and other nucleotides—Ubiquitous triggers of intercellular messenger release. Purinergic Signal 2015, 12, 25–57. [Google Scholar] [CrossRef]
- Mikolajewicz, N.; Mohammed, A.; Morris, M.; Komarova, S.V. Mechanically-stimulated ATP release from mammalian cells: Systematic review and meta-analysis. J. Cell Sci. 2018, 131, jcs.223354. [Google Scholar] [CrossRef] [Green Version]
- Lazarowski, E.R. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal 2012, 8, 359–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praetorius, H.A.; Leipziger, J. ATP release from non-excitable cells. Purinergic Signal 2009, 5, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Locovei, S.; Bao, L.; Dahl, G. Pannexin 1 in erythrocytes: Function without a gap. Proc. Natl. Acad. Sci. USA 2006, 103, 7655–7659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.-H.; Ravichandran, K.S.; Bayliss, D.A. Intrinsic properties and regulation of Pannexin 1 channel. Channels 2014, 8, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taruno, A. ATP Release Channels. Int. J. Mol. Sci. 2018, 19, 808. [Google Scholar] [CrossRef] [Green Version]
- Taruno, A.; Vingtdeux, V.; Ohmoto, M.; Ma, Z.; Dvoryanchikov, G.; Li, A.; Adrien, L.; Zhao, H.; Leung, S.; Abernethy, M.; et al. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 2013, 495, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Cotrina, M.L.; Lin, J.H.-C.; Alves-Rodrigues, A.; Liu, S.; Li, J.; Azmi-Ghadimi, H.; Kang, J.; Naus, C.C.G.; Nedergaard, M. Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. USA 1998, 95, 15735–15740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, M.M.; Quinton, P.M.; Haws, C.; Wine, J.J.; Grygorczyk, R.; Tabcharani, J.A.; Hanrahan, J.W.; Gunderson, K.L.; Kopito, R.R. Failure of the Cystic Fibrosis Transmembrane Conductance Regulator to Conduct ATP. Science 1996, 271, 1876–1879. [Google Scholar] [CrossRef]
- Boudreault, F.; Grygorczyk, R. Cell swelling-induced ATP release and gadolinium-sensitive channels. Am. J. Physiol. Cell Physiol. 2002, 282, C219–C226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Okada, T.; Islam, R.; Sabirov, R. Molecular Identities and ATP Release Activities of Two Types of Volume-Regulatory Anion Channels, VSOR and Maxi-Cl. Curr. Top. Membr. 2018, 81, 125–176. [Google Scholar] [CrossRef]
- Matsuura, H.; Kojima, A.; Fukushima, Y.; Xie, Y.; Mi, X.; Sabirov, R.Z.; Okada, Y. Positive Inotropic Effects of ATP Released via the Maxi-Anion Channel in Langendorff-Perfused Mouse Hearts Subjected to Ischemia-Reperfusion. Front. Cell Dev. Biol. 2021, 9, 597997. [Google Scholar] [CrossRef] [PubMed]
- Mim, C.; Perkins, G.; Dahl, G. Structure versus function: Are new conformations of pannexin 1 yet to be resolved? J. Gen. Physiol. 2021, 153. [Google Scholar] [CrossRef]
- Tan, J.J.; Ponomarchuk, O.; Grygorczyk, R.; Boudreault, F. Wide field of view quantitative imaging of cellular ATP release. Am. J. Physiol. Cell Physiol. 2019, 317, C566–C575. [Google Scholar] [CrossRef]
- Grygorczyk, R.; Boudreault, F.; Tan, J.J.; Ponomarchuk, O.; Sokabe, M.; Furuya, K. Mechanosensitive ATP release in the lungs: New insights from real-time luminescence imaging studies. Curr. Top. Membr. 2019, 83, 45–76. [Google Scholar] [CrossRef] [PubMed]
- Ponomarchuk, O.; Boudreault, F.; Orlov, S.N.; Grygorczyk, R. Calcium is not required for triggering volume restoration in hypotonically challenged A549 epithelial cells. Pflügers Archiv. 2016, 468, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, F.; Grygorczyk, R. Cell swelling-induced ATP release is tightly dependent on intracellular calcium elevations. J. Physiol. 2004, 561, 499–513. [Google Scholar] [CrossRef]
