
The ATP-Releasing Maxi-Cl Channel: Its Identity, Molecular Partners, and Physiological/Pathophysiological Implications


 ,
, 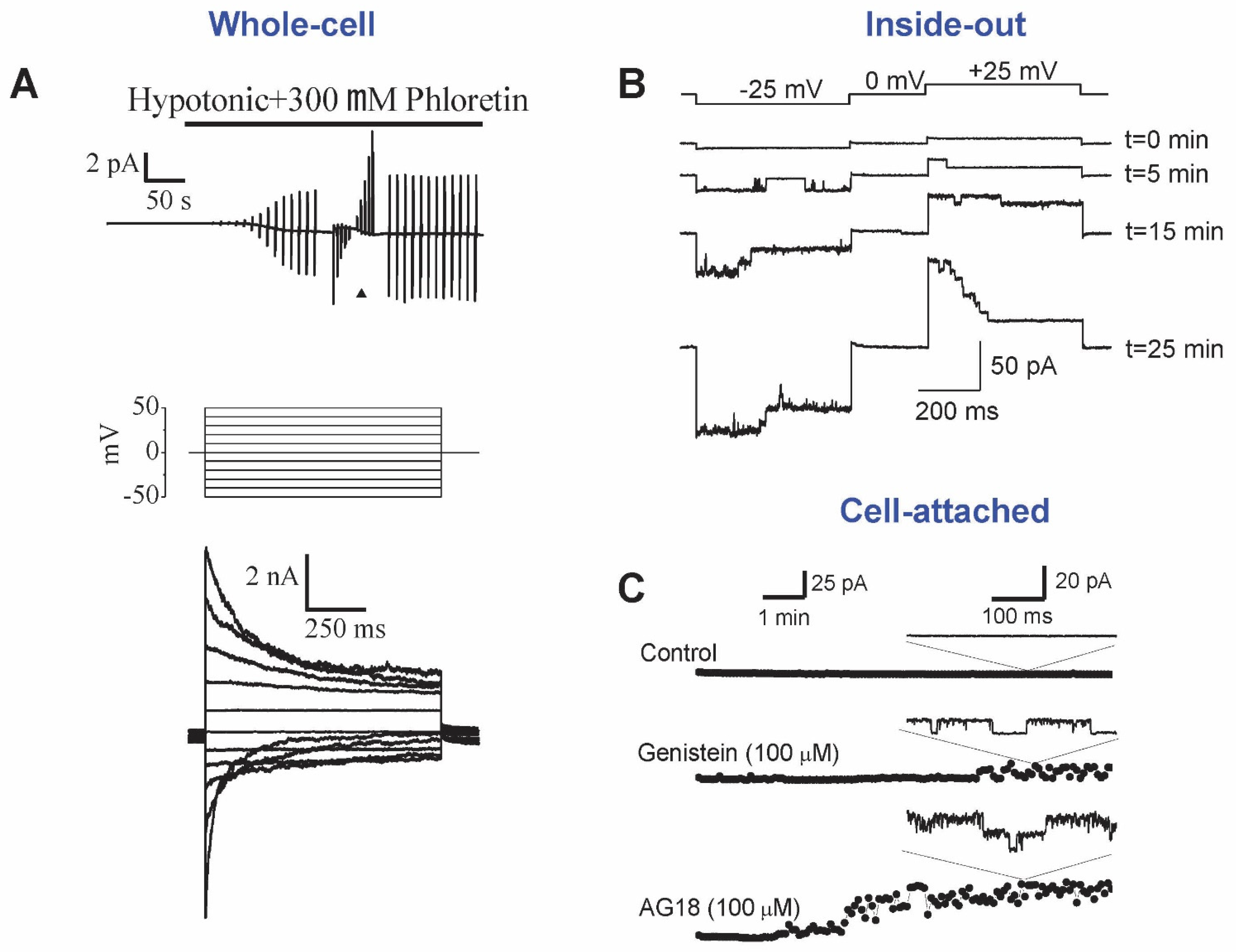{kind=link}
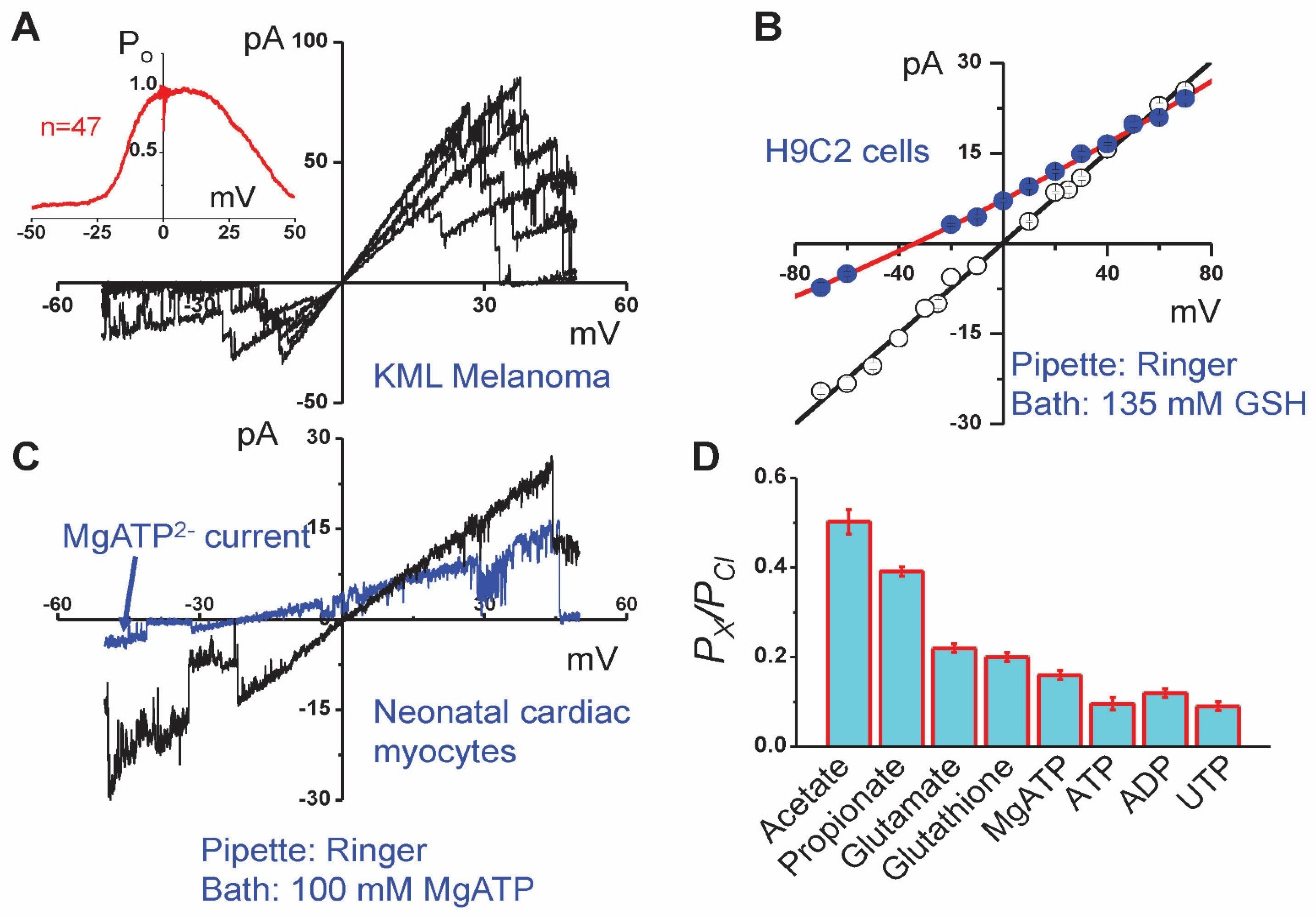{kind=link}
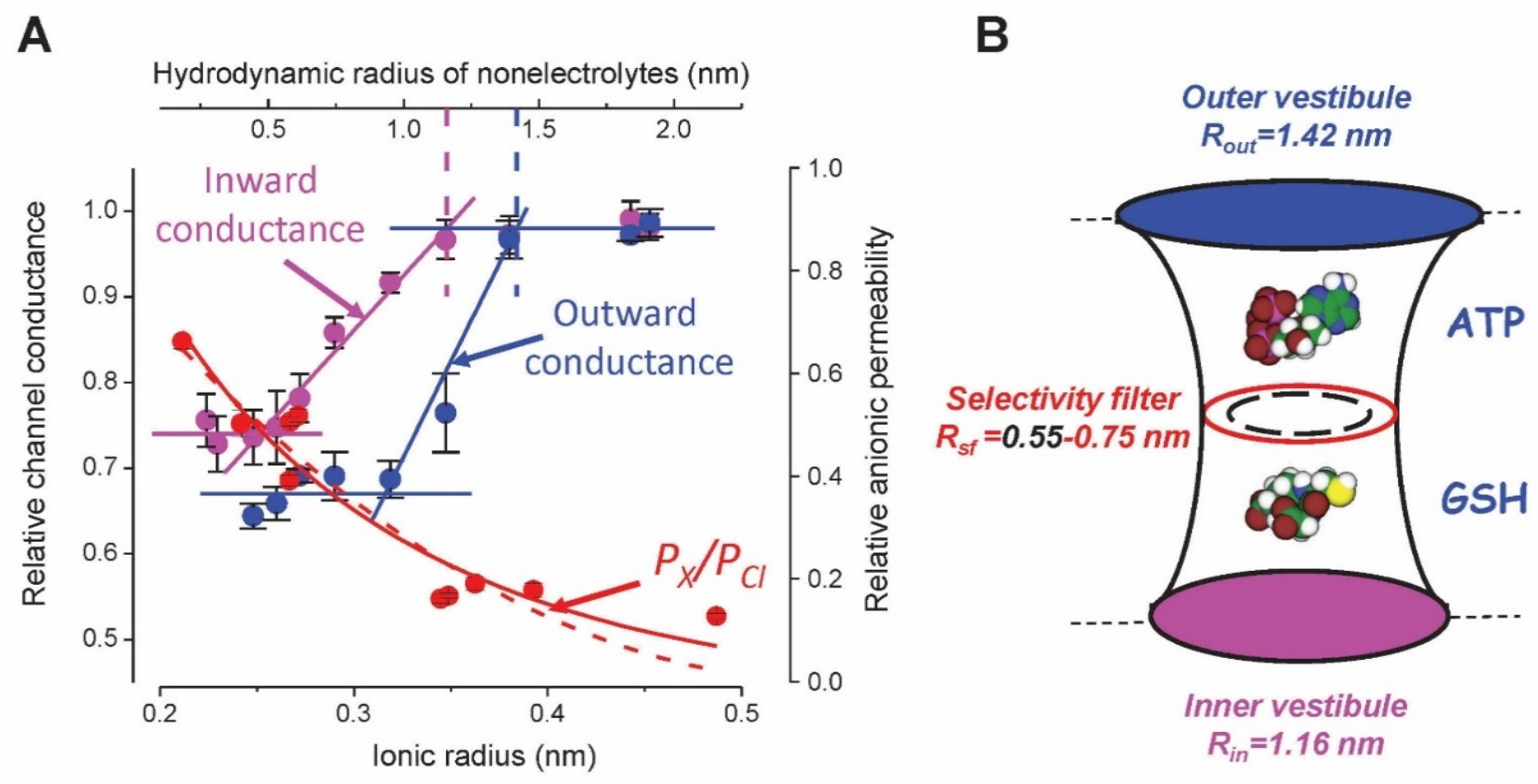{kind=link}
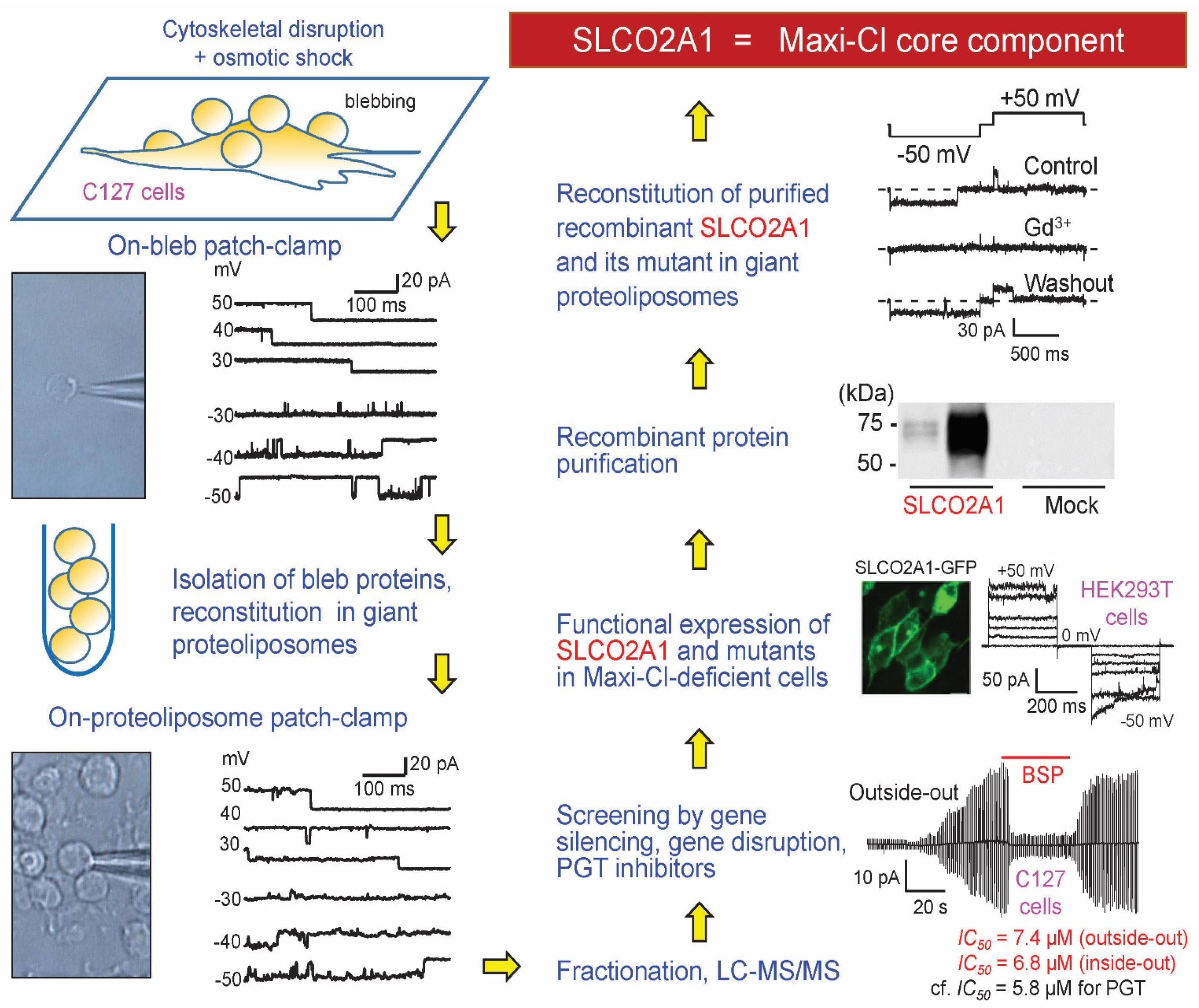{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Phenotype and Biophysical Profile of Maxi-Cl
3. Activation Stimuli of Maxi-Cl
4. Molecular Identity of Maxi-Cl: Rejected Candidates
5. Molecular Identity of Maxi-Cl: The Pore Component
6. Molecular Identity of Maxi-Cl: The Regulatory Components
7. Physiological/Pathophysiological Implications of Maxi-Cl
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zamaraeva, M.V.; Sabirov, R.Z.; Maeno, E.; Ando-Akatsuka, Y.; Bessonova, S.V.; Okada, Y. Cells die with increased cytosolic ATP during apoptosis: A bioluminescence study with intracellular luciferase. Cell Death Differ. 2005, 12, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, H.J.; Pouli, A.E.; Ainscow, E.K.; Jouaville, L.S.; Rizzuto, R.; Rutter, G.A. Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. Potential role for strategically located mitochondria. J. Biol. Chem. 1999, 274, 13281–13291. [Google Scholar] [CrossRef] [Green Version]
- Gribble, F.M.; Loussouarn, G.; Tucker, S.J.; Zhao, C.; Nichols, C.G.; Ashcroft, F.M. A novel method for measurement of submembrane ATP concentration. J. Biol. Chem. 2000, 275, 30046–30049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamaraeva, M.V.; Sabirov, R.Z.; Manabe, K.; Okada, Y. Ca(2+)-dependent glycolysis activation mediates apoptotic ATP elevation in HeLa cells. Biochem. Biophys. Res. Commun. 2007, 363, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Brake, A.J.; Julius, D. Signaling by extracellular nucleotides. Annu. Rev. Cell Dev. Biol. 1996, 12, 519–541. [Google Scholar] [CrossRef]
- Okada, Y.; Okada, T.; Islam, M.R.; Sabirov, R.Z. Molecular identities and ATP release activities of two types of volume-regulatory anion channels, VSOR and Maxi-Cl. Curr. Top. Membr. 2018, 81, 125–176. [Google Scholar] [CrossRef]
- Zimmermann, H. Two novel families of ectonucleotidases: Molecular structures, catalytic properties and a search for function. Trends Pharmacol. Sci. 1999, 20, 231–236. [Google Scholar] [CrossRef]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine release, metabolism, and signaling in the inflammatory response. Annu. Rev. Immunol. 2019, 37, 325–347. [Google Scholar] [CrossRef]
- Dwyer, K.M.; Kishore, B.K.; Robson, S.C. Conversion of extracellular ATP into adenosine: A master switch in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 509–524. [Google Scholar] [CrossRef]
- Burnstock, G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 2007, 87, 659–797. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G. Purinergic signalling: Its unpopular beginning, its acceptance and its exciting future. Bioessays 2012, 34, 218–225. [Google Scholar] [CrossRef]
- Sabirov, R.Z.; Okada, Y. ATP-conducting maxi-anion channel: A new player in stress-sensory transduction. Jpn. J. Physiol. 2004, 54, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Sabirov, R.Z.; Okada, Y. ATP release via anion channels. Purinergic Signal. 2005, 1, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Sabirov, R.Z.; Okada, Y. The maxi-anion channel: A classical channel playing novel roles through an unidentified molecular entity. J. Physiol. Sci. 2009, 59, 3–21. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Banerjee, J.; Leung, C.T.; Peterson-Yantorno, K.; Stamer, W.D.; Civan, M.M. Mechanisms of ATP release, the enabling step in purinergic dynamics. Cell. Physiol. Biochem. 2011, 28, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Lazarowski, E.R.; Boucher, R.C.; Harden, T.K. Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol. Pharmacol. 2003, 64, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Lazarowski, E.R.; Sesma, J.I.; Seminario-Vidal, L.; Kreda, S.M. Molecular mechanisms of purine and pyrimidine nucleotide release. Adv. Pharmacol. 2011, 61, 221–261. [Google Scholar] [CrossRef] [PubMed]
- Lazarowski, E.R. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 2012, 8, 359–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taruno, A.; Vingtdeux, V.; Ohmoto, M.; Ma, Z.; Dvoryanchikov, G.; Li, A.; Adrien, L.; Zhao, H.; Leung, S.; Abernethy, M.; et al. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 2013, 495, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, J.; Dartt, D.A.; Trinkaus-Randall, V.; Pintor, J.; Civan, M.M.; Delamere, N.A.; Fletcher, E.L.; Salt, T.E.; Grosche, A.; Mitchell, C.H. Purines in the eye: Recent evidence for the physiological and pathological role of purines in the RPE, retinal neurons, astrocytes, Muller cells, lens, trabecular meshwork, cornea and lacrimal gland. Exp. Eye Res. 2014, 127, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, Y.; Hiasa, M.; Sakamoto, S.; Omote, H.; Nomura, M. Vesicular nucleotide transporter (VNUT): Appearance of an actress on the stage of purinergic signaling. Purinergic Signal. 2017, 13, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Taruno, A. ATP release channels. Int. J. Mol. Sci. 2018, 19, 808. [Google Scholar] [CrossRef] [Green Version]
