
Functional Effects In Silico Prediction for Androgen Receptor Ligand-Binding Domain Novel I836S Mutation

 and
and 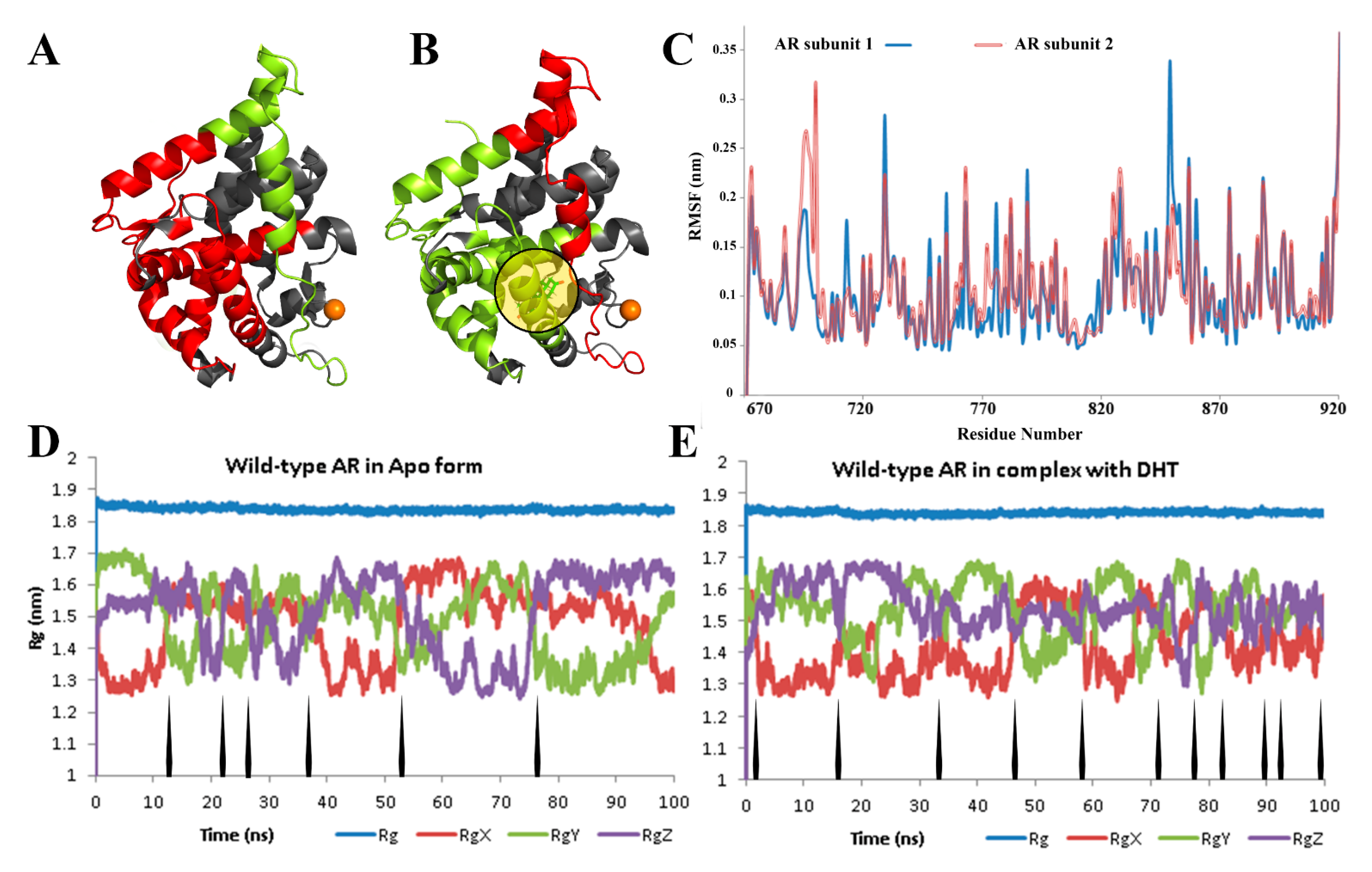{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
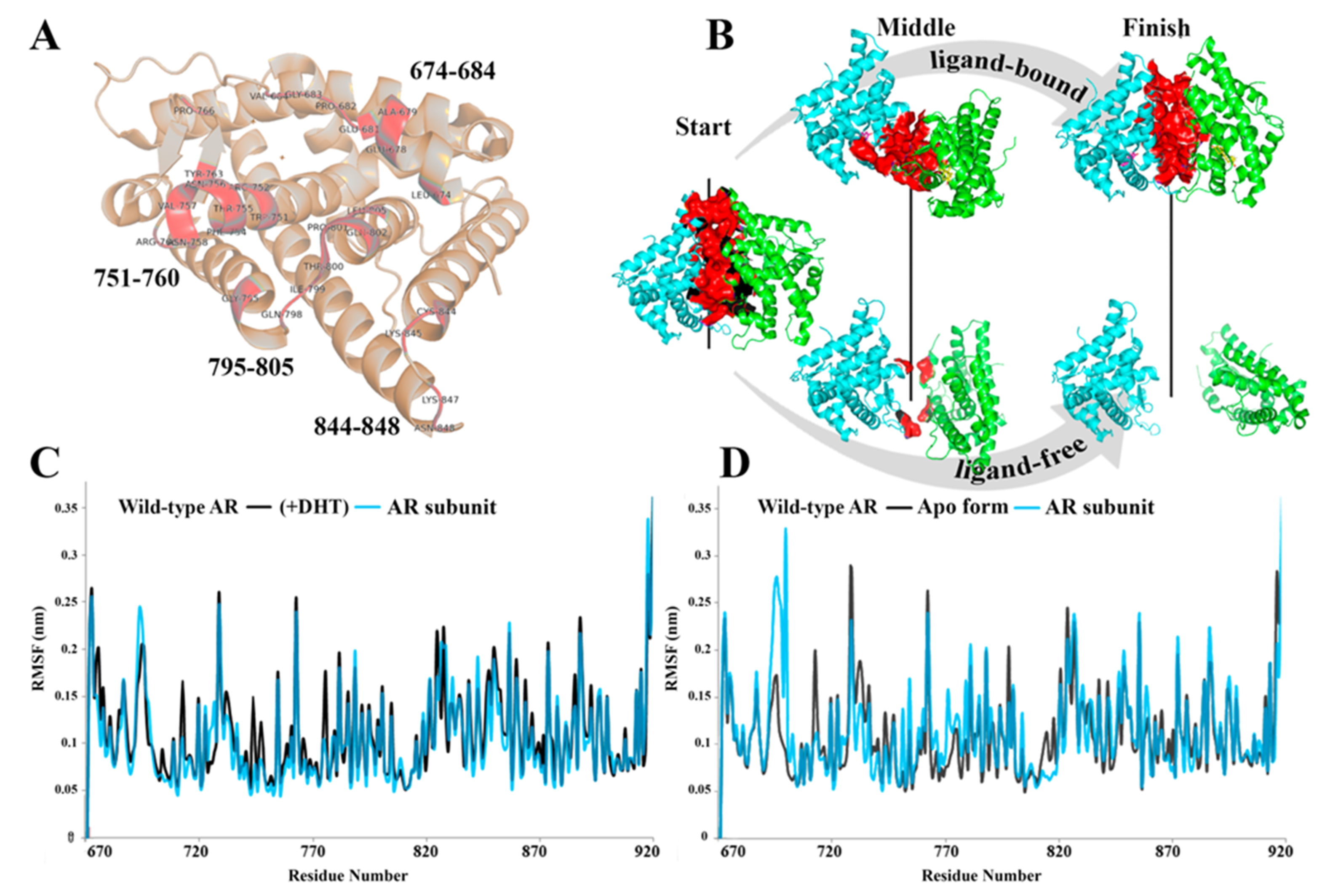
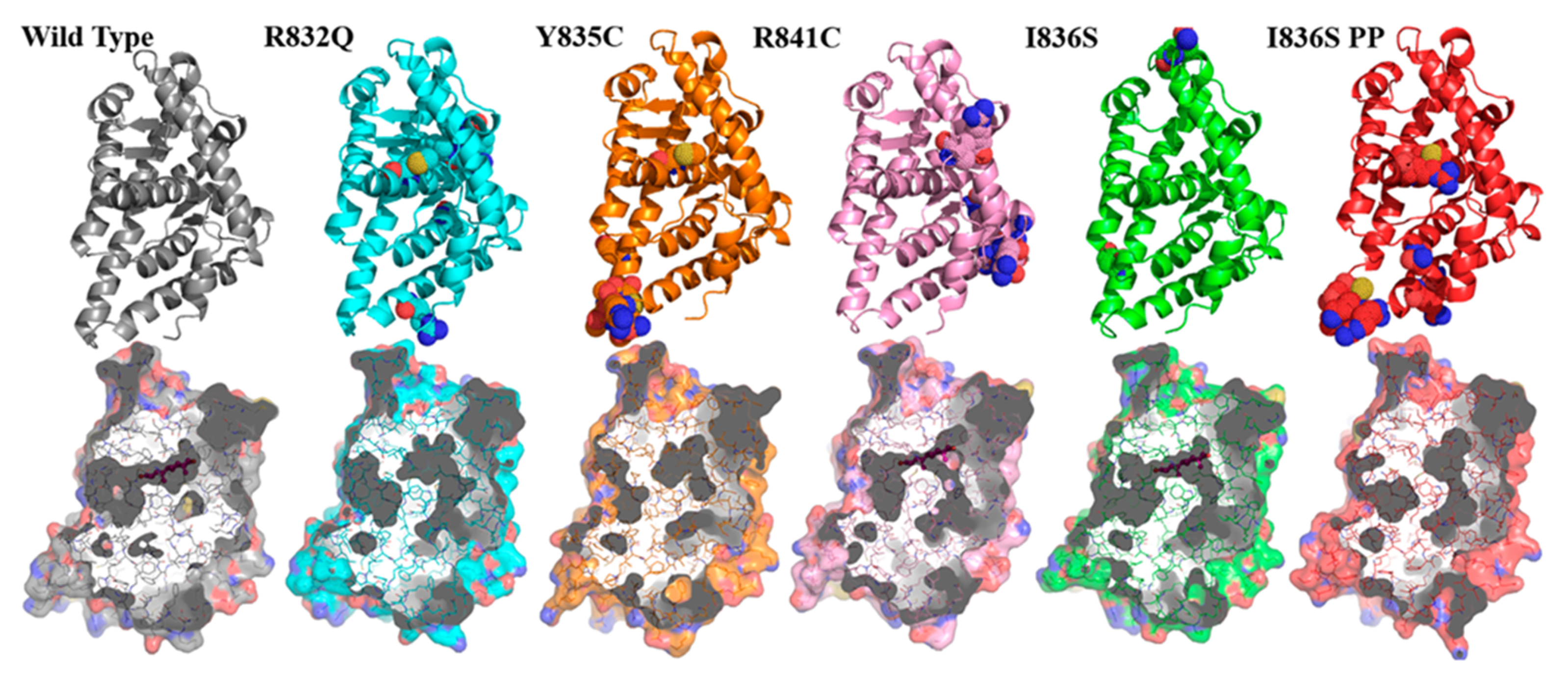
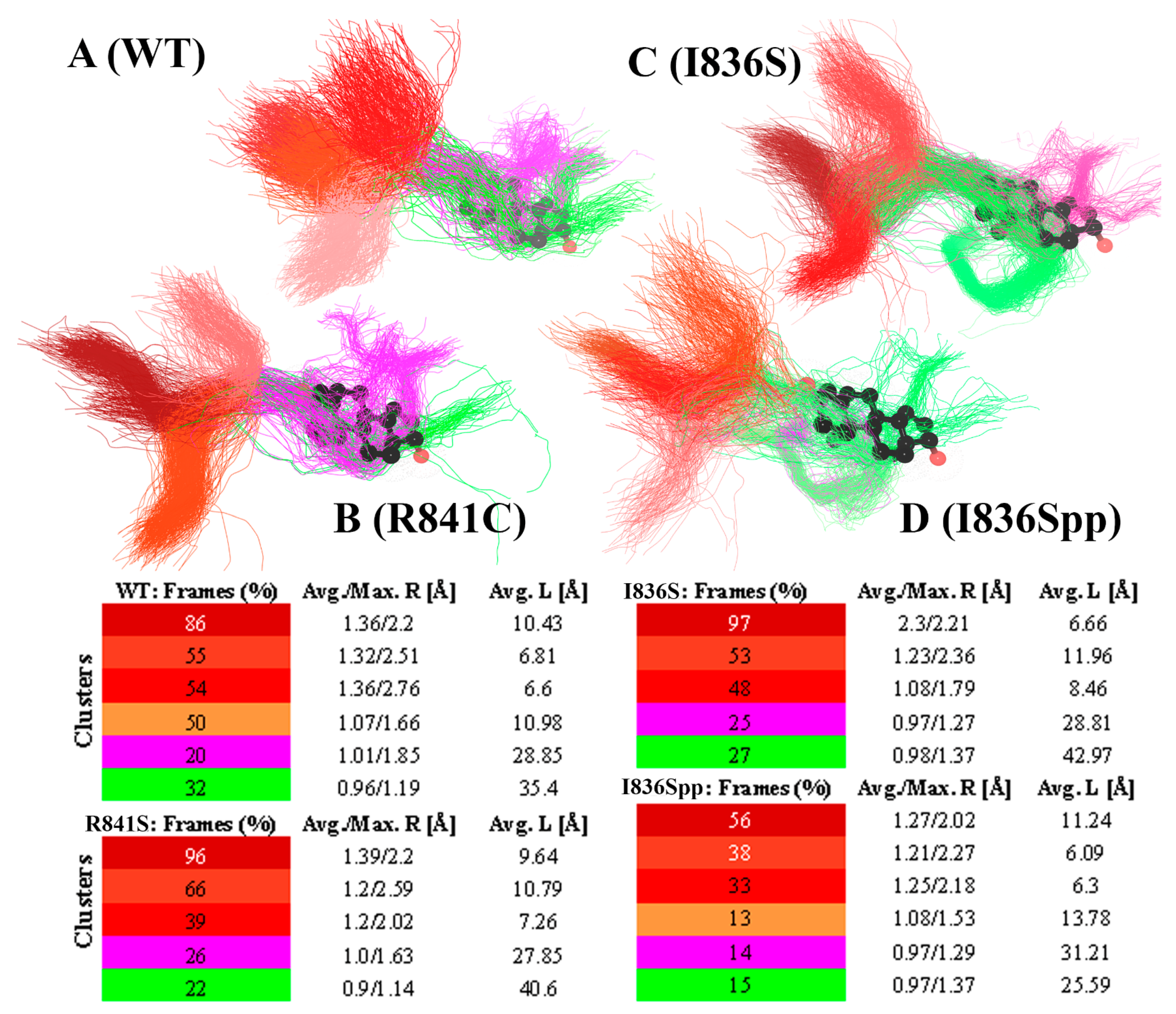
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oakes, M.B.; Eyvazzadeh, A.D.; Quint, E.; Smith, Y.R. Complete androgen insensitivity syndrome—A review. J. Pediatr. Adolesc. Gynecol. 2008, 21, 305–310. [Google Scholar] [CrossRef]
- Galani, A.; Kitsiou-Tzeli, S.; Sofokleous, C.; Kanavakis, E.; Kalpini-Mavrou, A. Androgen insensitivity syndrome: Clinical features and molecular defects. Hormones (Athens) 2008, 7, 217–229. [Google Scholar] [CrossRef]
- Bevan, C.L.; Hoare, S.; Claessens, F.; Heery, D.M.; Parker, M.G. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol. Cell Biol. 1999, 19, 8383–8392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrevoets, C.A.; Doesburg, P.; Steketee, K.; Trapman, J.; Brinkmann, A.O. Functional interactions of the AF-2 activation domain core region of the human androgen receptor with the amino-terminal domain and with the transcriptional coactivator TIF2 (transcriptional intermediary factor2). Mol. Endocrinol. 1998, 12, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, T.; Palvimo, J.J.; Jänne, O.A. Heterodimerization is mainly responsible for the dominant negative activity of amino-terminally truncated rat androgen receptor forms. FEBS Lett. 1998, 430, 393–396. [Google Scholar] [CrossRef] [Green Version]