- Halter, M.; Elliott, J.T.; Hubbard, J.B.; Tona, A.; Plant, A.L. Cell volume distributions reveal cell growth rates and division times. J. Theor. Biol. 2009, 257, 124–130. [Google Scholar] [CrossRef]
- Boudreault, F.; Grygorczyk, R. Evaluation of rapid volume changes of substrate-adherent cells by conventional microscopy 3D imaging. J. Microsc. 2004, 215, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, T.; Ogawa, R.; Feril, L.B.; Kagiya, G.; Fuse, H.; Kondo, T. Enhancement of ultrasound-mediated gene transfection by membrane modification. J. Gene Med. 2003, 5, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Bernecker, C.; Köfeler, H.; Pabst, G.; Trötzmüller, M.; Kolb, D.; Strohmayer, K.; Trajanoski, S.; Holzapfel, G.A.; Schlenke, P.; Dorn, I. Cholesterol Deficiency Causes Impaired Osmotic Stability of Cultured Red Blood Cells. Front. Physiol. 2019, 10, 1529. [Google Scholar] [CrossRef] [PubMed]
- Furuya, K.; Sokabe, M.; Grygorczyk, R. Real-time luminescence imaging of cellular ATP release. Methods 2014, 66, 330–344. [Google Scholar] [CrossRef]
- Feranchak, A.P.; Lewis, M.A.; Kresge, C.; Sathe, M.; Bugde, A.; Luby-Phelps, K.; Antich, P.P.; Fitz, J.G. Initiation of Purinergic Signaling by Exocytosis of ATP-containing Vesicles in Liver Epithelium. J. Biol. Chem. 2010, 285, 8138–8147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, H.H.; Ellsworth, M.L.; Sprague, R.S.; Dacey, R.G. Red blood cell regulation of microvascular tone through adenosine triphosphate. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1294–H1298. [Google Scholar] [CrossRef]
- Ellsworth, M.L.; Sprague, R.S. Regulation of blood flow distribution in skeletal muscle: Role of erythrocyte-released ATP. J. Physiol. 2012, 590, 4985–4991. [Google Scholar] [CrossRef]
- Ellsworth, M.L.; Forrester, T.; Ellis, C.; Dietrich, H.H. The erythrocyte as a regulator of vascular tone. Am. J. Physiol. 1995, 269, H2155–H2161. [Google Scholar] [CrossRef]
- Forrester, T. An estimate of adenosine triphosphate release into the venous effluent from exercising human forearm muscle. J. Physiol. 1972, 224, 611–628. [Google Scholar] [CrossRef] [Green Version]
- González-Alonso, J.; Olsen, D.B.; Saltin, B. Erythrocyte and the Regulation of Human Skeletal Muscle Blood Flow and Oxygen Delivery: Role of circulating ATP. Circ. Res. 2002, 91, 1046–1055. [Google Scholar] [CrossRef] [Green Version]
- Kalsi, K.K.; Chiesa, S.T.; Trangmar, S.J.; Ali, L.; Lotlikar, M.D.; González-Alonso, J. Mechanisms for the control of local tissue blood flow during thermal interventions: Influence of temperature-dependent ATP release from human blood and endothelial cells. Exp. Physiol. 2017, 102, 228–244. [Google Scholar] [CrossRef]
- González-Alonso, J.; Kalsi, K.K. The ubiquitous ATP molecule: Could it be the elusive thermal mediator igniting skin perfusion and sweating in the heat-stressed human? J. Physiol. 2015, 593, 2399. [Google Scholar] [CrossRef] [Green Version]
- Grygorczyk, R.; Orlov, S.N. Effects of Hypoxia on Erythrocyte Membrane Properties—Implications for Intravascular Hemolysis and Purinergic Control of Blood Flow. Front. Physiol. 2017, 8, 1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprague, R.S.; Ellsworth, M.L.; Stephenson, A.H.; Kleinhenz, M.E.; Lonigro, A.J. Deformation-induced ATP release from red blood cells requires CFTR activity. Am. J. Physiol. 1998, 275, H1726–H1732. [Google Scholar] [CrossRef]