- Praetorius, H.A.; Leipziger, J. ATP release from non-excitable cells. Purinergic Signal. 2009, 5, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Unwin, R.; Inscho, E.W.; Leipziger, J.; Kishore, B.K. Extracellular nucleotides and P2 receptors in renal function. Physiol. Rev. 2020, 100, 211–269. [Google Scholar] [CrossRef]
- Lohman, A.W.; Isakson, B.E. Differentiating connexin hemichannels and pannexin channels in cellular ATP release. FEBS Lett. 2014, 588, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Yeung, A.K.; Patil, C.S.; Jackson, M.F. Pannexin-1 in the CNS: Emerging concepts in health and disease. J. Neurochem. 2020, 154, 468–485. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.L.; Mat Nor, M.N.; Guo, C.X.; Mugisho, O.O.; Coutinho, F.P.; Rupenthal, I.D.; Green, C.R. Connexin therapeutics: Blocking connexin hemichannel pores is distinct from blocking pannexin channels or gap junctions. Neural Regen. Res. 2021, 16, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Navis, K.E.; Fan, C.Y.; Trang, T.; Thompson, R.J.; Derksen, D.J. Pannexin 1 channels as a therapeutic target: Structure, inhibition, and outlook. ACS Chem. Neurosci. 2020, 11, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Andrejew, R.; Oliveira-Giacomelli, A.; Ribeiro, D.E.; Glaser, T.; Arnaud-Sampaio, V.F.; Lameu, C.; Ulrich, H. The P2X7 receptor: Central hub of brain diseases. Front. Mol. Neurosci. 2020, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Panicucci, C.; Raffaghello, L.; Bruzzone, S.; Baratto, S.; Principi, E.; Minetti, C.; Gazzerro, E.; Bruno, C. eATP/P2X7R Axis: An orchestrated pathway triggering inflammasome activation in muscle diseases. Int. J. Mol. Sci. 2020, 21, 5963. [Google Scholar] [CrossRef]
- Ma, Z.; Tanis, J.E.; Taruno, A.; Foskett, J.K. Calcium homeostasis modulator (CALHM) ion channels. Pflug. Arch. 2016, 468, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Mousawi, F.; Li, D.; Roger, S.; Li, J.; Yang, X.; Jiang, L.H. Adenosine triphosphate release and P2 receptor signaling in Piezo1 channel-dependent mechanoregulation. Front. Pharmacol. 2019, 10, 1304. [Google Scholar] [CrossRef] [Green Version]
- Sabirov, R.Z.; Merzlyak, P.G.; Islam, M.R.; Okada, T.; Okada, Y. The properties, functions, and pathophysiology of maxi-anion channels. Pflug. Arch. 2016, 468, 405–420. [Google Scholar] [CrossRef]
- Elorza-Vidal, X.; Gaitan-Penas, H.; Estevez, R. Chloride channels in astrocytes: Structure, roles in brain homeostasis and implications in disease. Int. J. Mol. Sci. 2019, 20, 1034. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Okada, T.; Sato-Numata, K.; Islam, M.R.; Ando-Akatsuka, Y.; Numata, T.; Kubo, M.; Shimizu, T.; Kurbannazarova, R.S.; Marunaka, Y.; et al. Cell volume-activated and volume-correlated anion channels in mammalian cells: Their biophysical, molecular, and pharmacological properties. Pharmacol. Rev. 2019, 71, 49–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Sabirov, R.Z.; Sato-Numata, K.; Numata, T. Cell death induction and protection by activation of ubiquitously expressed anion/cation channels. Part 1: Roles of VSOR/VRAC in cell volume regulation, release of double-edged signals and apoptotic/necrotic cell death. Front. Cell Dev. Biol. 2021, 8, 614040. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J.; Pusch, M. CLC chloride channels and transporters: Structure, function, physiology, and disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef]
- Blount, P.; Iscla, I. Life with bacterial mechanosensitive channels, from discovery to physiology to pharmacological target. Microbiol. Mol. Biol. Rev. 2020, 84, e00055-00019. [Google Scholar] [CrossRef]
- Blatz, A.L.; Magleby, K.L. Single voltage-dependent chloride-selective channels of large conductance in cultured rat muscle. Biophys. J. 1983, 43, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Riquelme, G.; Stutzin, A.; Barros, L.F.; Liberona, J.L. A chloride channel from human placenta reconstituted into giant liposomes. Am. J. Obstet. Gynecol. 1995, 173, 733–738. [Google Scholar] [CrossRef]
- Strange, K.; Emma, F.; Jackson, P.S. Cellular and molecular physiology of volume-sensitive anion channels. Am. J. Physiol. 1996, 270, C711–C730. [Google Scholar] [CrossRef]