- Karvonen, U.; Kallio, P.J.; Jänne, O.A.; Palvimo, J.J. Interaction of androgen receptors with androgen response element in intact cells. Roles of amino- and carboxyl-terminal regions and the ligand. J. Biol. Chem. 1997, 272, 15973–15979. [Google Scholar] [CrossRef] [Green Version]
- Langley, E.; Kemppainen, J.A.; Wilson, E.M. Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J. Biol. Chem. 1998, 273, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shizu, R.; Yokobori, K.; Perera, L.; Pedersen, L.; Negishi, M. Ligand induced dissociation of the AR homodimer precedes AR monomer translocation to the nucleus. Sci. Rep. 2019, 9, 16734. [Google Scholar] [CrossRef]
- Meehan, K.L.; Sadar, M.D. Androgens and androgen receptor in prostate and ovarian malignancies. Front. Biosci. 2003, 8, d780–d800. [Google Scholar] [CrossRef]
- Deng, Q.; Zhang, Z.; Wu, Y.; Yu, W.Y.; Zhang, J.; Jiang, Z.M.; Zhang, Y.; Liang, H.; Gui, Y.T. Non-Genomic Action of Androgens is Mediated by Rapid Phosphorylation and Regulation of Androgen Receptor Trafficking. Cell Physiol. Biochem. 2017, 43, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Gioeli, D.; Paschal, B.M. Post-translational modification of the androgen receptor. Mol. Cell Endocrinol. 2012, 352, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Myers, M.; Brown, M. Formation of the androgen receptor transcription complex. Mol. Cell 2002, 9, 601–610. [Google Scholar] [CrossRef]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat 2012, 33, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Sirokha, D.; Gorodna, O.; Lozhko, D.; Livshyts, G.; Zelinska, N.; Livshits, L. Novel