- Sridharan, M.; Bowles, E.A.; Richards, J.P.; Krantic, M.; Davis, K.L.; Dietrich, K.A.; Stephenson, A.H.; Ellsworth, M.L.; Sprague, R.S. Prostacyclin receptor-mediated ATP release from erythrocytes requires the voltage-dependent anion channel. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H553–H559. [Google Scholar] [CrossRef]
- Sridharan, M.; Adderley, S.P.; Bowles, E.A.; Egan, T.; Stephenson, A.H.; Ellsworth, M.L.; Sprague, R.S. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1146–H1152. [Google Scholar] [CrossRef] [Green Version]
- Shaskey, D.J.; Green, G.A. Sports Haematology. Sports Med. 2000, 29, 27–38. [Google Scholar] [CrossRef]
- Mao, T.-Y.; Fu, L.-L.; Wang, J.-S. Hypoxic exercise training causes erythrocyte senescence and rheological dysfunction by depressed Gardos channel activity. J. Appl. Physiol. 2011, 111, 382–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mairbäurl, H.; Ruppe, F.A.; Bärtsch, P. Role of Hemolysis in Red Cell Adenosine Triphosphate Release in Simulated Exercise Conditions In Vitro. Med. Sci. Sports Exerc. 2013, 45, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Sikora, J.; Orlov, S.N.; Furuya, K.; Grygorczyk, R. Hemolysis is a primary ATP-release mechanism in human erythrocytes. Blood 2014, 124, 2150–2157. [Google Scholar] [CrossRef] [Green Version]
- Huisjes, R.; Bogdanova, A.; Van Solinge, W.; Schiffelers, R.; Kaestner, L.; Van Wijk, R. Squeezing for Life—Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.L.Y. Intravascular hemolysis: The sacrifice of few…. Blood 2014, 124, 2011–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, A.W.; Gill, D.M.; Pal, S.K.; Agarwal, N. The future of immune checkpoint cancer therapy after PD-1 and CTLA-4. Immunotherapy 2017, 9, 681–692. [Google Scholar] [CrossRef]
- Kourie, H.R.; Awada, G.G.; Awada, A. The second wave of immune checkpoint inhibitor tsunami: Advance, challenges and perspectives. Immunotherapy 2017, 9, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef]
- Allard, D.; Allard, B.; Stagg, J. On the mechanism of anti-CD39 immune checkpoint therapy. J. Immunother. Cancer 2019, 8, e000186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vultaggio-Poma, V.; Sarti, A.C.; Di Virgilio, F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells 2020, 9, 2496. [Google Scholar] [CrossRef]
- Campos-Contreras, A.D.R.; Díaz-Muñoz, M.; Vázquez-Cuevas, F.G. Purinergic Signaling in the Hallmarks of Cancer. Cells 2020, 9, 1612. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [Green Version]
- Allard, D.; Chrobak, P.; Allard, B.; Messaoudi, N.; Stagg, J. Targeting the CD73-adenosine axis in immuno-oncology. Immunol. Lett. 2019, 205, 31–39. [Google Scholar] [CrossRef]
- Yu, F.T.H.; Chen, X.; Wang, J.; Qin, B.; Villanueva, F.S. Low Intensity Ultrasound Mediated Liposomal Doxorubicin Delivery Using Polymer Microbubbles. Mol. Pharm. 2016, 13, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Xu, Y.; Gao, C.; Zhou, Y.; Zhang, N.; Dai, Z. Ultrasound contrast agent microbubbles with ultrahigh loading capacity of camptothecin and floxuridine for enhancing tumor accumulation and combined chemotherapeutic efficacy. NPG Asia Mater. 2018, 10, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Amate, M.; Goldgewicht, J.; Sellamuthu, B.; Stagg, J.; Yu, F.T. The effect of ultrasound pulse length on microbubble cavitation induced antibody accumulation and distribution in a mouse model of breast cancer. Nanotheranostics 2020, 4, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-L.; Hsieh, H.-Y.; Lu, L.-A.; Kang, C.-W.; Wu, M.-F.; Lin, C.