- Riquelme, G. Apical Maxi-chloride channel from human placenta: 12 years after the first electrophysiological recordings. Biol. Res. 2006, 39, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Bell, P.D.; Komlosi, P.; Zhang, Z.R. ATP as a mediator of macula densa cell signalling. Purinergic Signal. 2009, 5, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riquelme, G. Placental chloride channels: A review. Placenta 2009, 30, 659–669. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.J.; Kelly, S.P.; Nurse, C.A.; Wood, C.M. A maxi Cl(−) channel in cultured pavement cells from the gills of the freshwater rainbow trout Oncorhynchus mykiss. J. Exp. Biol. 2001, 204, 1783–1794. [Google Scholar] [CrossRef]
- Woll, K.H.; Leibowitz, M.D.; Neumcke, B.; Hille, B. A high-conductance anion channel in adult amphibian skeletal muscle. Pflug. Arch. 1987, 410, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Hussy, N. Calcium-activated chloride channels in cultured embryonic Xenopus spinal neurons. J. Neurophysiol. 1992, 68, 2042–2050. [Google Scholar] [CrossRef]
- Stumpff, F.; Martens, H.; Bilk, S.; Aschenbach, J.R.; Gabel, G. Cultured ruminal epithelial cells express a large-conductance channel permeable to chloride, bicarbonate, and acetate. Pflug. Arch. 2009, 457, 1003–1022. [Google Scholar] [CrossRef] [Green Version]
- Stumpff, F.; Georgi, M.I.; Mundhenk, L.; Rabbani, I.; Fromm, M.; Martens, H.; Gunzel, D. Sheep rumen and omasum primary cultures and source epithelia: Barrier function aligns with expression of tight junction proteins. J. Exp. Biol. 2011, 214, 2871–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgi, M.I.; Rosendahl, J.; Ernst, F.; Gunzel, D.; Aschenbach, J.R.; Martens, H.; Stumpff, F. Epithelia of the ovine and bovine forestomach express basolateral maxi-anion channels permeable to the anions of short-chain fatty acids. Pflug. Arch. 2014, 466, 1689–1712. [Google Scholar] [CrossRef]
- Stumpff, F. A look at the smelly side of physiology: Transport of short chain fatty acids. Pflug. Arch. 2018, 470, 571–598. [Google Scholar] [CrossRef] [PubMed]
- Groschner, K.; Kukovetz, W.R. Voltage-sensitive chloride channels of large conductance in the membrane of pig aortic endothelial cells. Pflug. Arch. 1992, 421, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Olesen, S.P.; Bundgaard, M. Chloride-selective channels of large conductance in bovine aortic endothelial cells. Acta Physiol. Scand. 1992, 144, 191–198. [Google Scholar] [CrossRef]
- Sabirov, R.Z.; Dutta, A.K.; Okada, Y. Volume-dependent ATP-conductive large-conductance anion channel as a pathway for swelling-induced ATP release. J. Gen. Physiol. 2001, 118, 251–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.R.; Uramoto, H.; Okada, T.; Sabirov, R.Z.; Okada, Y. Maxi-anion channel and pannexin 1 hemichannel constitute separate pathways for swelling-induced ATP release in murine L929 fibrosarcoma cells. Am. J. Physiol. Cell Physiol. 2012, 303, C924–C935. [Google Scholar] [CrossRef] [Green Version]
- Bell, P.D.; Lapointe, J.Y.; Sabirov, R.; Hayashi, S.; Peti-Peterdi, J.; Manabe, K.; Kovacs, G.; Okada, Y. Macula densa cell signaling involves ATP release through a maxi anion channel. Proc. Natl. Acad. Sci. USA 2003, 100, 4322–4327. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.K.; Sabirov, R.Z.; Uramoto, H.; Okada, Y. Role of ATP-conductive anion channel in ATP release from neonatal rat cardiomyocytes in ischaemic or hypoxic conditions. J. Physiol. 2004, 559, 799–812. [Google Scholar] [CrossRef]
- Dutta, A.K.; Korchev, Y.E.; Shevchuk, A.I.; Hayashi, S.; Okada, Y.; Sabirov, R.Z. Spatial distribution of maxi-anion channel on cardiomyocytes detected by smart-patch technique. Biophys. J. 2008, 94, 1646–1655. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.T.; Tashmukhamedov, B.A.; Inoue, H.; Okada, Y.; Sabirov, R.Z. Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 2006, 54, 343–357. [Google Scholar] [CrossRef]
- Liu, H.T.; Sabirov, R.Z.; Okada, Y. Oxygen-glucose deprivation induces ATP release via maxi-anion channels in astrocytes. Purinergic Signal. 2008, 4, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Sabirov, R.Z.; Sheiko, T.; Liu, H.; Deng, D.; Okada, Y.; Craigen, W.J. Genetic demonstration that the plasma membrane maxianion channel and voltage-dependent anion channels are unrelated proteins. J. Biol. Chem. 2006, 281, 1897–1904. [Google Scholar] [CrossRef] [Green Version]
- Toychiev, A.H.; Sabirov, R.Z.; Takahashi, N.; Ando-Akatsuka, Y.; Liu, H.; Shintani, T.; Noda, M.; Okada, Y. Activation of maxi-anion channel by protein tyrosine dephosphorylation. Am. J. Physiol. Cell Physiol. 2009, 297, C990–C1000. [Google Scholar] [CrossRef] [PubMed]
- Kurbannazarova, R.S.; Bessonova, S.V.; Okada, Y.; Sabirov, R.Z. Swelling-activated anion channels are essential for volume regulation of mouse thymocytes. Int. J. Mol. Sci. 2011, 12, 9125–9137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarze, W.; Kolb, H.A. Voltage-dependent kinetics of an anionic channel of large unit conductance in macrophages and myotube membranes. Pflug. Arch. 1984, 402, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Geletyuk, V.I.; Kazachenko, V.N. Single Cl(−) channels in molluscan neurones: Multiplicity of the conductance states. J. Membr. Biol. 1985, 86, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.P.; Supplisson, S.; Mayer, E. Chloride channels in myocytes from rabbit colon are regulated by a pertussis toxin-sensitive G protein. Am. J. Physiol. 1993, 264, G774–G785. [Google Scholar] [CrossRef]