missense mutation in ligand binding domain of AR gene identified in patient with androgen insensitivity syndrome from Ukraine. Clin. Case Rep. 2021, 9, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.; Schoof, E.M.; Kim, J.; Robin, X.; Miller, M.L.; Diella, F.; Palma, A.; Cesareni, G.; Jensen, L.J.; Linding, R. KinomeXplorer: An integrated platform for kinome biology studies. Nature Methods 2014, 11, 603–604. [Google Scholar] [CrossRef]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Zhou, J.; Zhang, Y.; Shi, Y.; Peng, D.; Xue, Y. GPS 5.0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genom. Proteom. Bioinform. 2020, 18, 72–80. [Google Scholar] [CrossRef]
- Patrick, R.; Kobe, B.; Lê Cao, K.-A.; Bodén, M. PhosphoPICK-SNP: Quantifying the effect of amino acid variants on protein phosphorylation. Bioinformatics 2017, 33, 1773–1781. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA, 2017.
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef]
- García, A.E. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 1992, 68, 2696–2699. [Google Scholar] [CrossRef]
- Falsafi-Zadeh, S.; Karimi, Z.; Galehdari, H. VMD DisRg: New User-Friendly Implement for calculation distance and radius of gyration in VMD program. Bioinformation 2012, 8, 341–343. [Google Scholar] [CrossRef] [Green Version]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Jurcik, A.; Bednar, D.; Byska, J.; Marques, S.M.; Furmanova, K.; Daniel, L.; Kokkonen, P.; Brezovsky, J.; Strnad, O.; Stourac, J.; et al. CAVER Analyst 2.0: Analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 2018, 34, 3586–3588. [Google Scholar] [CrossRef] [Green Version]