-Y. Low-pressure pulsed focused ultrasound with microbubbles promotes an anticancer immunological response. J. Transl. Med. 2012, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulner, S.; Prodeus, A.; Gariepy, J.; Hynynen, K.; Goertz, D.E. Enhancing Checkpoint Inhibitor Therapy with Ultrasound Stimulated Microbubbles. Ultrasound Med. Biol. 2019, 45, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Tang, J.; Yang, J.; Zhu, B.; Wang, X.; Luo, Y.; Yang, H.; Jang, F.; Zou, J.; Liu, Z.; et al. Tumor perfusion enhancement by ultrasound stimulated microbubbles potentiates PD-L1 blockade of MC38 colon cancer in mice. Cancer Lett. 2021, 498, 121–129. [Google Scholar] [CrossRef]
- Belcik, J.T.; Davidson, B.P.; Xie, A.; Wu, M.D.; Yadava, M.; Qi, Y.; Liang, S.; Chon, C.R.; Ammi, A.Y.; Field, J.; et al. Augmentation of Muscle Blood Flow by Ultrasound Cavitation Is Mediated by ATP and Purinergic Signaling. Circulation 2017, 135, 1240–1252. [Google Scholar] [CrossRef] [Green Version]
- Hauben, M.; Hung, E.Y.; Hanretta, K.C.; Bangalore, S.; Snow, V. Safety of Perflutren Ultrasound Contrast Agents: A Disproportionality Analysis of the US FAERS Database. Drug Saf. 2015, 38, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Tse, W.-K.; Leung, P.T. Theory of light emission in sonoluminescence as thermal radiation. Phys. Rev. E 2006, 73, 056302. [Google Scholar] [CrossRef] [Green Version]
- Mathias, W.; Tsutsui, J.M.; Tavares, B.G.; Fava, A.M.; Aguiar, M.O.; Borges, B.C.; Oliveira, M.T.; Soeiro, A.; Nicolau, J.; Ribeiro, H.B.; et al. Sonothrombolysis in ST-Segment Elevation Myocardial Infarction Treated with Primary Percutaneous Coronary Intervention. J. Am. Coll. Cardiol. 2019, 73, 2832–2842. [Google Scholar] [CrossRef]
- Carson, A.R.; McTiernan, C.F.; Lavery, L.; Grata, M.; Leng, X.; Wang, J.; Chen, X.; Villanueva, F.S. Ultrasound-Targeted Microbubble Destruction to Deliver siRNA Cancer Therapy. Cancer Res. 2012, 72, 6191–6199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Shimoda, M.; Wang, M.-Y.; Ding, J.; Noguchi, H.; Matsumoto, S.; Grayburn, P.A. Regeneration of pancreatic islets in vivo by ultrasound-targeted gene therapy. Gene Therapy 2010, 17, 1411–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotopoulis, S.; Dimcevski, G.; Gilja, O.H.; Hoem, D.; Postema, M. Treatment of human pancreatic cancer using combined ultrasound, microbubbles, and gemcitabine: A clinical case study. Med Phys. 2013, 40, 072902. [Google Scholar] [CrossRef] [PubMed]
- Kopechek, J.A.; Carson, A.R.; McTiernan, C.F.; Chen, X.; Klein, E.C.; Villanueva, F.S. Cardiac Gene Expression Knockdown Using Small Inhibitory RNA-Loaded Microbubbles and Ultrasound. PLoS ONE 2016, 11, e0159751. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Pacella, J.J.; Villanueva, F.S. Dynamic Behavior of Microbubbles during Long Ultrasound Tone-Burst Excitation: Mechanistic Insights into Ultrasound-Microbubble Mediated Therapeutics Using High-Speed Imaging and Cavitation Detection. Ultrasound Med. Biol. 2016, 42, 528–538. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Leeman, J.E.; Wang, J.; Pacella, J.J.; Villanueva, F.S. New Insights into Mechanisms of Sonothrombolysis Using Ultra-High-Speed Imaging. Ultrasound Med. Biol. 2014, 40, 258–262. [Google Scholar] [CrossRef]
- Yu, F.T.; Chen, X.; Straub, A.C.; Pacella, J.J. The Role of Nitric Oxide during Sonoreperfusion of Microvascular Obstruction. Theranostics 2017, 7, 3527–3538. [Google Scholar] [CrossRef]