- Sabirov, R.Z.; Okada, Y. Wide nanoscopic pore of maxi-anion channel suits its function as an ATP-conductive pathway. Biophys. J. 2004, 87, 1672–1685. [Google Scholar] [CrossRef] [Green Version]
- Hurnak, O.; Zachar, J. Selectivity of maxi chloride channels in the L6 rat muscle cell line. Gen. Physiol. Biophys. 1995, 14, 91–105. [Google Scholar] [PubMed]
- Soejima, M.; Kokubun, S. Single anion-selective channel and its ion selectivity in the vascular smooth muscle cell. Pflug. Arch. 1988, 411, 304–311. [Google Scholar] [CrossRef]
- Dutta, A.K.; Okada, Y.; Sabirov, R.Z. Regulation of an ATP-conductive large-conductance anion channel and swelling-induced ATP release by arachidonic acid. J. Physiol. 2002, 542, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Sabirov, R.Z.; Kurbannazarova, R.S.; Melanova, N.R.; Okada, Y. Volume-sensitive anion channels mediate osmosensitive glutathione release from rat thymocytes. PLoS ONE 2013, 8, e55646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabirov, R.Z.; Merzlyak, P.G. Plasmalemmal VDAC controversies and maxi-anion channel puzzle. Biochim. Biophys. Acta 2012, 1818, 1570–1580. [Google Scholar] [CrossRef] [Green Version]
- Falke, L.C.; Misler, S. Activity of ion channels during volume regulation by clonal N1E115 neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1989, 86, 3919–3923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalonen, T. Single-channel characteristics of the large-conductance anion channel in rat cortical astrocytes in primary culture. Glia 1993, 9, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Schwiebert, E.M.; Mills, J.W.; Stanton, B.A. Actin-based cytoskeleton regulates a chloride channel and cell volume in a renal cortical collecting duct cell line. J. Biol. Chem. 1994, 269, 7081–7089. [Google Scholar] [CrossRef]
- Peti-Peterdi, J.; Morishima, S.; Bell, P.D.; Okada, Y. Two-photon excitation fluorescence imaging of the living juxtaglomerular apparatus. Am. J. Physiol. Ren. Physiol. 2002, 283, F197–F201. [Google Scholar] [CrossRef]
- Komlosi, P.; Fintha, A.; Bell, P.D. Unraveling the relationship between macula densa cell volume and luminal solute concentration/osmolality. Kidney Int. 2006, 70, 865–871. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, K.; Takuwa, N. Bombesin activates large-conductance chloride channels in Swiss 3T3 fibroblasts. Biochem. Biophys. Res. Commun. 1991, 177, 292–298. [Google Scholar] [CrossRef]
- Bajnath, R.B.; Groot, J.A.; de Jonge, H.R.; Kansen, M.; Bijman, J. Calcium ionophore plus excision induce a large conductance chloride channel in membrane patches of human colon carcinoma cells HT-29cl.19A. Experientia 1993, 49, 313–316. [Google Scholar] [CrossRef]
- Dezaki, K.; Tsumura, T.; Maeno, E.; Okada, Y. Receptor-mediated facilitation of cell volume regulation by swelling-induced ATP release in human epithelial cells. Jpn. J. Physiol. 2000, 50, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Kajita, H.; Kotera, T.; Shirakata, Y.; Ueda, S.; Okuma, M.; Oda-Ohmae, K.; Takimoto, M.; Urade, Y.; Okada, Y. A maxi Cl(−) channel coupled to endothelin B receptors in the basolateral membrane of guinea-pig parietal cells. J. Physiol. 1995, 488, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Schwiebert, E.M.; Karlson, K.H.; Friedman, P.A.; Dietl, P.; Spielman, W.S.; Stanton, B.A. Adenosine regulates a chloride channel via protein kinase C and a G protein in a rabbit cortical collecting duct cell line. J. Clin. Investig. 1992, 89, 834–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.P.; Supplisson, S.; Torres, R.; Sachs, G.; Mayer, E. Characterization of large-conductance chloride channels in rabbit colonic smooth muscle. J. Physiol. 1992, 448, 355–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, S.P.; Valverde, M.A. Novel plasma membrane action of estrogen and antiestrogens revealed by their regulation of a large conductance chloride channel. FASEB J. 1994, 8, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.; Bahamonde, M.I.; Lock, H.; Munoz, F.J.; Hardy, S.P.; Posas, F.; Valverde, M.A. Okadaic acid-sensitive activation of Maxi Cl(−) channels by triphenylethylene antioestrogens in C1300 mouse neuroblastoma cells. J. Physiol. 2001, 536, 79–88. [Google Scholar] [CrossRef]
- Pahapill, P.A.; Schlichter, L.C. Cl(−) channels in intact human T lymphocytes. J. Membr. Biol. 1992, 125, 171–183. [Google Scholar] [CrossRef]
- Nam, J.H.; Zheng, H.F.; Earm, K.H.; Ko, J.H.; Lee, I.J.; Kang, T.M.; Kim, T.J.; Earm, E.; Kim, S.J. Voltage-dependent slowly activating anion current regulated by temperature and extracellular pH in mouse B cells. Pflug. Arch. 2006, 452, 707–717. [Google Scholar] [CrossRef]