- Pavelka, A.; Sebestova, E.; Kozlikova, B.; Brezovsky, J.; Sochor, J.; Damborsky, J. CAVER: Algorithms for Analyzing Dynamics of Tunnels in Macromolecules. IEEE/ACM Trans. Comput Biol. Bioinform. 2016, 13, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Grino, P.B.; Griffin, J.E.; Wilson, J.D. Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology 1990, 126, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Guiochon-Mantel, A.; Delabre, K.; Lescop, P.; Milgrom, E. The Ernst Schering Poster Award. Intracellular traffic of steroid hormone receptors. J. Steroid Biochem. Mol. Biol. 1996, 56, 3–9. [Google Scholar] [CrossRef]
- Prescott, J.; Coetzee, G.A. Molecular chaperones throughout the life cycle of the androgen receptor. Cancer Lett. 2006, 231, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Schaufele, F.; Carbonell, X.; Guerbadot, M.; Borngraeber, S.; Chapman, M.S.; Ma, A.A.K.; Miner, J.N.; Diamond, M.I. The structural basis of androgen receptor activation: Intramolecular and intermolecular amino-carboxy interactions. Proc. Natl. Acad. Sci. USA 2005, 102, 9802–9807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anne Etgen, D.W.P. Molecular Mechanisms of Hormone Actions on Behavior, 1st ed.; Academic Press: Cambridge, MA, USA, 2009; p. 1100. [Google Scholar]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.X.; Lane, M.V.; Kemppainen, J.A.; French, F.S.; Wilson, E.M. Specificity of ligand-dependent androgen receptor stabilization: Receptor domain interactions influence ligand dissociation and receptor stability. Mol. Endocrinol. 