- Abrahao, A.; Meng, Y.; Llinas, M.; Huang, Y.; Hamani, C.; Mainprize, T.; Aubert, I.; Heyn, C.; Black, S.E.; Hynynen, K.; et al. First-in-human trial of blood–brain barrier opening in amyotrophic lateral sclerosis using MR-guided focused ultrasound. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Deprez, J.; Lajoinie, G.; Engelen, Y.; De Smedt, S.; Lentacker, I. Opening doors with ultrasound and microbubbles: Beating biological barriers to promote drug delivery. Adv. Drug Deliv. Rev. 2021, 172, 9–36. [Google Scholar] [CrossRef]
- Kooiman, K.; Roovers, S.; Langeveld, S.A.; Kleven, R.T.; Dewitte, H.; O’Reilly, M.A.; Escoffre, J.-M.; Bouakaz, A.; Verweij, M.D.; Hynynen, K.; et al. Ultrasound-Responsive Cavitation Nuclei for Therapy and Drug Delivery. Ultrasound Med. Biol. 2020, 46, 1296–1325. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; E Kumon, R.; Deng, C.X. Mechanisms of microbubble-facilitated sonoporation for drug and gene delivery. Ther. Deliv. 2014, 5, 467–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfield, B.; Chen, X.; Watkins, S.; Villanueva, F.S. Biophysical insight into mechanisms of sonoporation. Proc. Natl. Acad. Sci. USA 2016, 113, 9983–9988. [Google Scholar] [CrossRef] [Green Version]
- Beekers, I.; Vegter, M.; Lattwein, K.R.; Mastik, F.; Beurskens, R.; Van Der Steen, A.F.; De Jong, N.; Verweij, M.D.; Kooiman, K. Opening of endothelial cell–cell contacts due to sonoporation. J. Control. Release 2020, 322, 426–438. [Google Scholar] [CrossRef]
- Karshafian, R.; Bevan, P.D.; Williams, R.; Samac, S.; Burns, P.N. Sonoporation by Ultrasound-Activated Microbubble Contrast Agents: Effect of Acoustic Exposure Parameters on Cell Membrane Permeability and Cell Viability. Ultrasound Med. Biol. 2009, 35, 847–860. [Google Scholar] [CrossRef]
- Hu, Y.; Wan, J.M.; Yu, A.C.H. Membrane Perforation and Recovery Dynamics in Microbubble-Mediated Sonoporation. Ultrasound Med. Biol. 2013, 39, 2393–2405. [Google Scholar] [CrossRef] [PubMed]
- Kumon, R.; Aehle, M.; Sabens, D.; Parikh, P.; Han, Y.; Kourennyi, D.; Deng, C. Spatiotemporal Effects of Sonoporation Measured by Real-Time Calcium Imaging. Ultrasound Med. Biol. 2009, 35, 494–506. [Google Scholar] [CrossRef] [Green Version]
- van Rooij, T.; Skachkov, I.; Beekers, I.; Lattwein, K.R.; Voorneveld, J.D.; Kokhuis, T.J.; Bera, D.; Luan, Y.; van der Steen, A.F.; de Jong, N.; et al. Viability of endothelial cells after ultrasound-mediated sonoporation: Influence of targeting, oscillation, and displacement of microbubbles. J. Control. Release 2016, 238, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Haugse, R.; Langer, A.; Murvold, E.; Costea, D.; Gjertsen, B.; Gilja, O.; Kotopoulis, S.; De Garibay, G.R.; McCormack, E. Low-Intensity Sonoporation-Induced Intracellular Signalling of Pancreatic Cancer Cells, Fibroblasts and Endothelial Cells. Pharmaceutics 2020, 12, 1058. [Google Scholar] [CrossRef] [PubMed]
- Price, R.J.; Skyba, D.M.; Kaul, S.; Skalak, T.C. Delivery of Colloidal Particles and Red Blood Cells to Tissue Through Microvessel Ruptures Created by Targeted Microbubble Destruction with Ultrasound. Circulation 1998, 98, 1264–1267. [Google Scholar] [CrossRef] [Green Version]
- Everbach, E.C.; Makin, I.R.; Azadniv, M.; Meltzer, R.S. Correlation of ultrasound-induced hemolysis with cavitation detector output in vitro. Ultrasound Med. Biol. 1997, 23, 619–624. [Google Scholar] [CrossRef]