- Schwiebert, E.M.; Light, D.B.; Fejes-Toth, G.; Naray-Fejes-Toth, A.; Stanton, B.A. A GTP-binding protein activates chloride channels in a renal epithelium. J. Biol. Chem. 1990, 265, 7725–7728. [Google Scholar] [CrossRef]
- McGill, J.M.; Basavappa, S.; Fitz, J.G. Characterization of high-conductance anion channels in rat bile duct epithelial cells. Am. J. Physiol. 1992, 262, G703–G710. [Google Scholar] [CrossRef]
- McGill, J.M.; Gettys, T.W.; Basavappa, S.; Fitz, J.G. GTP-binding proteins regulate high conductance anion channels in rat bile duct epithelial cells. J. Membr. Biol. 1993, 133, 253–261. [Google Scholar] [CrossRef]
- Islam, M.R.; Okada, T.; Merzlyak, P.G.; Toychiev, A.H.; Ando-Akatsuka, Y.; Sabirov, R.Z.; Okada, Y. Annexin A2-S100A10 represents the regulatory component of Maxi-Cl channel dependent on protein tyrosine dephosphorylation and intracellular Ca2+. Cell. Physiol. Biochem. 2020, 54, 538–555. [Google Scholar] [CrossRef]
- De Pinto, V. Renaissance of VDAC: New insights on a protein family at the interface between mitochondria and cytosol. Biomolecules 2021, 11, 107. [Google Scholar] [CrossRef]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium 2021, 94, 102356. [Google Scholar] [CrossRef] [PubMed]
- Colombini, M. Voltage gating in the mitochondrial channel, VDAC. J. Membr. Biol. 1989, 111, 103–111. [Google Scholar] [CrossRef]
- Colombini, M. VDAC: The channel at the interface between mitochondria and the cytosol. Mol. Cell. Biochem. 2004, 256–257, 107–115. [Google Scholar] [CrossRef]
- Colombini, M. Voltage gating in VDAC: Toward a molecular mechanism. In Ion Channel Reconstitution; Miller, C., Ed.; Plenum Press: New York, NY, USA, 1986; pp. 533–550. [Google Scholar]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Rostovtseva, T.; Colombini, M. ATP flux is controlled by a voltage-gated channel from the mitochondrial outer membrane. J. Biol. Chem. 1996, 271, 28006–28008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostovtseva, T.K.; Bezrukov, S.M. ATP transport through a single mitochondrial channel, VDAC, studied by current fluctuation analysis. Biophys. J. 1998, 74, 2365–2373. [Google Scholar] [CrossRef] [Green Version]
- Rostovtseva, T.K.; Komarov, A.; Bezrukov, S.M.; Colombini, M. Dynamics of nucleotides in VDAC channels: Structure-specific noise generation. Biophys. J. 2002, 82, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Dermietzel, R.; Hwang, T.K.; Buettner, R.; Hofer, A.; Dotzler, E.; Kremer, M.; Deutzmann, R.; Thinnes, F.P.; Fishman, G.I.; Spray, D.C.; et al. Cloning and in situ localization of a brain-derived porin that constitutes a large-conductance anion channel in astrocytic plasma membranes. Proc. Natl. Acad. Sci. USA 1994, 91, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Buettner, R.; Papoutsoglou, G.; Scemes, E.; Spray, D.C.; Dermietzel, R. Evidence for secretory pathway localization of a voltage-dependent anion channel isoform. Proc. Natl. Acad. Sci. USA 2000, 97, 3201–3206. [Google Scholar] [CrossRef] [PubMed]
- Bahamonde, M.I.; Valverde, M.A. Voltage-dependent anion channel localises to the plasma membrane and peripheral but not perinuclear mitochondria. Pflug. Arch. 2003, 446, 309–313. [Google Scholar] [CrossRef]
- Bahamonde, M.I.; Fernandez-Fernandez, J.M.; Guix, F.X.; Vazquez, E.; Valverde, M.A. Plasma membrane voltage-dependent anion channel mediates antiestrogen-activated maxi Cl- currents in C1300 neuroblastoma cells. J. Biol. Chem. 2003, 278, 33284–33289. [Google Scholar] [CrossRef] [Green Version]
- Elinder, F.; Akanda, N.; Tofighi, R.; Shimizu, S.; Tsujimoto, Y.; Orrenius, S.; Ceccatelli, S. Opening of plasma membrane voltage-dependent anion channels (VDAC) precedes caspase activation in neuronal apoptosis induced by toxic stimuli. Cell Death. Differ. 2005. [Google Scholar] [CrossRef] [Green Version]
- Akanda, N.; Elinder, F. Biophysical properties of the apoptosis-inducing plasma membrane voltage-dependent anion channel. Biophys. J. 2006, 90, 4405–4417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Mizuno, A. A novel human Cl(−) channel family related to Drosophila flightless locus. J. Biol. Chem. 2004, 279, 22461–22468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M. The Drosophila tweety family: Molecular candidates for large-conductance Ca2+-activated Cl(−) channels. Exp. Physiol. 2006, 91, 141–147. [Google Scholar] [CrossRef]
- Okada, Y.; Sato, K.; Toychiev, A.H.; Suzuki, M.; Dutta, A.K.; Inoue, H.; Sabirov, R. The puzzles of volume-activated anion channels. In Physiology and Pathology of Chloride Transporters and Channels in the Nervous System. From Molecules to Diseases; Alvarez-Leefmans, F.J., Delpire, E., Eds.; Elsevier: San Diego, CA, USA, 2009; pp. 283–306. [Google Scholar]
- Okada, T.; Islam, M.R.; Merzlyak, P.G.; Sabirov, R.; Okada, Y. Attempts to identify the gene encoding maxi-anion channel. J. Physiol. Sci. 2012, 62, S85. [Google Scholar]