1995, 9, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Pereira De Jésus-Tran, K.; Côté, P.-L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006, 15, 987–999. [Google Scholar] [CrossRef] [Green Version]
- Nadal, M.; Prekovic, S.; Gallastegui, N.; Helsen, C.; Abella, M.; Zielinska, K.; Gay, M.; Vilaseca, M.; Taulès, M.; Houtsmuller, A.B.; et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017, 8, 14388. [Google Scholar] [CrossRef] [PubMed]
- Lusher, S.J.; Raaijmakers, H.C.A.; Vu-Pham, D.; Kazemier, B.; Bosch, R.; McGuire, R.; Azevedo, R.; Hamersma, H.; Dechering, K.; Oubrie, A.; et al. X-ray structures of progesterone receptor ligand binding domain in its agonist state reveal differing mechanisms for mixed profiles of 11β-substituted steroids. J. Biol. Chem. 2012, 287, 20333–20343. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.P.; Sigler, P.B. Atomic structure of progesterone complexed with its receptor. Nature 1998, 393, 392–396. [Google Scholar] [CrossRef]
- Su, L.; Cheng, J.; Yin, X.; Liu, G.; Lu, Z.; Sheng, H.; Cai, Y.; Shi, Q.; Liu, L. Clinical and molecular characteristics in 15 patients with androgen receptor gene mutations from South China. Andrologia 2017, 49. [Google Scholar] [CrossRef]
- Yaegashi, N.; Uehara, S.; Senoo, M.; Sato, J.; Fujiwara, J.; Funato, T.; Sasaki, T.; Yajima, A. Point mutations in the steroid-binding domain of the androgen receptor gene of five Japanese patients with androgen insensitivity syndrome. Tohoku J. Exp. Med. 1999, 187, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.M.; Griffin, J.E.; Wilson, J.D.; Marcelli, M.; Zoppi, S.; McPhaul, M.J. Immunoreactive androgen receptor expression in subjects with androgen resistance. J. Clin. Endocrinol. Metab. 