- Sun, T.; Samiotaki, G.; Wang, S.; Acosta, C.; Chen, C.C.; Konofagou, E.E. Acoustic cavitation-based monitoring of the reversibility and permeability of ultrasound-induced blood-brain barrier opening. Phys. Med. Biol. 2015, 60, 9079–9094. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Liu, H.; Mayer, M.; Deng, C.X. Spatiotemporally controlled single cell sonoporation. Proc. Natl. Acad. Sci. USA 2012, 109, 16486–16491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, P.; Xu, L.; Hu, Y.; Zhong, W.; Cai, P.; Du, L.; Jin, L.; Yu, A.C. Sonoporation-Induced Depolarization of Plasma Membrane Potential: Analysis of Heterogeneous Impact. Ultrasound Med. Biol. 2014, 40, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Leow, R.S.; Hu, Y.; Wan, J.M.F.; Yu, A.C.H. Single-site sonoporation disrupts actin cytoskeleton organization. J. R. Soc. Interface 2014, 11, 20140071. [Google Scholar] [CrossRef] [Green Version]
- Leow, R.S.; Wan, J.M.F.; Yu, A.C.H. Membrane blebbing as a recovery manoeuvre in site-specific sonoporation mediated by targeted microbubbles. J. R. Soc. Interface 2015, 12, 20150029. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Zhou, Q.; Wan, J.M.F.; Yu, A.C.H. Sonoporation generates downstream cellular impact after membrane resealing. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zeghimi, A.; Escoffre, J.M.; Bouakaz, A. Role of endocytosis in sonoporation-mediated membrane permeabilization and uptake of small molecules: A electron microscopy study. Phys. Biol. 2015, 12, 066007. [Google Scholar] [CrossRef] [Green Version]
- Meijering, B.D.M.; Juffermans, L.J.M.; van Wamel, A.; Henning, R.; Zuhorn, I.; Emmer, M.; Versteilen, A.M.G.; Paulus, W.J.; van Gilst, W.; Kooiman, K.; et al. Ultrasound and Microbubble-Targeted Delivery of Macromolecules Is Regulated by Induction of Endocytosis and Pore Formation. Circ. Res. 2009, 104, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Carson, A.R.; McTiernan, C.F.; Lavery, L.; Hodnick, A.; Grata, M.; Leng, X.; Wang, J.; Chen, X.; Modzelewski, R.A.; Villanueva, F.S. Gene Therapy of Carcinoma Using Ultrasound-Targeted Microbubble Destruction. Ultrasound Med. Biol. 2011, 37, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, Z.I.; Burks, S.R.; Frank, J.A. Focused ultrasound with microbubbles induces sterile inflammatory response proportional to the blood brain barrier opening: Attention to experimental conditions. Theranostics 2018, 8, 2245–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcik, J.T.; Mott, B.H.; Xie, A.; Zhao, Y.; Kim, S.; Lindner, N.J.; Ammi, A.; Linden, J.M.; Lindner, J.R. Augmentation of Limb Perfusion and Reversal of Tissue Ischemia Produced by Ultrasound-Mediated Microbubble Cavitation. Circ. Cardiovasc. Imaging 2015, 8, e002979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccetti, F.; Belcik, T.; Latifi, Y.; Xie, A.; Ozawa, K.; Brown, E.; Davidson, B.P.; Packwood, W.; Ammi, A.; Huke, S.; et al. Flow Augmentation in the Myocardium by Ultrasound Cavitation of Microbubbles: Role of Shear-Mediated Purinergic Signaling. J. Am. Soc. Echocardiogr. 2020, 33, 1023–1031.e2. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grygorczyk, R.; Boudreault, F.; Ponomarchuk, O.; Tan, J.J.; Furuya, K.; Goldgewicht, J.; Kenfack, F.D.; Yu, F. Lytic Release of Cellular ATP: Physiological Relevance and Therapeutic Applications. Life 2021, 11, 700. https://doi.org/10.3390/life11070700
Grygorczyk R, Boudreault F, Ponomarchuk O, Tan JJ, Furuya K, Goldgewicht J, Kenfack FD, Yu F. Lytic Release of Cellular ATP: Physiological Relevance and Therapeutic Applications. Life. 2021; 11(7):700. https://doi.org/10.3390/life11070700
Chicago/Turabian StyleGrygorczyk, Ryszard, Francis Boudreault, Olga Ponomarchuk, Ju Jing Tan, Kishio Furuya, Joseph Goldgewicht, Falonne Démèze Kenfack, and François Yu. 2021. "Lytic Release of Cellular ATP: Physiological Relevance and Therapeutic Applications" Life 11, no. 7: 700. https://doi.org/10.3390/life11070700