- Islam, M.R.; Merzlyak, P.G.; Okada, T.; Sabirov, R.Z.; Okada, Y. Searching for molecular basis of maxi-anion channel. J. Physiol. Sci. 2013, 63, S133. [Google Scholar]
- Merzlyak, P.G.; Okada, Y.; Sabirov, R.Z. Maxi-anion channel activity in membrane blebs and after reconstitution in artificial membranes. J. Physiol. Sci. 2010, 60, S123. [Google Scholar]
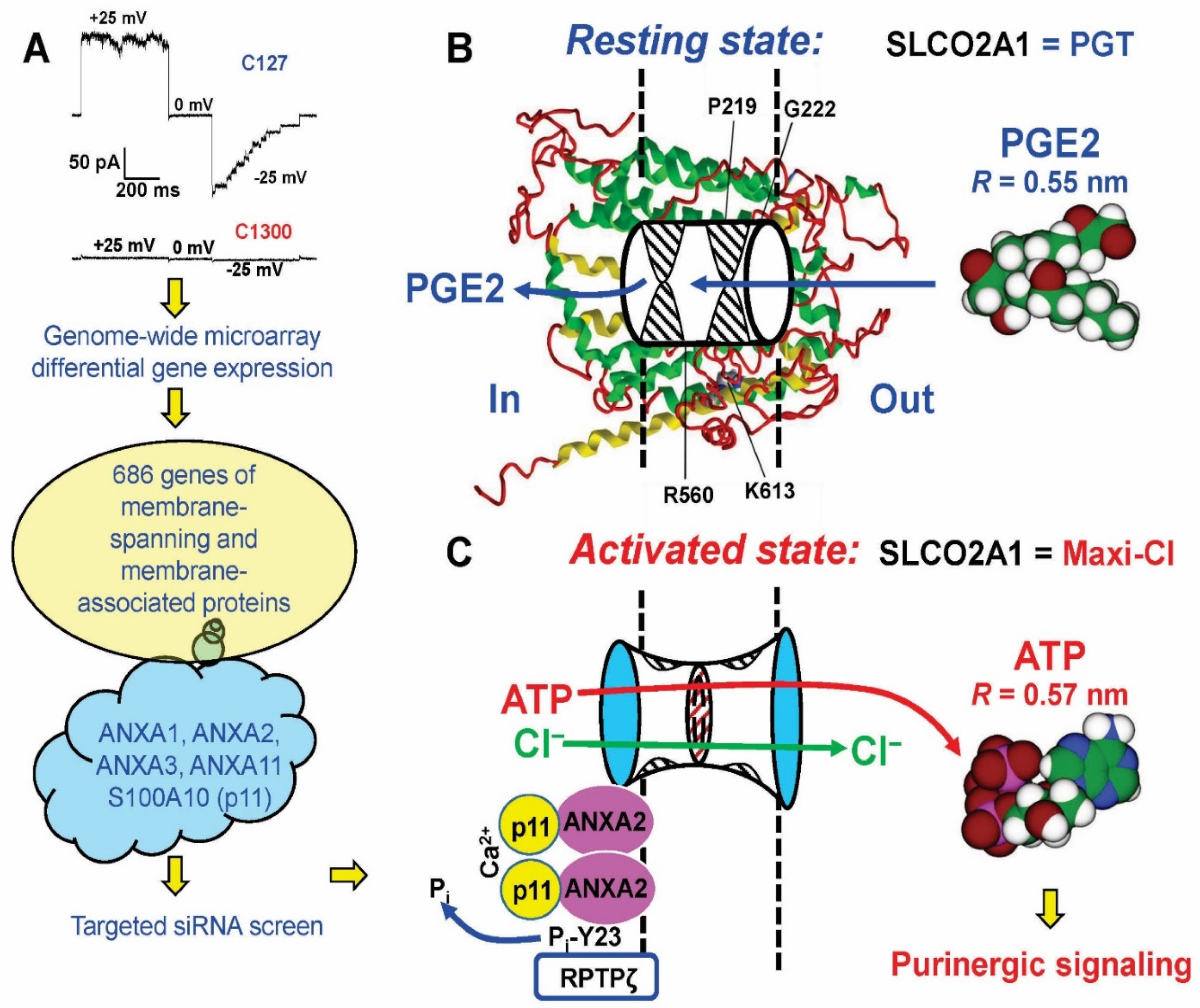
- Sabirov, R.Z.; Merzlyak, P.G.; Okada, T.; Islam, M.R.; Uramoto, H.; Mori, T.; Makino, Y.; Matsuura, H.; Xie, Y.; Okada, Y. The organic anion transporter SLCO2A1 constitutes the core component of the Maxi-Cl channel. EMBO J. 2017, 36, 3309–3324. [Google Scholar] [CrossRef]
- Kanai, N.; Lu, R.; Satriano, J.A.; Bao, Y.; Wolkoff, A.W.; Schuster, V.L. Identification and characterization of a prostaglandin transporter. Science 1995, 268, 866–869. [Google Scholar] [CrossRef]
- Chan, B.S.; Bao, Y.; Schuster, V.L. Role of conserved transmembrane cationic amino acids in the prostaglandin transporter PGT. Biochemistry 2002, 41, 9215–9221. [Google Scholar] [CrossRef]
- Zhang, Z.; Xia, W.; He, J.; Zhang, Z.; Ke, Y.; Yue, H.; Wang, C.; Zhang, H.; Gu, J.; Hu, W.; et al. Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am. J. Hum. Genet. 2012, 90, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; He, J.W.; Fu, W.Z.; Zhang, C.Q.; Zhang, Z.L. Two novel mutations in the SLCO2A1 gene in a Chinese patient with primary hypertrophic osteoarthropathy. Gene 2014, 534, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Schuster, V.L. Molecular mechanisms of prostaglandin transport. Annu. Rev. Physiol. 1998, 60, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Schuster, V.L. Prostaglandin transport. Prostaglandins Other Lipid Mediat. 2002, 68–69, 633–647. [Google Scholar] [CrossRef]
- Chang, H.Y.; Locker, J.; Lu, R.; Schuster, V.L. Failure of postnatal ductus arteriosus closure in prostaglandin transporter-deficient mice. Circulation 2010, 121, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Schuster, V.L.; Chi, Y.; Lu, R. The prostaglandin transporter: Eicosanoid reuptake, control of signaling, and development of high-affinity inhibitors as drug candidates. Trans. Am. Clin. Clim. Assoc. 2015, 126, 248–257. [Google Scholar]
- Minor, D.L., Jr. Channel surfing uncovers a dual-use transporter. EMBO J. 2017, 36, 3272–3273. [Google Scholar] [CrossRef]
- Picollo, A.; Malvezzi, M.; Accardi, A. TMEM16 proteins: Unknown structure and confusing functions. J. Mol. Biol. 2015, 427, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Picollo, A.; Pusch, M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 2005, 436, 420–423. [Google Scholar] [CrossRef]
- Scheel, O.; Zdebik, A.A.; Lourdel, S.; Jentsch, T.J. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 2005, 436, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Accardi, A.; Picollo, A. CLC channels and transporters: Proteins with borderline personalities. Biochim. Biophys. Acta 2010, 1798, 1457–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahlke, C.; Kortzak, D.; Machtens, J.P. Molecular physiology of EAAT anion channels. Pflug. Arch. 2016, 468, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Ohana, E.; Yang, D.; Shcheynikov, N.; Muallem, S. Diverse transport modes by the solute carrier 26 family of anion transporters. J. Physiol. 2009, 587, 2179–2185. [Google Scholar] [CrossRef]
- Riquelme, G.; Llanos, P.; Tischner, E.; Neil, J.; Campos, B. Annexin 6 modulates the maxi-chloride channel of the apical membrane of syncytiotrophoblast isolated from human placenta. J. Biol. Chem. 2004, 279, 50601–50608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaria-Kisiel, L.; Rintala-Dempsey, A.C.; Shaw, G.S. Calcium-dependent and -independent interactions of the S100 protein family. Biochem. J. 2006, 396, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, M.A.; Glenney, J.R. Regulation of calpactin I phospholipid binding by calpactin I light-chain binding and phosphorylation by p60v-src. Biochem. J. 1987, 247, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Gerke, V.; Prenen, J.; Szucs, G.; Heinke, S.; Weber, K.; Droogmans, G. Annexin II modulates volume-activated chloride currents in vascular endothelial cells. J. Biol. Chem. 1996, 271, 30631–30636. [Google Scholar] [CrossRef] [Green Version]
- Borthwick, L.A.; McGaw, J.; Conner, G.; Taylor, C.J.; Gerke, V.; Mehta, A.; Robson, L.; Muimo, R. The formation of the cAMP/protein kinase A-dependent annexin 2-S100A10 complex with cystic fibrosis conductance regulator protein (CFTR) regulates CFTR channel function. Mol. Biol. Cell 2007, 18, 3388–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borthwick, L.A.; Riemen, C.; Goddard, C.; Colledge, W.H.; Mehta, A.; Gerke, V.; Muimo, R. Defective formation of PKA/CnA-dependent annexin 2-S100A10/CFTR complex in deltaF508 cystic fibrosis cells. Cell. Signal. 2008, 20, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Muimo, R. Regulation of CFTR function by annexin A2-S100A10 complex in health and disease. Gen. Physiol. Biophys. 2009, 28, F14–F19. [Google Scholar] [PubMed]