1992, 75, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, J.; Yong, E.L. Ligand- and coactivator-mediated transactivation function (AF2) of the androgen receptor ligand-binding domain is inhibited by the cognate hinge region. J. Biol. Chem. 2001, 276, 7493–7499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duff, J.; McEwan, I.J. Mutation of Histidine 874 in the Androgen Receptor Ligand-Binding Domain Leads to Promiscuous Ligand Activation and Altered p160 Coactivator Interactions. Mol. Endocrinol. 2005, 19, 2943–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayevsky, A.; Sirokha, D.; Samofalova, D.; Lozhko, D.; Gorodna, O.; Prokopenko, I.; Livshits, L. Functional Effects In Silico Prediction for Androgen Receptor Ligand-Binding Domain Novel I836S Mutation. Life 2021, 11, 659. https://doi.org/10.3390/life11070659
Rayevsky A, Sirokha D, Samofalova D, Lozhko D, Gorodna O, Prokopenko I, Livshits L. Functional Effects In Silico Prediction for Androgen Receptor Ligand-Binding Domain Novel I836S Mutation. Life. 2021; 11(7):659. https://doi.org/10.3390/life11070659
Chicago/Turabian StyleRayevsky, Alexey, Dmytro Sirokha, Dariia Samofalova, Dmytro Lozhko, Olexandra Gorodna, Inga Prokopenko, and Liudmyla Livshits. 2021. "Functional Effects In Silico Prediction for Androgen Receptor Ligand-Binding Domain Novel I836S Mutation" Life 11, no. 7: 659. https://doi.org/10.3390/life11070659