- Cahalan, M.D.; Lewis, R.S. Role of potassium and chloride channels in volume regulation by T lymphocytes. Soc. Gen. Physiol. Ser. 1988, 43, 281–301. [Google Scholar]
- Schlichter, L.C.; Grygorczyk, R.; Pahapill, P.A.; Grygorczyk, C. A large, multiple-conductance chloride channel in normal human T lymphocytes. Pflug. Arch. 1990, 416, 413–421. [Google Scholar] [CrossRef]
- Hurnak, O.; Zachar, J. Maxi chloride channels in L6 myoblasts. Gen. Physiol. Biophys. 1992, 11, 389–400. [Google Scholar]
- Mitchell, C.H.; Wang, L.; Jacob, T.J. A large-conductance chloride channel in pigmented ciliary epithelial cells activated by GTPgammaS. J. Membr. Biol. 1997, 158, 167–175. [Google Scholar] [CrossRef]
- Bernucci, L.; Umana, F.; Llanos, P.; Riquelme, G. Large chloride channel from pre-eclamptic human placenta. Placenta 2003, 24, 895–903. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Kim, S.U. Ion channels in cultured adult human Schwann cells. Glia 1991, 4, 534–539. [Google Scholar] [CrossRef]
- Quasthoff, S.; Strupp, M.; Grafe, P. High conductance anion channel in Schwann cell vesicles from rat spinal roots. Glia 1992, 5, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.J.; Nordeen, M.H.; Howell, K.E.; Caldwell, J.H. A large-conductance anion channel of the Golgi complex. Biophys. J. 2002, 83, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Do, C.W.; Peterson-Yantorno, K.; Mitchell, C.H.; Civan, M.M. cAMP-activated maxi-Cl(−) channels in native bovine pigmented ciliary epithelial cells. Am. J. Physiol. Cell Physiol. 2004, 287, C1003–C1011. [Google Scholar] [CrossRef] [Green Version]
- Do, C.W.; Civan, M.M. Swelling-activated chloride channels in aqueous humour formation: On the one side and the other. Acta Physiol. 2006, 187, 345–352. [Google Scholar] [CrossRef]
- Becq, F.; Fanjul, M.; Mahieu, I.; Berger, Z.; Gola, M.; Hollande, E. Anion channels in a human pancreatic cancer cell line (Capan-1) of ductal origin. Pflug. Arch. 1992, 420, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.T.; Cook, D.I.; Gage, P.W.; Young, J.A. Voltage sensitive, high-conductance chloride channels in the luminal membrane of cultured pulmonary alveolar (type II) cells. Pflug. Arch. 1985, 404, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Vallejos, C.; Riquelme, G. The maxi-chloride channel in human syncytiotrophoblast: A pathway for taurine efflux in placental volume regulation? Placenta 2007, 28, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Stea, A.; Nurse, C.A. Chloride channels in cultured glomus cells of the rat carotid body. Am. J. Physiol. 1989, 257, C174–C181. [Google Scholar] [CrossRef]
- Khakh, B.S.; North, R.A. Neuromodulation by extracellular ATP and P2X receptors in the CNS. Neuron 2012, 76, 51–69. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Pelleg, A. Cardiac purinergic signalling in health and disease. Purinergic Signal. 2015, 11, 1–46. [Google Scholar] [CrossRef]
- Liu, H.T.; Toychiev, A.H.; Takahashi, N.; Sabirov, R.Z.; Okada, Y. Maxi-anion channel as a candidate pathway for osmosensitive ATP release from mouse astrocytes in primary culture. Cell Res. 2008. [Google Scholar] [CrossRef]
- Matsuura, H.; Kojima, A.; Fukushima, Y.; Xie, Y.; Mi, X.; Sabirov, R.Z.; Okada, Y. Positive inotropic effects of ATP released via the maxi-anion channel in Langendorff-perfused mouse hearts subjected to ischemia-reperfusion. Front. Cell Dev. Biol. 2021, 9, 597997. [Google Scholar] [CrossRef]
- Ternovsky, V.I.; Okada, Y.; Sabirov, R.Z. Sizing the pore of the volume-sensitive anion channel by differential polymer partitioning. FEBS Lett. 2004, 576, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, T.; Nakamura, Y.; Umeno, J. Recent advances in studies of SLCO2A1 as a key regulator of the delivery of prostaglandins to their sites of action. Pharmacol. Ther. 2021, 223, 107803. [Google Scholar] [CrossRef] [PubMed]
- Umeno, J.; Hisamatsu, T.; Esaki, M.; Hirano, A.; Kubokura, N.; Asano, K.; Kochi, S.; Yanai, S.; Fuyuno, Y.; Shimamura, K.; et al. A hereditary enteropathy caused by mutations in the SLCO2A1 gene, encoding a prostaglandin transporter. PLoS Genet. 2015, 11, e1005581. [Google Scholar] [CrossRef]
- Hosoe, N.; Ohmiya, N.; Hirai, F.; Umeno, J.; Esaki, M.; Yamagami, H.; Onodera, K.; Bamba, S.; Imaeda, H.; Yanai, S.; et al. Chronic enteropathy associated with SLCO2A1 gene [CEAS]-characterisation of an enteric disorder to be considered in the differential diagnosis of Crohn’s disease. J. Crohns. Colitis. 2017, 11, 1277–1281. [Google Scholar] [CrossRef]
- Tsuzuki, Y.; Aoyagi, R.; Miyaguchi, K.; Ashitani, K.; Ohgo, H.; Yamaoka, M.; Ishizawa, K.; Kayano, H.; Hisamatsu, T.; Umeno, J.; et al. Chronic enteropathy associated with SLCO2A1 with pachydermoperiostosis. Intern. Med. 2020, 59, 3147–3154. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, A.; Wada, Y.; Togo, K.; Mizukami, K.; Fuyuno, Y.; Umeno, J.; Fujioka, S.; Fukuda, K.; Okamoto, K.; Ogawa, R.; et al. Characteristic facial appearance was the key to diagnosing chronic enteropathy associated with SLCO2A1-associated primary hypertrophic osteoarthropathy. Intern. Med. 2020, 59, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Guda, K.; Fink, S.P.; Milne, G.L.; Molyneaux, N.; Ravi, L.; Lewis, S.M.; Dannenberg, A.J.; Montgomery, C.G.; Zhang, S.; Willis, J.; et al. Inactivating mutation in the prostaglandin transporter gene, SLCO2A1, associated with familial digital clubbing, colon neoplasia, and NSAID resistance. Cancer Prev. Res. 2014, 7, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Jimbo, K.; Okuno, T.; Ohgaki, R.; Nishikubo, K.; Kitamura, Y.; Sakurai, Y.; Quan, L.; Shoji, H.; Kanai, Y.; Shimizu, T.; et al. A novel mutation in the SLCO2A1 gene, encoding a prostaglandin transporter, induces chronic enteropathy. PLoS ONE 2020, 15, e0241869. [Google Scholar] [CrossRef]
- Nakanishi, T.; Ohno, Y.; Aotani, R.; Maruyama, S.; Shimada, H.; Kamo, S.; Oshima, H.; Oshima, M.; Schuetz, J.D.; Tamai, I. A novel role for OATP2A1/SLCO2A1 in a murine model of colon cancer. Sci. Rep. 2017, 7, 16567. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.; Pereira, C.; Farinha, M.; Medeiros, R.; Dinis-Ribeiro, M. Prostaglandin E2 pathway is dysregulated in gastric adenocarcinoma in a Caucasian population. Int. J. Mol. Sci. 2020, 21, 7680. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, S.; Ruzzo, W.L. Spatial modeling of prostate cancer metabolic gene expression reveals extensive heterogeneity and selective vulnerabilities. Sci. Rep. 2020, 10, 3490. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Zhu, Q.; Wang, Z.; Shanahan, C.M.; Bensen, J.T.; Fontham, E.T.H.; Smith, G.J.; Pop, E.A.; Azabdaftari, G.; Mohler, J.L.; et al. Differential associations of SLCO transporters with prostate cancer aggressiveness between African Americans and European Americans. Cancer Epidemiol. Biomark. Prev. 2021. [Google Scholar] [CrossRef]
- Cai, C.; Long, Y.; Li, Y.; Huang, M. Coexisting of COX7A2L-ALK, LINC01210-ALK, ATP13A4-ALK and acquired SLCO2A1-ALK in a lung adenocarcinoma with rearrangements loss during the treatment of crizotinib and ceritinib: A case report. Onco. Targets Ther. 2020, 13, 8313–8316. [Google Scholar] [CrossRef]
- Nakata, R.; Nakamura, Y.; Hosomi, S.; Okuda, H.; Nishida, Y.; Sugita, N.; Itani, S.; Nadatani, Y.; Otani, K.; Tanaka, F.; et al. Slco2a1 deficiency exacerbates experimental colitis via inflammasome activation in macrophages: A possible mechanism of chronic enteropathy associated with SLCO2A1 gene. Sci. Rep. 2020, 10, 4883. [Google Scholar] [CrossRef]
- Inagaki, M.; Nishimura, T.; Nakanishi, T.; Shimada, H.; Noguchi, S.; Akanuma, S.I.; Tachikawa, M.; Hosoya, K.I.; Tamai, I.; Nakashima, E.; et al. Contribution of prostaglandin transporter OATP2A1/SLCO2A1 to placenta-to-maternal hormone signaling and labor induction. iScience 2020, 23, 101098. [Google Scholar] [CrossRef]
- Nakanishi, T.; Sakiyama, S.; Takashima, H.; Honda, R.; Shumba, M.N.; Nakamura, Y.; Kasahara, K.; Tamai, I. Toxicological implication of prostaglandin transporter SLCO2A1 inhibition by cigarette smoke in exacerbation of lung inflammation. Toxicol. Appl. Pharmacol. 2020, 405, 115201. [Google Scholar] [CrossRef]
- Norita, K.; Asanuma, K.; Koike, T.; Okata, T.; Fujiya, T.; Abe, Y.; Nakagawa, K.; Hatta, W.; Uno, K.; Nakamura, T.; et al. Impaired mucosal integrity in proximal esophagus is involved in development of proton pump inhibitor-refractory nonerosive reflux disease. Digestion 2020, 1–11. [Google Scholar] [CrossRef]
- Theocharidis, G.; Baltzis, D.; Roustit, M.; Tellechea, A.; Dangwal, S.; Khetani, R.S.; Shu, B.; Zhao, W.; Fu, J.; Bhasin, S.; et al. Integrated skin transcriptomics and serum multiplex assays reveal novel mechanisms of wound healing in diabetic foot ulcers. Diabetes 2020, 69, 2157–2169. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabirov, R.Z.; Islam, M.R.; Okada, T.; Merzlyak, P.G.; Kurbannazarova, R.S.; Tsiferova, N.A.; Okada, Y. The ATP-Releasing Maxi-Cl Channel: Its Identity, Molecular Partners, and Physiological/Pathophysiological Implications. Life 2021, 11, 509. https://doi.org/10.3390/life11060509
Sabirov RZ, Islam MR, Okada T, Merzlyak PG, Kurbannazarova RS, Tsiferova NA, Okada Y. The ATP-Releasing Maxi-Cl Channel: Its Identity, Molecular Partners, and Physiological/Pathophysiological Implications. Life. 2021; 11(6):509. https://doi.org/10.3390/life11060509
Chicago/Turabian StyleSabirov, Ravshan Z., Md. Rafiqul Islam, Toshiaki Okada, Petr G. Merzlyak, Ranokhon S. Kurbannazarova, Nargiza A. Tsiferova, and Yasunobu Okada. 2021. "The ATP-Releasing Maxi-Cl Channel: Its Identity, Molecular Partners, and Physiological/Pathophysiological Implications" Life 11, no. 6: 509. https://doi.org/10.3390/life11060509